Abstract
Bromination of bis(3,4-dimethoxyphenyl)methanone (5) gave four products (6–9) with mono, di, tri, and tetra Br under different conditions. Reduction and demethylation reactions of product 9 with tetra Br were performed, consecutively and a natural product, 5,5′-methylene bis(3,4-dibrombenzene-1,2-diol) (1), was obtained with a 53% yield. Five derivatives, (13–17) (bromophenols), of 1 were also synthesised. The antioxidant and radical scavenging activities of bromophenols 1 and 13–17 were determined by employing various in vitro assays such as 1,1-diphenyl-2-picryl-hydrazyl free radical (DPPH•), 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid (ABTS•+), N,N-dimethyl-p-phenylenediamine dihydrochloride radical cation (DMPD•+), and superoxide anion radical (O2•-) scavenging, reducing ability determination by the Fe3+-Fe2+ and Cu2+-Cu+ cupric reducing antioxidant capacity (CUPRAC) transformation methods, hydrogen peroxide scavenging, and ferrous ion (Fe2+) chelating activities. Moreover, these activities were compared to those of synthetic standard antioxidant compounds such as butylated hydroxyanisole (BHA), butylated hydroxytoluene (BHT), α-tocopherol, and trolox. The results showed that the synthesised bromophenols had effective antioxidant power.
Introduction
Naturally occurring organohalogen compounds are abundantly found in marine life [Citation1]. Bromophenols, frequently isolated from red algae of the family Rhodomelaceae, have some important biological activities [Citation1, Citation2].
2,2′,3,3′-Tetrabromo-4,4′,5,5′-tetrahydroxydiphenylmethane (1) and 3-bromo-4-(2,3-dibromo-4,5-dihydroxybenzyl)-5-methoxymethylpyrocatechol (2) were first isolated in 1977 [Citation3]. Since then they have been obtained from different sources many times [Citation4–10]. Compounds 1 and 2 exhibit important biological activities such as enzyme inhibition, cytotoxicity, feeding deterrent, and anti-microbial activities [Citation4–9]. It was reported that bromophenol 1 extracted from red algae is an inhibitor of protein tyrosine phosphatase, and it can be used to treat and prevent diabetes mellitus and obesity [Citation11–13] ().
Figure 1. Some naturally occurring bromophenols.
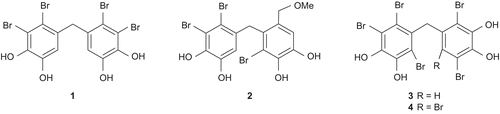
Other diphenylmethane derivatives, 2,2′,3,5′,6-Pentabromo-3′,4,4′,5-tetrahydroxydiphenylmethane (3) and bis(2,3,6-tribromo-4,5-dihydroxyphenyl)methane (4), exhibit significant aldose reductase inhibitory activity [Citation14] (). The bromophenols, 4 and 2-(2,3,6-tribromo-4,5-dihydroxybenzyl)cyclohexanone, exhibit antioxidant activity [Citation15,Citation16]. To our knowledge, the first synthesis of the bromophenol 1 was carried out in low yield (0.3%) by Lundgren et al. [Citation17]. Two synthesis of bromophenol 1 have been published recently [Citation18,Citation19].
Recently, interest in finding antioxidants for foods, cosmetics or medicines has increased considerably. Among the various antioxidants, phenolic compounds are reported to act by quenching oxygen-derived free radicals through donating a hydrogen atom or an electron to the free radical. Many phenolic compounds have been reported to possess potent antioxidant activity and also to have anticarcinogenic, antimutagenic, antibacterial, antiviral, or anti-inflammatory activities. Their physiological and pharmacological functions may originate from their antioxidant properties. The antioxidant activities are related to the structures of phenolic compounds [Citation20,Citation21]. Compound 2 [Citation22] has also been studied. It is thought that the bromophenol 1 will have important biological activities and some of its new derivatives will show useful antioxidant activities so methods of synthesis of bromophenol 1 will be very important. Therefore, the synthesis and antioxidant activities of the bromophenol 1 and some of its derivatives were investigated in the present study.
Materials and methods
All chemicals and solvents are commercially available and were used after distillation or treatment with drying agents. Column chromatography (CC): silica gel (SiO2; 60 mesh, Merck-Darmstadt, Germany). Preparative thick-layer chromatography (PLC): 1 mm of SiO2 60 PF (Merck) on glass plates. Mp: cap. melting-point apparatus (BUCHI 530 - Flawil, Switzerland); uncorrected. IR Spectra: solns. in 0.1 mm cells with a Mattson 1000 FT-IR spectrophotometer (Cambrige, England). 1H- and 13C- NMR spectra: 200 (50) and 400 (100)-MHz Varian spectrometer (Danbury, USA); δ in ppm; Me4Si as the internal standard. Elemental analyses: Leco CHNS-932 apparatus (Michigan, USA). Antioxidant activities of samples was determined in a spectrophotometer (Shimadzu, UV-1208, Japan).
Synthesis
Bis(3,4-dimethoxyphenyl)methanone (5) was obtained from the reaction of 3,4-dimethoxybenzoic acid and veratrole in polyphosphoric acid (PPA) [Citation23]. Reactions of compound 5 under different conditions were investigated for the synthesis of the bromophenol 1 and its new derivatives. Aromatic compounds give selective bromination with ceric ammonium nitrate (CAN)/LiBr [Citation24]. By applying the same procedure, bromination of 5 with 1.1 and 2.2 equivalents of LiBr/CAN gave mono- and dibromides respectively. An AB-system in the aromatic region was present in the 1H-NMR spectrum of monobromide while it was absent in that of dibromide. Two protons (H-5 and H-6) of one aromatic ring in monobrominated 6 should give AB-system because of their being vicinal protons. Mono- and dibromides exhibit seventeen and nine lines respectively, in the 13C-NMR spectra. According to their NMR spectra, the monobromide is unsymmetric while the dibromide is symmetric. Isomeric structures are possible for mono- and dibromides. Their melting points are consistent with that of known bromides [Citation25,Citation26]. Therefore, their structures should be monobromide 6 and dibromide 7 (, ).
Scheme 1. Bromination of compound 5.
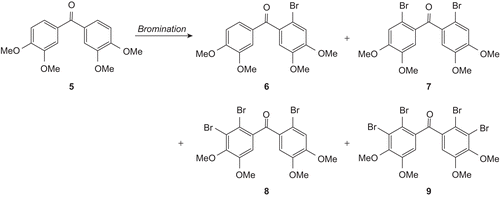
For the synthesis of the tribromide 8 and tetrabromide 9, the reactions of 5 with 3.3 and excess equivalents of LiBr/CAN at room temperature were performed. In these reactions, only the dibromide 7 was obtained. A complex mixture of products was found to be obtained from the reaction of 5 with excess equivalents (such as 7 equivalent) of LiBr/CAN at reflux temperature (in MeCN). Reactions of 5 with molecular bromine in CHCl3 at room and reflux temperatures gave 7, 8, and 9 (, ). The aromatic hydrogen atoms of 8 resonate at 7.15, 7.06, and 6.96 ppm as singlet(s) with relative intensities of 1:1:1, while the aromatic hydrogen atoms of 9 resonate at 7.04 ppm as s. The NMR data of 8 and 9 are consistent with the proposed structures. To make sure of the positions of the aromatic hydrogen atoms of 9, the exact structure of compound 9 was determined by X-ray diffraction analysis ().
Figure 2. (A) The molecular structure of tetrabromide 9 showing the atom numbering scheme. Thermal ellipsoids are drawn at the 40% probability level. (B) packing diagram and H bonding geometry along the a-axis. (symmetry code a: 2-x. 1-y.1-z).
