Abstract
The binding and inhibition strength of a series of benzimidazo[1,2-c][1,2,3]thiadiazole-7-sulphonamides were determined for recombinant human carbonic anhydrase isoforms I, II, and IX. The inhibition strength was determined by a stop-flow method to measure carbon dioxide hydration. Inhibitor-enzyme binding was determined by two biophysical techniques – isothermal titration calorimetry and thermal shift assay. The co-crystal structure was determined by X-ray crystallography. Comparing the results obtained using three different inhibition and binding methods increased the accuracy of compound affinity ranking and the ability to determine compound inhibitory specificity towards a particular carbonic anhydrase isoform. In most cases, all three methods yielded the same results despite using very different approaches to measure the binding and inhibition reactions. Some of the compounds studied are submicromolar inhibitors of the isoform IX, a prominent cancer target.
Introduction
An important task in the drug design for many diseases is the inhibition of selected carbonic anhydrase (CA) isozymes, as previously reviewed [Citation1]. Inhibitor binding to CAs can be measured either by enzymatic inhibition methods or by biophysical binding methods [Citation2]. If the binding reaction is stoichiometric and inhibits the enzyme, then both approaches should yield similar results. In this study, the enzymatic inhibition method was compared with the results obtained using two biophysical techniques, isothermal titration calorimetry [Citation3] and a thermal shift assay (ThermoFluor, differential scanning fluorimetry) [Citation4]. The inhibition methods are more widely used than the biophysical methods, but determining the binding reactions by several techniques will reduce the uncertainty in the measurements.
CAs (EC 4.2.1.1) are metalloenzymes that catalyse the reversible hydration of carbon dioxide to bicarbonate [Citation5]. To date 16 CA isoforms have been identified in mammals: CA I, II, III, VII, and XIII are cytosolic proteins, CA VA and CA VB are mitochondrial, CA VI is secreted, and CA IV, IX, XII, XIV, and XV are membrane-associated proteins (CA XV is not expressed in humans). CA VIII, CA X, and CA XI are acatalytic carbonic anhydrase-related proteins (CA-RPs) which have lost their zinc ion binding capabilities due to histidine mutations in their active sites [Citation5,Citation6].
CAs I and II are major cytoplasmic isoforms, participating in respiration and acid/base homeostasis. CA I is mainly expressed in human red blood cells, and also detected in several other tissues such as in the colon, while CA II is a ubiquitous isoform with very high catalytic activity [Citation7]. CA IX is one of the fastest isozymes [Citation8] and has a unique distribution in tissues and organs when compared with other isozymes. The expression of CA IX is very limited in normal tissues (it is detected in epithelial tissues of the gastrointestinal tract), but is frequently expressed in many different tumours [Citation9–11]. In addition to the catalytic domain common to all isozymes, CA IX has a transmembrane domain, a short intracellular fragment at the C-terminus, and a proteoglycan-like domain at the N-terminus. Another exceptional feature of CA IX is dimerisation – CA IX forms covalently linked dimers outside of the cell membrane [Citation12].
Searching for novel inhibitors of human carbonic anhydrase IX (hCA IX) is an important task because of their potential use as anticancer compounds. In the current study the binding and inhibition of human (h) CA I, hCA II and hCA IX by benzimidazo[1,2-c][1,2,3]thiadiazole-7-sulphonamides [Citation13] was studied by means of enzyme inhibition. These results were compared to biophysical methods of binding determination, and related to the co-crystal structure of the enzyme-inhibitor complex.
Materials and methods
Protein preparation
The cDNA of hCA I was purchased from RZPD Deutsches Ressourcenzentrum für Genomforschung (Berlin, Germany). For recombinant protein production, a nucleotide sequence encoding nearly full-length hCA I (amino acids 3-261), was inserted into the BamHI site of pET-15b vector (Novagen, WI) fusing a 6xHis-tag to the N terminus of the protein.
For protein expression, the plasmid pET-15b-hCA I was transformed into Escherichia coli strain BL21 (DE3). An overnight culture of plasmid-harbouring cells was inoculated into fresh Luna broth (LB) medium containing 60 μM ZnCl2 and cultured at 37°C until the A550 was 0.5 – 0.8. Expression of the target protein was induced with 0.2 mM IPTG. Cells cultured at 30°C in the presence of 0.4 mM ZnCl2 were harvested 4 h after induction and lysed by sonication. Soluble protein was purified using a Sepharose-IDA-Ni+2 affinity column (GE Healthcare Bio-Sciences AB, Uppsala), followed by anion exchange chromatography on S-Sepharose (Amersham, GE Healthcare Bio-Sciences AB, Uppsala). Eluted protein was dialysed against a storage buffer: 20 mM N-(2-hydroxyethyl)-piperazine-N’-2-ethanesulphonic acid (HEPES) (pH 7.8), 0.05 M NaCl and 0.2 mM dithiothreitol (DTT), lyophilised and stored at -20°C. The hCA I preparations were analysed by SDS-PAGE to check for purity and found to be higher than 95%. Protein concentrations were determined by UV-VIS spectrophotometry using an extinction coefficient ϵ280 = 44920 M−1cm−1 and confirmed by the standard Bradford method. The catalytic activity of purified hCA I was measured in 10 mM HEPES (pH 7.5), 50 mM Na2SO4 buffer, containing 10% acetonitrile (the standard buffer), using p-nitrophenyl acetate as a substrate [Citation14]. The enzyme activity was confirmed to be in the range of 240–285 pmol/(min × μg).