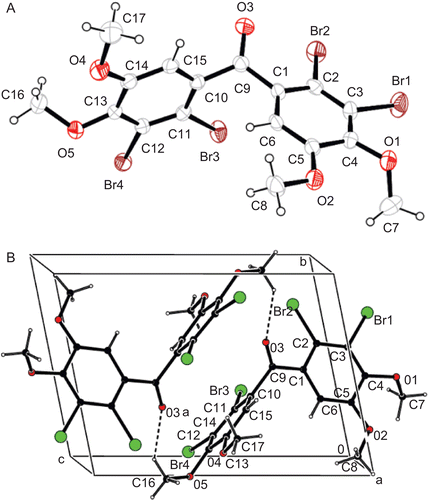
The compound 9 crystallises in the centrosymmetric space group P-1 (no:2) with Z = 2. The C-Br bonds range between 1.880(4)-1.884(4) å. Due to the strong steric hindrance between Br(3), Br(2) and O(3) of the ketone [C(9)-O(3) = 1.207(5) å] atoms, the aromatic rings are extremely twisted along the C(1)-C(9)-C(10) molecular axis. Moreover, methoxy and ketone moieties are joined by two C(16)·O(3) (C(16)-H•..O(3)a = 3.286(3)å, C(16)-H•..O(8)a =131°, symmetry code (a); 2-x, 1-y,1-z) H bonds, which lead to the formation of a centrosymmetric dimer of the molecule in the crystal unit cell ().
Table I. Bromination reactions of the compound 5 (1.0 eq.) at different conditions and % yield of the product(s).
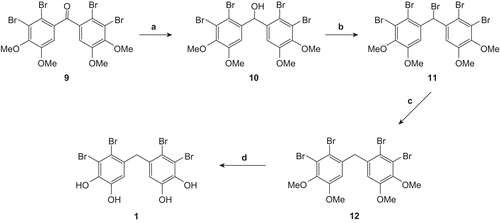
The compound 9 is a precursor of the natural product 1. The reduction of the 9 with NaBH4 in tetrahydrofuran (THF)-MeOH gave the alcohol 10 which was converted to the pentabromide 11 by treatment with PBr3-NEt3. In the synthesis of the natural product 1, selective reduction of benzylic bromide in 11 was the crucial step. Both benzylic and aromatic bromide may be reduced with different reducing agents [27,28]. Therefore, we assumed that (n-Bu)3SnH could be used in the selective reduction of 11 [Citation29]. As expected, the reduction of 11 (1.28 equiv.) with (n-Bu)3SnH (1.00 equiv.) selectively gave compound 12 in high yield and ether cleavage of 12 with BBr3 lead to the natural product 1 (). This work corresponded to published papers of Li et al. and Oh et al.[Citation18, Citation19].
Scheme 2. Reagents and conditions: (a) NaBH4/THF-MeOH. 0°C -RT. 12 h; then dilute HCl. 93%; (b) PBr3/NEt3. benzene. 0°C -RT. 12 h. 93%; (c) n-Bu3SnH. AIBN/THF. reflux.77%; (d) BBr3/CH2Cl2. 0°C -RT. 24 h; then MeOH. 97%.
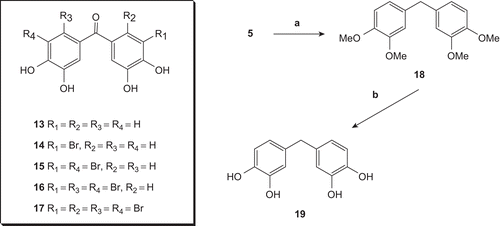
As known, phenol and bromophenols are biologically active compounds [Citation1–13,Citation20,Citation21]. Besides their biological importance, bromophenols are also useful for chemical purposes for instance they are used as precursors in synthetic organic chemistry [Citation30]. Bromophenols 13–17 were synthesised from the corresponding compounds 5–9 by ether cleavage (). Natural products 1 and 13 contain OH-Br and OH-CO, respectively, while the compounds 14–17 contain HO, Br and CO as functional groups. The comparison of the antioxidant properties of compounds 1 and 13–17 with that of phenol without Br and CO will also be important. Consequently, the Wolff-Kishner reduction and demethylation of ketone 5 were performed, consecutively, and known [Citation8,Citation9,Citation31] compound 19 was synthesised via 18 by an alternative method ().
Scheme 3. Reagents and conditions: (a) KOH-NH2NH2/(OHCH2)2. 110-190°C. 6 h. 86%; (b) BBr3. CH2Cl2.96%.
Antioxidant properties
The antioxidant properties of bromophenols 1, 13–17, and 19 were investigated using different antioxidant assays. The Fe3+-Fe2+ reducing power of bromophenols 1, 13–17, and 19 was measured by the direct reduction of Fe3+(CN−)6 to Fe2+(CN−)6, and was determined by measuring absorbance that resulted from the formation of the Perl′s Prussian blue complex following the addition of excess ferric ions (Fe3+). This method is based on the reduction of (Fe3+) ferricyanide in stoichiometric excess relative to the antioxidants [Citation32]. On the other hand, in order to determine the cupric ion (Cu2+) reducing ability of bromophenols 1, 13–17, and 19, the method proposed by Apak et al. was used with slight modification [Citation33]. Ferrous ion (Fe2+) chelating activity of bromophenols 1, 13–17, and 19 was measured by inhibiting the formation of Fe2+-ferrozine complex after treatment of the test material with Fe2+. The Fe2+-chelating ability of bromophenols 1, 13–17, and 19 was monitored using the absorbance of ferrous iron-ferrozine complex at 562 nm [Citation34].
The total radical scavenging capacity of bromophenols 1, 13–17, and 19 was determined and compared to that of BHA (butylated hydroxyanisole), BHT (butylated hydroxytoluene), α-tocopherol, and trolox using the DPPH· (1,1-diphenyl-2-picryl-hydrazyl free radical), ABTS•+ [2,2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid) radical cation], DMPD•+ (N,N-dimethyl-p-phenylenediamine dihydrochloride radical cation), and O2•- (superoxide anion radical) radical scavenging methods. The DPPH· solution, a deep violet colour, and the radical scavenging activity of antioxidant compounds can be measured spectrophotometrically at 517 nm by the loss of the absorbance as the pale yellow non-radical form (DPPH-H) produced. The method previously described by Gülçin [35] was used to assess the DPPH· free radical scavenging capacity of bromophenols 1, 13–17, and 19. The DPPH radical shows absorbance at 517 nm, but its absorption decreases upon reduction by an antioxidant or a radical. When a hydrogen atom or electron was transferred to the odd electron in DPPH·, the absorbance at 517 nm decreased proportional to the increases in the non-radical forms of DPPH [Citation35].
The second test is based on the ability of antiradical molecules to quench the ABTS•+, a blue-green chromophore with characteristic absorption at 734 nm; the addition of antioxidants to the preformed radical cation reduces it to ABTS, determining decolourisation [Citation21]. In this method, an antioxidant is added to a pre-formed ABTS radical solution, and after a period of time the remaining ABTS•+ is quantified spectrophotometrically at 734 nm [Citation21]. The ABTS•+ was produced by reacting 2 mM ABTS in distilled water with 2.45 mM potassium persulphate (K2S2O8) [Citation36].
The superoxide anion radical activity of bromophenols 1, 13–17, and 19 was determined as described by Zhishen and co-workers [Citation37] with slight modification [Citation38]. Superoxide radicals were generated in riboflavin, methionine illuminated, and assayed by the reduction of nitroblue tetrazolium (NBT) to form blue formazan. All solutions were prepared in 0.05 M phosphate buffer (pH 7.8). The photo-induced reactions were performed using fluorescent lamps (20 W) [Citation37].
Finally, antiradical capacity was analysed by DMPD•+ assay. The DMPD radical scavenging ability of bromophenols 1, 13–17, and 19 was determined according to Fogliano et al. [Citation39] with some modification [Citation33]. In the presence of Fe3+, a coloured DMPD radical cation is generated; antioxidant compounds able to transfer a hydrogen atom to DMPD•+ cause a decolouration of the solution measured by the decrease in absorbance at 505 nm [Citation33].
The percentage scavenging effects of bromophenols 1, 13–17, and 19 and standard compounds were calculated using the following equation (1):
where AC is the absorbance of the control and AS is the absorbance in the presence of synthesised bromophenol compounds. The control contains only FeCl2 and ferrozine [Citation34].
Standard procedure for selective bromination of bromophenol derivatives with LiBr/CAN
(2-Bromo-4,5-dimethoxyphenyl)(3,4-dimethoxyphenyl)methanone (6): To a solution of 5 (0.45 g, 1.5 mmol) and LiBr (0.14 g, 1.65 mmol) in CH3CN (6.5 mL) a solution of CAN (0.91 g, 1.65 mmol) in CH3CN (6.5 mL) was added dropwise at room temperature (RT) and under nitrogen (N2) over 15 min. After the solution was stirred at RT and under N2 for 12 hours (h), water (10 mL) was added and the mixture was extracted with ethyl acetate (3 × 30 mL). The organic phase was washed with solutions (5%, 2 × 15 mL) of NaHCO3, water (2 × 15 mL), and brine (15 mL), then dried over Na2SO4 and the solvents evaporated. Monobromide 6 (0.55 g, 95%), the sole product, was crystallised from ethyl acetate/CHCl3 as colourless crystals. Mp 132-134°C (lit. Mp 134°C) [Citation25]. 1H-NMR (CDCl3, 200 MHz): δ 7.52 (d, 4J = 1.9 Hz, 1H); 7.23 (dd, A part of AB-system, 4J = 1.9, 3J = 8.4 Hz, 1H,); 7.04 (s, 1H); 6.84 (s, 1H); 6.82 (d, B part of AB-system, 3J = 8.4 Hz, 1H); 3.89 (s, OCH3, 9H), 3.8 (s, OCH3, 3H). 13C-NMR (CDCl3, 50 MHz): δ 196.2 (CO); 155.9 (C); 152.6 (C); 151.3 (C); 150.2 (C); 134.8 (C); 131.5 (C); 128.2 (CH); 117.8 (CH); 114.1 (CH); 113.4 (CH); 112.6 (C); 112.0 (CH); 58.3 (OCH3); 58.2 (OCH3); 58.2 (OCH3); 58.1 (OCH3). IR (CH2Cl2, cm−1): 3084, 3011, 2945, 2910, 2845, 1663, 1595, 1562, 1510, 1470, 1445, 1377, 1331, 1265, 1206, 1173, 1140, 1061, 1020, 936, 862. Anal. calcd for C17H17BrO5: C 53.54, H 4.46; found: C 53.25, H 4.46%.
Bis(2-bromo-4,5-dimethoxyphenyl)methanone (7): This reaction was performed according to standard procedure described for 6. In this reaction, the moles of 5, CAN and LiBr used were 1.5, 3.3 and 3.3, respectively. The dibromide 7 (0.63 g, 93%) was the sole product and was crystallised from ethyl acetate/hexane as colourless crystals. Mp 175-177°C (lit. Mp (172.5-173.2°C)[Citation18] and (176-177°C) [Citation26]). 1H-NMR (CDCl3, 400 MHz): δ 7.041 (s, 2H); 7.039 (s, 2H); 3.93 (s, OCH3, 6H); 3.86 (s, OCH3, 6H). 13C-NMR (CDCl3, 100 MHz): δ 194.6 (CO); 152.1 (C); 148.5 (C); 131.9 (C); 116.4 (CH); 114.0 (CH); 113.4 (C); 56.6 (OCH3); 56.4 (OCH3). IR (CH2Cl2, cm−1): 3084, 3063, 3003, 2984, 2945, 2918, 2839, 2594, 2039, 1663, 1595, 1562, 1510, 1464, 1437, 1385, 1339, 1265, 1213, 1173, 1146, 1067, 1028, 941. Anal. calcd for C17H16Br2O5: C 44.35, H 3.48; found: C 44.43, H 3.42%. Reaction of 5 with excess equivalents (such as 7 equiv) of LiBr/CAN at room and under reflux gave only the dibromide 7 and a complex mixture of products, respectively.
Reaction of ketone 5 with bromine under different conditions
Bromination by 2.2 equivalent bromine at room temperature: To a stirred solution of ketone 5 (0.5 g, 1.66 mmol) in CHCl3 (25 mL) was added a solution of bromine (0.583 g, 3.64 mmol, 2.2 eq.) in CHCl3 (20 mL) dropwise at RT over 5 min. The mixture was stirred at the same temperature for three days, the solvent evaporated, and chromatography of the residue on silica gel (SiO2, 70 g) with ethyl acetate/hexane (1:9) gave the dibromide 7 (0.595 g, 78%) and monobromide 6 (0.07 g, 11%), respectively.
Bromination by 10 equivalent bromine at room temperature: To a stirred solution of ketone 5 (2 g, 6.6 mmol) in CHCl3 (40 mL) was added a solution of bromine (10.59 g, 66.2 mmol, 10 eq.) in CHCl3 (40 mL) dropwise at RT over 10 min. After the reaction mixture was stirred at RT for 15 days, water (150 mL) and NaHCO3 solution (5%, 150 mL) were added consecutively. The organic phase was separated and the water phase extracted with CHCl3 (2 × 50 mL). The combined organic phase was washed with Na2S2O3 solution (saturated, 180 mL) and brine (180 mL). Then it was dried over Na2S2O4 and the solvent was evaporated. The residue was subjected to column chromatography on silica gel (SiO2, 100g, Merck-Darmstadt, Germany) and eluted using a solvent gradient (5–20% EtOAc in hexane) to give the tribromide 8 (0.14 g, 4%) and tetrabromide 9 (2.85 g, 70%). Both bromides 8 and 9 were crystallised from ethyl acetate/hexane.
(2-Bromo-4,5-dimethoxyphenyl)(2,3-dibromo-4,5-dimethoxyphenyl)methanone (8): Colourless crystals, Mp 158-159°C. 1H-NMR (400 MHz, CDCl3): δ 7.16 (s, 1H); 7.06 (s, 1H); 6.96 (s, 1H); 3.94 (s, OCH3, 3H); 3.91 (s, OCH3, 3H); 3.86 (s, OCH3, 6H). 13C-NMR (100 MHz, CDCl3): δ 194.0 (CO); 152.89 (C); 152.85 (C); 149.7 (C); 148.5 (C); 138.1 (C); 130.1 (C); 123.3 (C); 116.9 (CH); 114.66 (CH); 114.57 (C); 114.54 (C); 113.1 (CH); 61.0 (OCH3); 56.63 (OCH3); 56.62 (OCH3); 56.5 (OCH3). IR (CH2Cl2, cm−1): 3090, 3011, 2970, 2945, 2845, 1668, 1595, 1510, 1464, 1424, 1377, 1339, 1312, 1265, 1206, 1167, 1080, 1047, 1007, 936. Anal. calcd for C17H15Br3O5: C 37.85, H 2.78; found: C 37.88, H 2.80%.
Bis(2,3-dibromo-4,5-dimethoxyphenyl)methanone (9). Colourless crystals, Mp 110-111°C. (lit. Mp 120.7-121.4°C) [Citation18] 1H-NMR (400 MHz, CDCl3): δ 7.03 (s, 2H); 3.91 (s, OCH3, 6H); 3.86 (s, OCH3, 6H). 13C-NMR (100 MHz, CDCl3); δ 194.0 (CO); 152.7 (C); 150.5 (C); 136.6 (C); 123.8 (C); 115.5 (C); 114.1 (CH); 61.0 (OCH3); 56.7 (OCH3). IR (CH2Cl2, cm−1): 3084, 3057, 3011, 2978, 2945, 2845, 2567, 1682, 1576, 1543, 1470, 1424, 1371, 1312, 1265, 1206, 1167, 1100, 1053, 1007, 936. Anal. calcd for C17H14Br4O5: C 33.01, H 2.27; found: C 33.04. H, 2.28%.
Bromination by 5.2 equivalent bromine at reflux temperature. To a stirring solution of ketone 5 (7.1 g, 23.5 mmol) in CHCl3 (50 mL) was added a solution of bromine (6.1 mL, 65.2 eq.) in CHCl3 (40 mL) dropwise at RT over 5 min. Then the reaction mixture was heated and refluxed for 30 h. After the mixture was cooled, the solvent was evaporated. Chromatography of the residue on silica gel (SiO2, 100g, Merck - Darmstadt, Germany) with ethyl acetate/hexane (1:9) gave the dibromide 7 (2.05 g, 19%), tribromide 8 (4.69 g, 37%), and tetrabromide 9 (3.49 g, 24%), consecutively.
Bis(2,3-dibromo-4,5-dimethoxyphenyl)methanol (10):. A solution (60 mL) of tetrabromide 9 (1.99 g, 3.22 mmol) in THF/methanol (1:1) was cooled to 0°C. NaBH4 (0.125 g) was carefully added over 5 min and the resulting mixture was stirred at the same temperature for 15 min. After the cold bath was removed, it was stirred at RT for 1 day. Water (30 mL) was added and then the mixture was acidified with HCl (5%) until its pH was 3. The solvent (methanol and THF) was removed by rotary evaporation and it was extracted with CH2Cl2 (2 × 50 mL). The combined organic phase was dried over Na2SO4 and the solvent was carefully evaporated. The product effervesced while the solvent was removed. Tetrabromo alcohol 10 (1.86 g, 93%) was obtained as a white amorphous solid and crystallised from ethyl acetate/CHCl3. Mp 175-176°C. 1H-NMR (400 MHz, CDCl3): δ 6.88 (s, 2H); 6.33 (bs, H-CO, 1H); 3,85 (s, OCH3, 6H); 3.77 (s, OCH3, 6H); 2.99 (bs, OH, 1H). 13C-NMR (100 MHz, CDCl3): δ 152.8 (C); 147.4 (C); 138.6 (CH); 126.6 (C); 117.3 (C); 111.6 (C); 76.6 (OCH); 60.8 (OCH3); 56.5 (OCH3). IR (CH2Cl2, cm−1): 3400, 2938, 2845, 1583, 1551, 1467, 1421, 1373, 1279, 1189, 1161, 1066, 1037, 1005, 853, 821, 736, 699. Anal. calcd for C17H16Br4O5: C 32.94, H 2.60; found: C 32.77; H 2.61%.
5,5′-(Bromomethylene)bis(3,4-dibromo-1,2-dimethoxybenzene) (11): Tetrabromo alcohol 10 (0.99 g, 1.6 mmol) was dissolved in benzene (25 mL) and NEt3 (0.178 g, 1.76 mmol) was added. After the mixture was cooled to 0°C, a solution of PBr3 (0.48 g, 1.1 eq.) in benzene (10 mL) was added dropwise in the dark over 5 min and the mixture was stirred for 12 h. The mixture was left at RT during the stirring. After the solvent was evaporated, water (40 mL) and ether (50 mL) were added, consecutively. The organic phase was separated and the water phase was extracted with ether (2 × 50 mL). The combined organic phases were washed with HCl (10%, 5 mL), NaHCO3 (saturated, 10 mL), and water (50 mL), dried over Na2SO4, and the solvent was evaporated. Pentabromide 11 (1.02 g, 93%) was obtained and crystallised from CH2Cl2 as white crystals. Mp 148-149°C. 1H-NMR (400 MHz, CDCl3): δ 7.14 (s, 2H); 6.87 (s, H-CBr, 1H); 3.87 (s, OCH3, 6H); 3.82 (s, OCH3, 6H).13C-NMR (100 MHz, CDCl3): δ 152.7 (C); 148.0 (C); 136.2 (C); 122.9 (C); 117.6 (C); 114.2 (CH); 60.9 (OCH3); 56.7 (CHBr); 56.5 (OCH3). IR (CH2Cl2, cm−1): 3003, 2967, 2936, 2844, 1583, 1546, 1467, 1421, 1373, 1321, 1295, 1275, 1261, 1198, 1160, 1056, 1005, 989, 826, 737. Anal. calcd for C17H15Br5O4: C 29.90, H 2.21; found: C 29.93, H 2.21%.
Bis(2,3-dibromo-4,5-dimethoxyphenyl)methane (12): To a solution in THF (80 mL) of the pentabromide 11 (1 g, 1.46 mmol) was added a solution of n-Bu3SnH (0.33 g, 1.14 mmol) in THF (15 mL) at RT. After the solution was heated and refluxed for 15 min, azobisisobutyronitrile (AIBN) (0.05 g, Merck-Schuchardt) was added and refluxed for 3 days. The mixture was cooled to RT and the solvent was evaporated. The reaction mixture was subjected to column chromatography on silica gel (SiO2, 100g, Merck-Darmstadt, Germany) and eluted with EtOAc/hexane (1:10). Tetrabromomethane 12 (0.68 g, 77%) and unreacted pentabromide 11 (0.16 g, 0.23 mmol) were isolated, consecutively. It was crystallised from CH2Cl2 as white crystals. Mp 150-151°C (lit. Mp (159-160°C) [Citation3] and (149.9-150.2°C) [Citation18]). 1H-NMR (400 MHz, CDCl3): δ 6.58 (s, 2H); 4.24 (s, CH2, 2H); 3.85 (s, OCH3, 6H); 3.74 (s, OCH3, 6H). 13C-NMR (100 MHz, CDCl3): δ 152.8 (C); 146.7 (C); 136.2 (C); 122.3 (C); 118.1 (C); 113.6 (CH); 60.8 (OCH3); 56.5 (OCH3); 45.5 (CH2). Anal. calcd for C17H16Br4O4: C 33.81, H, 2.67; found: C 33.75, H 2.67%.
Standard procedure for demethylation of compounds with OMe by ether cleavage synthesis of 5,5′-methylene bis(3,4-dibromobenzene-1,2-diol) (1)
A solution of tetrabromomethane 12 (0.76 g, 1.25 mmol) in CH2Cl2 (12 mL) was cooled to 0°C and then a solution of BBr3 (0.8 mL, 8.3 mmol) in CH2Cl2 (8.5 mL) was added dropwise under N2 (g) over 5 min. After the cold bath was removed, the mixture was stirred at RT and under N2 for one day. Methanol (30 mL) was slowly added over 15 min and then the solvent was evaporated. After water (40 mL) and EtOAc (50 mL) were added, the mixture was shaken. The organic phase was separated and the water phase was extracted with EtOAc (2 × 30 mL). The combined organic phases were dried over Na2SO4 and the solvent was evaporated. The natural product tetrabromotetrol 1 (0.7 g, 97%) was obtained and crystallised from Et2O as pale orange crystals. Mp 204-206°C (lit. Mp (196-199°C) [Citation17]. and (199.0-199.8 °C) [Citation18]). 1H-NMR (200 MHz, CD3COCD3): δ 8.95 (s, OH, 2H); 8.34 (s, OH, 2H); 6.57 (s, 2H); 4.05 (s, CH2, 2H). 13C-NMR (50 MHz, CD3COCD3): δ 147.6 (C); 145.9 (C); 134.4 (C); 118.7 (C and CH); 115.9 (C); 46.8 (CH2). Anal. calcd for C13H8Br4O4: C 28.5, H 1.47. found: C 28.5; H 1.46%.
Bis (3,4-dimethoxyphenyl)methane (18): To a solution of the methanone 5 (0.604 g, 2 mmol) in OHCH2CH2OH (4.5 mL) were added KOH (0.452 g) and hydrazine hydrate (1.1 mL), consecutively, at RT and under N2(g). After the mixture was heated to 110°C and stirred at this temperature for 1 h, it was then stirred at 190°C for 6 h. The mixture was cooled to RT, acidified with HCl (10%), and exracted with EtOAc (2 × 30 mL). The combined organic phases were dried over Na2SO4 and the solvent was evaporated. The residue was filtrated from small silica gel column (SiO2, 100g, Merck-Darmstadt, Germany) and 18 (0.50 g, 86%) was obtained. Mp 63-65°C (lit. Mp (68-69°C) [Citation23]). 1H-NMR (400 MHz, CDCl3): δ 6.8 (d, 3J = 8.07 Hz, 2H); 6.71 (dd, 3J = 8.07, 4J = 1.47 Hz, 2H); 6.7 (bs, 2H); 3.88 (s, CH2, 2H); 3.85 (s, OCH3, 3H); 3.82 (s, OCH3, 3H). 13C-NMR (100 MHz, CDCl3): δ 149.2 (C); 147.6 (C); 134.2 (C); 121 (CH); 112.4 (CH); 111.5 (CH); 56.1 (OCH3); 56 (OCH3); 41.2 (OCH3).
Synthesis of phenols 13–17 and 19 from 5-9 and 18: The standard procedure described above for the synthesis of 1 with BBr3 was applied to 5-9 and 18. From these reactions, phenols 13–17 and 19 were obtained.
Bis (2,3-dihydroxyphenyl) methanone (13): This was obtained and crystallised from ether as pale yellow crystals (98%); Mp 231-233°C (lit. for hydrated 13: 230-231°C) [Citation40]. 1H-NMR (400 MHz, CD3COCD3): δ 8.63 (s, 2 OH, 2H); 8.38 (s, 2 OH, 2H); 7.32 (d, J = 1.83 Hz, 2H); 7.2 (dd, 3J =8.06, 4J = 1.83 Hz, 2H); 6.92 (d, 3J = 8.06 Hz, 2H); 13C-NMR (100 MHz, CD3COCD3): δ 193.4 (CO); 149.5 (C); 144.9 (C); 130.7 (C); 123.5 (CH); 117.1 (CH); 114.7 (CH).
2-Bromo-4,5-dihydroxyphenyl)(3,4- dihydroxyphenyl) methanone(14): This was obtained as amorphous (95%). Mp 67-69°C. 1H-NMR (200 MHz, CD3COCD3): δ 8.95-8.2 (m, OH, 4H); 7.33 (d, 4J = 2.1 Hz, 1H); 7.17 (dd, A part of AB-system, 3J = 8.3, 3J = 2.1 Hz, 1H); 7.11 (s, 1H); 6.01 (d, B part of AB-system, 3J = 8.3 Hz, 1H); 6.86 (s, 1H). 13C-NMR (50 MHz, CD3COCD3): δ 195.9 (CO); 153.4 (C); 150.1 (C); 147.7 (C); 147.2 (C); 135.5 (C); 132.2 (C); 126.9 (CH); 122.1 (CH); 119.3 (C); 118.8 (CH); 118 (CH); 111.1 (C). IR (CH3OH, cm−1): 3399, 1589, 1505, 1442, 1414, 1369, 1295, 1220, 1187, 1137, 1114, 1056, 1033, 1016, 953, 881, 852, 827, 862, 783, 636. Anal. calcd for C13H9BrO5: C 48.03, H 2.79; found: C 48.04; H 2.80%.
Bis-(2-bromo-4,5-dihydroxyphenyl) methanone(15): This was obtained and crystallised from ether as brown crystals (91%). Mp 217-219°C. 1H-NMR (200 MHz, CD3COCD3): δ 8.93-8.5 (m, OH, 4H); 7.12 (s, 2H); 6.97 (s, 2H). 13C-NMR (50 MHz, CD3COCD3): δ 194.9 (CO); 151.8 (C); 147.1 (C); 134 (C); 123.1 (C); 121 (CH); 113.4 (C). IR (CH3OH, cm−1): 3230, 1589, 1503, 1416, 1358, 1212, 1179, 1038, 862, 795, 634. Anal. calcd for C13H8Br2O5: C 38.65, H 2. found: C 38.64; H 2.02%.
2-Bromo-4,5- dihydroxyphenyl(2,3-dibromo-4,5- dihydroxyphenyl) methanone (16): This was obtained as yellow amorphous solid (85%). Mp 126-128°C. 1H-NMR (200 MHz, CD3COCD3): δ 9.23 (m, OH, 1H); 8.95 (m, OH, 1H); 8.85 (m, OH, 1H); 8.52 (m, OH, 1H); 7.14 (s, 1H); 7 (s, 1H); 6.96 (s, 1H). 13C-NMR (50 MHz, CD3COCD3): δ 195 (CO); 152.2 (C); 149.3 (C); 147.2 (C); 147.1 (C); 136.4 (C); 132.8 (C); 123.6 (CH); 121.7 (CH); 118.6 (CH); 116.9 (C); 115.8 (C); 114.1 (C). IR (CH3OH, cm−1): 3270, 1648, 1589, 1504, 1467, 1394, 1330, 1283, 1216, 1184, 1055, 1012, 863, 774, 737. Anal. calcd for C13H7Br3O5: C 32.33, H 1.46; found: C 32.32; H 1.47%.
Bis-(2,3-dibromo-4,5-dihydroxyphenyl) methanone(17): This was obtained as amorphous solid (88%, brown). Mp 204-206°C. 1H-NMR (200 MHz, CD3COCD3): 9.25 (m, 2 OH, 2H), 9.05 (m, 2 OH, 2H), 7 (s, 2H). 13C-NMR (50 MHz, CD3COCD3): δ 195.0 (CO), 149.9 (C), 146.9 (C), 135.1 (C), 119.3 (CH), 117.4 (C), 116.5 (C). IR (CH3OH, cm−1): 3434, 2844, 2078, 1638, 1474, 1412, 1341, 1282, 1195, 1161, 1051, 1015, 857, 663. Anal. calcd for C13H6Br4O5: C 27.79, H 1.08; found: C 27.8; H 1.12%.
4,4′-Methylenedibenzen-1,2-diol (19): This was obtained as amorphous solid (86%). Mp 141-142°C. 1H-NMR (400 MHz, CD3COCD3): δ 7.66 (s, 2 OH, 2H); 7.59 (s, 2 OH, 2H); 6.72 (d, 3J = 8.04 Hz, 2H); 6.64 (d, 4J = 2.20 Hz, 2H); 6.53 (dd, 3J = 7.13, 4J = 1.85 Hz, 2H). 13C-NMR (100 MHz, CD3COCD3): δ 145.1 (C); 143.4 (C); 134 (C); 120.2 (CH); 116.1 (CH); 115.2 (CH); 40.5 (CH2). Anal. calcd for C13H12O4: C 67.23, H 5.21; found: C 67.45, H 5.2. (The NMR data are consisitent with literature [Citation41]).
X-Ray structure determination for 9
For the crystal structure determination, the single-crystals of tetrabromide 9 were used for data collection on a four-circle Rigaku R-AXIS RAPID-S diffractometer (equipped with a two-dimensional area imaging plate (IP) detector) [42]. The graphite-monochromatised Mo Kα radiation (λ = 0.71073 å) and oscillation scans technique with δω = 5° for one image were used for data collection. The lattice parameters were determined by the least-squares methods on the basis of all reflections with F2>2σ(F2). Integration of the intensities, correction for Lorentz and polarisation effects and cell refinement was performed using CrystalClear (Rigaku/MSC) software [Citation42]. The structures were solved by direct methods using using SHELXS-97 [43] and refined by a full-matrix least-squares procedure using the program SHELXL-97 [Citation43]. H atoms were positioned geometrically and refined using a riding model, fixing the aromatic C–H distances at 0.93 å and methyl C–H distances at 0.96 å [Uiso(H) = 1.2Ueq(C) and Uiso(H) = 1.5Ueq(methyl C)]. The final difference Fourier maps showed no peaks of chemical significance.
Crystal data for 9: C17H14Br4O5, crystal system, space group: triclinic, P-1; (no:2); unit cell dimensions: a = 9.834(4), b = 9.964(4), c = 11.822(5) å, α = 72.95(5) β = 81.6(6), γ = 63.33(5)°; volume: 989.5(3) å3; Z=2; calculated density: 2.07 mg/m3; absorption coefficient: 8.159 mm−1; F(000): 592; θ range for data collection 2.3–30.5°; refinement method: full-matrix least-square on F2; data/parameters: 6005/239; goodness-of-fit on F2: 1.321; final R indices [I>2σ(I)]: R1 = 0.084, wR2 = 0.125; R indices (all data): R1 = 0.153, wR2 = 0.138; largest diff. peak and hole: 0.513 and -0.478 e å−3; CCDC-710557.
Antioxidant assays
Fe3+ reducing power assay: The Fe3+(CN−)6 to Fe2+(CN−)6 reducing power of bromophenol derivatives 1, 13–17, and 19 was measured as described by Oyaizu [Citation44] with slight modification [Citation20]. For this purpose, 10 μg/mL of bromophenol derivatives 1, 13–17, and 19 in 0.75 mL of distilled water were mixed with 1.25 mL of sodium phosphate buffer (0.2 M, pH 6.6) and 1.25 mL of potassium ferricyanide [K3Fe(CN)6] (1%). The mixture was incubated at 50°C for 20 min. After 20 min of incubation, the reaction mixture was acidified with 1.25 mL of trichloroacetic acid (10%). Finally, 0.5 mL of FeCl3 (0.1%) was added to this solution, and the absorbance was measured at 700 nm in a spectrophotometer (UVmini -1240, Shimadadzo, Kyoto/Japan) [45].
Cupric ions (Cu2+) reducing assay: The cupric ion (Cu2+) reducing ability (CUPRAC assay) of bromophenol derivatives 1, 13–17, and 19 was determined according to the method proposed by Apak and co-workers [Citation46]. Briefly, 0.25 mL of CuCl2 solution (0.01 M), 0.25 mL ethanolic neocuproine solution (7.5 × 10−3 M) and 0.25 mL of NH4Ac buffer solution (1 M) were added to a test tube, followed by mixing with 10 μg/mL concentration of bromophenol derivatives 1, 13–17, and 19. Then, total volume was adjusted to 2 mL with distilled water, and mixed well. The tubes were stoppered and kept at room temperature. Absorbance was measured at 450 nm against a reagent blank 30 min later.
Chelating activity on ferrous ions (Fe2+): The ferrous ion (Fe2+) chelating activity was measured by inhibiting the formation of Fe2+-ferrozine complex after treatment of the test material with Fe2+, following the method described by Dinis and co-workers [Citation47]. The ferrous ion (Fe2+) chelating ability of bromophenol derivatives 1, 13–17, and 19 was monitored using the absorbance of the ferrous iron-ferrozine complex at 562 nm. Briefly, 10 μg/mL concentrations of bromophenol derivatives 1, 13–17, and 19 in 0.4 mL of methanol were added to a solution of FeCl2 (0.6 mM, 0.1 mL). The reaction was initiated by the addition of ferrozine (5 mM, 0.1 mL) dissolved in methanol. Then, the mixture was shaken vigorously and left at room temperature for 10 min. Absorbance of the solution was then measured spectrophotometrically at 562 nm [Citation48].
DPPH· scavenging activity: The DPPH radical scavenging activity of bromophenol derivatives 1, 13–17, and 19 was determined as previously described by Gülçin [Citation49]. Briefly, a solution (0.1 mM) of DPPH· was prepared in ethanol and 0.5 mL of this solution was added to 1.5 mL of bromophenol derivatives 1, 13–17, and 19 solutions in ethanol at different concentrations (10–30 μg/mL). These solutions were vortexed thoroughly, and incubated in the dark for 30 min. Another 30 min later, the absorbance was measured at 517 nm against blank samples lacking scavenger. A standard curve was prepared using different concentrations of DPPH·. The DPPH· scavenging capacity was expressed as mM in the reaction medium, and calculated from the calibration curve determined by linear regression (r2: 0.9845):
ABTS•+ scavenging activity: In the ABTS•+ scavenging method, an antioxidant is added to a pre-formed ABTS radical solution, and after a fixed time period, the remaining ABTS•+ is quantified spectrophotometrically at 734 nm [Citation50,Citation51]. The ABTS•+ was produced by reacting ABTS (2 mM) in H2O with potassium persulphate (2.45 mM, K2S2O8), stored in the dark at room temperature for 6 h. Then, 0.5 mL of ABTS•+ solution was added to 1.5 mL of bromophenol derivatives 1, 13–17, and 19 solution in ethanol at different concentrations (10–30 μg/ mL). The absorbance was recorded 30 min after mixing, and the percentage of radical scavenging was calculated for each concentration relative to a blank, containing no scavenger. The extent of decolourisation is calculated as the percentage reduction in absorbance [Citation52,Citation53]. For preparation of a standard curve, different concentrations of ABTS•+ (0.033-0.33 mM) were used. The ABTS•+ concentration (mM) in the reaction medium was calculated from the following calibration curve, determined by linear regression (r2: 0.9899):
DMPD•+ scavenging activity: The DMPD•+ scavenging ability of bromophenol derivatives 1, 13–17, and 19 was determined according to Fogliano et al. [Citation39]. DMPD (100 mM) was prepared by dissolving 209 mg of DMPD in 10 mL of deionised water, and 1 mL of this solution was added to 100 mL of 0.1 M acetate buffer (pH 5.25), and the coloured radical cation (DMPD•+) was obtained by adding 0.2 mL of a solution of 0.05 M ferric chloride (FeCl3). Different concentrations of bromophenol derivatives 1, 13–17, and 19 (10-30 μg/mL) were added to test tubes, and the total volumes were adjusted to 0.5 mL with distilled water. The absorbance was measured at 505 nm ten minutes later. A one millilitre aliquot of the DMPD•+ solution was directly added to the reaction mixture, and its absorbance was measured at 505 nm. The buffer solution was used as a blank sample. The DMPD•+ concentration (mM) in the reaction medium was calculated from the following calibration curve, determined by linear regression (r2: 0.9993):
Discussion
In the present study, we demonstrated the antioxidant and radical scavenging effects of bromophenols 1, 13–17, and 19 using different in vitro bioanalytical methodologies. Antioxidant capacity is widely used as a parameter for medicinal bioactive components. The antioxidant and radical scavenging activities of bromophenols 1, 13–17, and 19 were compared to those of BHA, BHT, α-tocopherol, and its water-soluble analogue trolox. These comparisons were performed using a series of in vitro tests including DPPH•, ABTS•+, DMPD•+ and O2•- radicals scavenging activities, and reducing power by two methods (Fe3+-Fe2+ transformation and CUPRAC assays), hydrogen peroxide scavenging and metal chelating on ferrous ion (Fe3+) activities.
The reducing power, reflects the electron donating capacity of bioactive compounds and is associated with antioxidant activity. Antioxidants can be reductants and inactivators of oxidants. The reducing capacity of a compound can be measured by the direct reduction of Fe[(CN)6]3 to Fe[(CN)6]2. Addition of free Fe3+ to the reduced product leads to the formation of the intense Perl′s Prussian blue complex Fe4[Fe(CN)6]3, which has strong absorbance at 700 nm. As can be seen from Table II, all of the bromophenols 1, 13–17, and 19 have powerful Fe3+ reducing ability. Similarly, the bromophenols showed marked cupric ion (Cu2+) reducing ability.
Ferrous ions are the most effective pro-oxidants in food systems; the chelating effect would be beneficial and removal of free iron ions from circulation could be a promising approach to prevent oxidative stress-induced diseases. When an iron ion is chelated, it may lose pro-oxidant properties. Iron, in nature, can be found as either ferrous (Fe2+) or ferric ions (Fe3+), with the latter form predominant in foods. Ferrous chelation may render important antioxidative effects by retarding metal-catalysed oxidation. Ferrous ion chelating activities of bromophenol derivatives 1, 13–17, and 19, BHA, BHT, α-tocopherol, and Trolox are shown in . This shows that the most powerful metal chelating properties were found in 15. In addition, the other bromophenols have marked Fe2+ chelating activity.
Table II. Comparison of ferric ions (Fe3+) reducing ability by Fe3+-Fe2+ transformation methods, Cu+ reducing ability by CUPRAC method, ferrous ion (Fe2+) chelating activity of bromophenol derivatives (1, 13–17 and 19) and standard antioxidant compounds such as BHA, BHT, α-tocopherol and trolox at the same concentration (10 μg/mL).
The free radical chain reaction is widely accepted as a common mechanism of lipid peroxidation. Radical scavengers may directly react with and quench peroxide radicals to terminate the peroxidation chain reactions, and improve the quality and stability of food products. Assays based upon the use of DPPH·, ABTS•+, DMPD•+, and O2•- radicals are among the most popular spectrophotometric methods for determination of the antioxidant capacity of molecules. These radicals can directly react with antioxidants. Additionally, these radical scavenging assays have been used to evaluate the antioxidant activity of compounds due to the simple, rapid, sensitive, and reproducible procedures. These tests are standard assays in antioxidant activity studies, and offer a rapid technique for screening the radical scavenging activity of specific compounds. As can be seen in , DPPH·, ABTS•+, DMPD•+, and O2•- radical scavenging activities of bromophenol derivatives were evaluated. In particular, the IC50 values of the bromophenol derivatives used in the DPPH·, ABTS•+, and O2•- assays are lower than those of standard antioxidant compounds.
Table III. Concentration required for 50% scavenging (IC50) of DPPH·. ABTS·+. DMPD·+ and O2·- radical scavenging activities of bromophenol derivatives (1. 13–17 and 19) and standard antioxidant compounds such as BHA. BHT. α-tocopherol and trolox (DPPH·: 1.1-diphenyl-2-picryl-hydrazyl free radical. ABTS·+: 2.2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid. DMPD·+: N.N-dimethyl-p-phenylenediamine dihydrochloride radical. O2·-: superoxide anion radicals.
Phenol derivatives 1, 13–17, and 19 showed high antioxidant activities. In most of the phenol derivatives, CO and Br are present as functional groups. Antioxidant activities of the phenols with CO are higher than those of the others. Among phenols with Br and without Br, these activities are almost the same, while they are similar in phenols that include different numbers of Br.
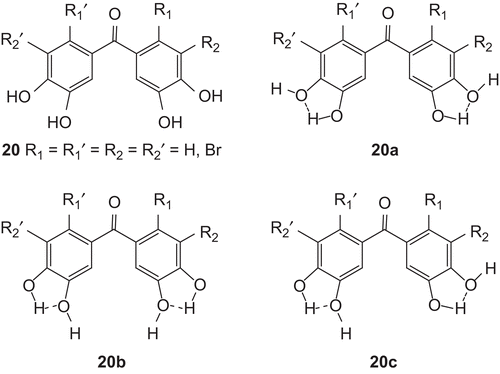
The reason for the higher antioxidant activities in the phenols with CO may be the CO group itself. Phenol derivatives have two OH groups on each ring as ortho or 3,4 and 3′,4′. As shown in , OH groups in one ring of the molecule may create intramolecular hydrogen bonds to each other.
Figure 3. Structures of hydrogen bonds between OH groups in the one ring of phenol 20.
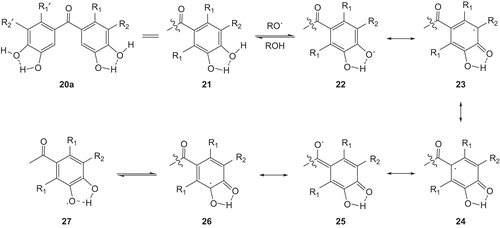
An antioxidant compound is an inhibitor used to control auto-oxidation. For inhibitory effects of these phenol derivatives, the following mechanism may be proposed (). From the structures of phenol 20, 20a may be taken and abbreviated as 21. The reaction of 21 with a radical (RO.) can give 22, which converts to structures 23-26 by resonance and 27 by equilibrium. The structure 25 is an enol radical of a radical with CO. Ten resonance structures may be given for 20 in total because five resonance structures, 22-26, are written for one ring of 20. Therefore, phenols with CO will be better inhibitors than those without CO.
Scheme 4. Stabilization of radicals by phenol 20.
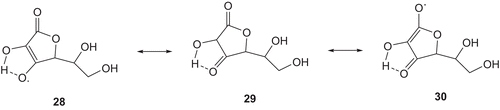
Ascorbic acid (vitamin C) is also a natural antioxidant and, like it forms a resonance-stabilised radical () [Citation54]. The structure 30 is an enol structure of ester CO. It is reported that CO groups [Citation55] stabilise radicals and ketone [Citation56,Citation57] is formed in the reduction of aryl ketone.
Scheme 5. Stabilization of radicals by ascorbic acid (vitamin C).
Declaration of interest
The authors are indebted to Atatürk University and TüBİTAK (107T348) for financial support and to Cavit Kazaz, Barış Anıl and Fevzi Topal for technical assistance.
Related Research Data
References
- Gribble GW. Naturally-occurring organohalogen compounds - a survey. J Nat Prod 1992;55:1353–1395.
- Gribble GW. The diversity of naturally occurring organobromine compounds. Chem Soc Rev 1999;28:335–346.
- Kurata K, Amiya T. 2 New bromophenols from red alga, Rhodomela larix. Chem Lett 1977;12:1435–1438.
- Lee HS, Lee TH, Lee JH, Chae CS, Chung SC, Shin DS, Shin J, Oh KB. Inhibition of the pathogenicity of Magnaporthe grisea by bromophenols, isocitrate lyase inhibitors, from the red alga Odonthalia corymbifera. J Agric Food Chem 2007;55:6923–6928.
- Xu X, Song F, Wang S, Li S, Xiao F, Zhao J, Yang Y, Shang S, Yang L, Shi J. Dibenzyl bromophenols with diverse dimerization patterns from the brown alga Leathesia nana. J Nat Prod 2004;67:1661–1666.
- Yuan Z, Han L, Fan X, Li S, Shi D, Sun J, Yang Y, Shi J. Cytotoxic constituents from red alga Gymnogongrus flabelliformis. Zhongcaoyao 2007;38:652–655.
- Kurata K, Taniguchii K, Takashima K, Hayashi I, Suzuki M. Feeding-deterrent bromophenols from Odonthalia corymbifera. Phytochemistry 1997;45:485–487.
- Oh KB, Lee JH, Chung SC, Shin J, Shin HJ, Kim HK, Lee HS. Antimicrobial activities of the bromophenols from the red alga Odonthalia corymbifera and some synthetic derivatives. Bioorg Med Chem Lett 2008;18:104–108.
- Xu N, Fan X, Yan X, Li X, Niu R, Tseng CK. Antibacterial bromophenols from the marine red alga Rhodomela confervoides. Phytochemistry 2003;62:1221–1224.
- Fan X, Xu NJ, Shi JG. Bromophenols from the red alga Rhodomela confervoides. J Nat Prod 2003;66:455–458.
- Fan X, Ma C, Han L, Shi D, Liu Q. Extraction of bromophenol compounds from red algae and their uses in the treatment of diabetes and obesity. CN Patent 1,853,618 2006; Chem Abstr 2007;146:13018v.
- Shi D, Fan X, Han L, Xu F, Yuan Z. Application of oceanic bromphenol compound to prepare the medical preparations for preventing and treating malignant neoplasm. CN Patent 10,015,299, 2007.
- Shi DY, Xu F, He J, Li J, Fan X, Han LJ. Inhibition of bromophenols against PTP1B and anti-hyperglycemic effect of Rhodomela confervoides extract in diabetic rats. Chinese Sci Bull 2008;53:2476–2479.
- Wang W, Okada Y, Shi H, Wang Y, Okuyama T. Structures and aldose reductase inhibitory effects of bromophenols from the red alga Symphyocladia latiuscula. J Nat Prod 2005;68:620–622.
- Duan XJ, Li XM, Wang BG. Highly brominated mono- and bis-phenols from the marine red alga Symphyocladia latiuscula with radical-scavenging activity. J Nat Prod 2007;70:1210–1213.
- Choi JS, Park HJ, Jung HA, Chung HY, Jung JH, Choi WC. A cyclohexanonyl bromophenol from the red alga Symphyocladia latiuscula. J Nat Prod 2000;63:1705–1706.
- Lundgren L, Olsson K, Theander O. Synthesis of some bromophenols present in red algae. Acta Chem Scand B 1979;33:105–108.
- Li J, Guo SJ, Su H, Han LJ, Shi DY. Total synthesis of bis-(2,3-dibromo-4,5-dihydroxyphenyl)-methane as potent PTP1B inhibitor. Chin Chem Lett 2008;19:1290–1292.
- Oh KB, Lee JH, Lee JW, Yoon KM, Chung SC, Jeon HB, Shin J, Lee HS. Synthesis and antimicrobial activities of halogenated bis(hydroxyphenyl)methanes. Bioorg Med Chem Lett 2009;19:945–948.
- Gülçin İ. Comparison of in vitro antioxidant and antiradical activities of L-tyrosine and L-dopa. Amino Acids 2007;32:431–438.
- Gülçin İ, Elias R, Gepdiremen A, Boyer L. Antioxidant activity of lignans from fringe tree (Chionanthus virginicus L). Eur Food Res Technol 2006;223:759–765.
- Akbaba Y, Balaydın HT, Göksu S, şahin E, Menzek A. The first total synthesis of the biologically active, naturally occurring 3,4-dibromo-5-[2-bromo-3,4-dihydroxy-6-(methoxymethyl)benzyl]benzene-1,2-diol: an equivalent of regioselective O-demethylation in arylmethylethers. Helv Chim Acta 2010; DOI: 10.1002/hlca.200900300 (In press).
- Harig M, Neumann B, Stammler HG, Kuck D. 2,3,6,7,10,11-hexamethoxytribenzotriquinacene: Synthesis, solid-state structure, and functionalization of a rigid analogue of cyclotriveratrylene. Eur J Org Chem 2004;11:2381–2397.
- Roy SC, Guin C, Rana KK, Maiti G. An efficient chemo and regioselective oxidative nuclear bromination of activated aromatic compounds using lithium bromide and ceric ammonium nitrate. Tetrahedron Lett 2001;42: 6941–6942.
- Aboulezz AF, Quelet R. Derivatives of veratrole. Synthesis of homoveratric acid and some substituted diphenylmethane and fluorenone derivatives. J Chem UAR 1962;5:137–146.
- Garofano T, Oliverio A. The influence of o-substituents on the reactivity of aryl ketones. I. Influence of o-substituents on the reactivity of benzophenone. Ann Chim-Rome 1957;47:260–284.
- Goksu S, Secen H. Concise syntheses of 2-aminoindans via indan-2-ol. Tetrahedron 2005;61:6801–6807.
- Prashad M, Hu B, Har D, Repic O, Blacklock TJ, Acemoglu M. Efficient and practical syntheses of (R)-(5-amino-2,3-dihydro-1H-inden-2-yl)-carbamic acid methyl ester. Adv Synth Catal 2001;343:461–472.
- Menzek A, Gökmen M. Synthesis and rearrangement reactions of dihydrobenzhomobarrelene derivatives:influence of double bond on product distribution. J Chem Res Synop 2002;475–476.
- Kelebekli L. Regio- and stereospecific synthesis of a new benzoconduritol-C derivative. J Chem Res 2008;104–106.
- Hickey MJ, Coutts IGC, Tsang TLL, Pogson CI. Novel inhibitors of 3-phosphoglycerate kinase. Biochem Soc T 1995;23:S607–S607.
- Ak T, Gülçin İ . Antioxidant and radical scavenging properties of curcumin. Chem Biol Int 2008;174:27–37.
- Gülçin İ . Measurement of antioxidant ability of melatonin and serotonin by the DMPD and CUPRAC methods as trolox equivalent. J Enzym Inhib Med Chem 2008;23:871–876.
- Gülçin İ . Antioxidant and antiradical activities of L-Carnitine. Life Sci 2006;78:803–811.
- Gülçin İ . Antioxidant activity of caffeic acid (3,4-dihydroxycinnamic acid). Toxicology 2006;217:213–220.
- Gülçin İ Tel, AZ, Kirecci E. Antioxidant, antimicrobial, antifungal, and antiradical activities of Cyclotrichium niveum (Boiss) Manden and Scheng. Int J Food Propert 2008;11:450–471.
- Zhishen J, Mengcheng T, Jianming W. The determination of flavonoid contents on mulberry and their scavenging effects on superoxide radical. Food Chem 1999;64:555–559.
- Gülçin İ . Antioxidant activity of L-adrenaline:an activity-structure insight. Chem Biol Interact 2009;179:71–80.
- Fogliano V, Verde V, Randazzo G, Ritieni A. Method for measuring antioxidant activity and its application to monitoring the antioxidant capacity of wines. J Agric Food Chem 1999;47:1035–1040.
- Pearl IA. Reactions of vanillin and its derived compounds. XXIII. The synthesis of 4,4′-dihydroxy-3,3′-dimethoxybenzophenone. J Am Chem Soc 1954;76:3635–3637.
- Cabaton N, Zalko D, Rathahao E, Canlet C, Delous G, Chagnon MC, Cravedi JP, Perdu E. Biotransformation of bisphenol F by human and rat liver subcellular fractions. Toxicology in Vitro 2008;22:1697–1704.
- Rigaku/MSC, 9009 new Trails Drive, The Woodlands, TX 77381.
- Sheldrick GM. SHELXS97 and SHELXL97. Crystal structure solution and refinement programs. 1997.
- Oyaizu M. Studies on product of browning reaction prepared from glucoseamine. Jpn J Nut 1986;44:307–315.
- Gülçin İ Mshvildadze, V, Gepdiremen A, Elias R. Screening of antioxidant and antiradical activity of monodesmosides and crude extract from Leontice smirnowii tuber. Phytomedicine 2006;13:343–351.
- Apak R, Güçlü K, özyürek M, Karademir SE, Erça E. The cupric ion reducing antioxidant capacity and polyphenolic content of some herbal teas. Int J Food Sci Nut 2006;57:292–304.
- Dinis TCP, Madeira VMC, Almeida LM. Action of phenolic derivates (acetoaminophen, salycilate, and 5-aminosalycilate) as inhibitors of membrane lipid peroxidation and as peroxyl radical scavengers. Arch Biochem Biophys 1994;315:161–169.
- Gülçin İ, Daştan A. Synthesis of dimeric phenol derivatives and determination of in vitro antioxidant and radical scavenging activities. J Enzym Inhib Med Chem 2007;22:685–695.
- Gülçin İ. The antioxidant and radical scavenging activities of black pepper (Piper nigrum) seeds. Int J Food Sci Nut 2005;56:491–499.
- Gülçin İ,R, Elias, Gepdiremen A, Taoubi K, Köksal E. Antioxidant secoiridoids from fringe tree (Chionanthus virginicus L.). Wood Sci Technol 2009;43:195–212.
- Köksal E, Gülçin İ öztürk Sarinodot;kaya, SB, Bursal E. On the in vitro antioxidant activity of silymarin. J Enzym Inhib Med Chem 2009;24:395–405.
- Gülçin İ. Antioxidant properties of resveratrol: A structure-activity insight. Innovat Food Sci Emerg Techn 2010;11:210–218.
- Talaz O, Gülçin İ, Göksu S, Saracoglu N. Antioxidant activity of 5,10-dihydroindeno[1,2-b]indoles containing substituents on dihydroindeno part. Bioorg Med Chem 2009;17:6583–6589.
- Fessenden RJ, Fessenden JS. Organic Chemistry, 5th ed. Pacific. Grove, California, USA, Brooks/Cole Publishing Company, 1990;256–266.
- Akbulut N, Menzek A, Balci M. Termolysis and CoTPP-catalyzed rearrangement of endoperoxide derived from 2,3-dihydro-1(2H)-azulenone:a new endoperoxide-endoperoxide rearrangement. Tetrahedron Lett 1987;28:1689–1692.
- Narisada M, Watanabe F. Reductive alkylation of monoaromatic ketones. J Org Chem 1973;38:3887–3892.
- Menzek A, Karakaya MG, Kaya AA. Reductions of benzene derivatives whose benzylic positions bear oxygen atoms under mild conditions. Helv Chim Acta 2008;91:2299–2307.