The hCA II was expressed in Escherichia coli and purified as previously described [Citation15]. The hCA IX was produced using the baculovirus-insect cell expression system as previously reported [Citation8]. The catalytic domain with a proteoglycan domain form of the protein was used in this study. It represents hCA IX produced recombinantly that contains: the signal peptide, polyhistidine tag, thrombin site, proteoglycan domain, and the catalytic domain (residues 1–377 of the mature CA IX sequence and residues 38–414 of Swiss-Prot entry Q16790). The protein preparation contained a mixture of monomers and dimers through cysteine (Cys) residues that were separated by gel filtration as previously described [Citation8]. Thermal shift assay enabled the measurement of ligand binding to the monomeric and dimeric protein forms either in the same sample, or separated by gel filtration, because their melting temperatures were sufficiently different.
Catalytic activity measurement
An Applied Photophysics (Oxford) stopped-flow instrument was used for assaying the CA catalysed CO2 hydration activity [Citation16]. Phenol red (at a concentration of 0.2 mM) was used as an indicator, working at the absorbance maximum of 557 nm, with 10 mM Hepes (pH 7.5) as a buffer, 0.1 M Na2SO4 (for maintaining constant ionic strength) and following the CA-catalysed CO2 hydration reaction. The kinetic parameters and inhibition constants were determined with CO2 concentrations that ranged from 1.7 to 17 mM. For each inhibitor, at least six traces of the initial 5–10% of the reaction were used to determine the initial velocity. The uncatalysed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (10 mM) were prepared in distilled-deionised water with 10-20% (v/v) DMSO (which is not inhibitory at these concentrations) and subsequently dilutions up to 0.01 μM were performed with distilled-deionised water. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to the assay in order to allow for the formation of the E-I complex. The inhibition constants were obtained by a non-linear least-squares methods using PRISM 3 (Graph Pad Software, La Jolla), and represents the mean from at least three different determinations. The CA isozymes were obtained as described above and also as previously reported in [Citation17–20].
Thermal shift assay
The thermal shift assay (TSA, also called differential scanning fluorimetry) [Citation21, Citation22] was performed using the iCycler iQ Real Time Detection System (Bio-Rad, Hercules, CA), originally designed for polymerase chain reaction (PCR). Protein unfolding was monitored by measuring fluorescence of the solvatochromic fluorescent dye Dapoxyl™ sulphonic acid sodium salt (Invitrogen, Eugene). The dye was excited using an excitation filter 420/40X and emission was detected with a 490/20X filter. A constant temperature increment of 1 deg/min was applied.
Samples contained 10 µM protein, 0 to 200 µM ligand, 50 mM sodium phosphate buffer (pH 7), 50 mM NaCl and 100 µM Dapoxyl™ sulphonate at a total volume of 10 µL, covered with 2.5 µL of silicone oil DC 200 and 96-well iCycler iQ PCR plates were used.
Protein melting temperatures were determined by fitting the protein melting curve according to Equation 1 [Citation4]. The unfolding of the CA isozyme hCA I and hCA II yielded a single transition because the protein preparations were homogeneous. However, the hCA IX protein preparation from insect cells was composed of roughly equal percentages of monomer and dimer [Citation8]. Therefore hCA IX unfolding gave two transitions – the first at ~48°C, corresponding to monomer unfolding, and the second at ~67°C, corresponding to dimer unfolding. The melting temperatures (Tm) of both transitions were determined by fitting either the first or second transition of the single protein melting curve according to Equation 1:
where y(T) is calculated fluorescence as a function of temperature; ![]() is the fluorescence of the probe bound to folded native protein before transition, at Tm;
is the fluorescence of the probe bound to folded native protein before transition, at Tm; ![]() is the fluorescence of the probe bound to the unfolded protein after unfolding transition, at Tm; mF is the slope of the fluorescence dependence on temperature when the probe is bound to the native protein; mU is the slope of fluorescence dependence on temperature when the probe is bound to the unfolded protein;
is the fluorescence of the probe bound to the unfolded protein after unfolding transition, at Tm; mF is the slope of the fluorescence dependence on temperature when the probe is bound to the native protein; mU is the slope of fluorescence dependence on temperature when the probe is bound to the unfolded protein; ![]() is the enthalpy of protein unfolding, at Tm;
is the enthalpy of protein unfolding, at Tm; ![]() is the entropy of protein unfolding, at Tm; ΔUCP is the heat capacity of protein unfolding, assumed to be temperature-independent over the temperature range studied; R is the universal gas constant; T is the absolute temperature, in Kelvin.
is the entropy of protein unfolding, at Tm; ΔUCP is the heat capacity of protein unfolding, assumed to be temperature-independent over the temperature range studied; R is the universal gas constant; T is the absolute temperature, in Kelvin.
Binding constants were determined by following the increase in Tm for various ligand concentrations by using Equation 2, as previously described [Citation4,Citation15]. A typical experiment consisted of 12 ligand concentrations prepared by a 2 × or 1.5 × step dilution beginning from 200 µM. The total added ligand concentration (Lt) is related to the binding constant (![]() ) by the following relationship:
) by the following relationship:
where ![]() is the protein unfolding equilibrium constant, at Tm; Pt is the total protein concentration;
is the protein unfolding equilibrium constant, at Tm; Pt is the total protein concentration; ![]() is the ligand binding constant, at Tm;
is the ligand binding constant, at Tm; ![]() is the enthalpy of protein unfolding, at Tr; Tr is the protein melting temperature when no ligand is added;
is the enthalpy of protein unfolding, at Tr; Tr is the protein melting temperature when no ligand is added; ![]() is the entropy of protein unfolding, at Tr; ΔUCP is the heat capacity of protein unfolding, assumed to be temperature-independent over the temperature range studied;
is the entropy of protein unfolding, at Tr; ΔUCP is the heat capacity of protein unfolding, assumed to be temperature-independent over the temperature range studied; ![]() is the enthalpy of ligand binding, at T0; T0 is the temperature of ligand binding (usually 37°C);
is the enthalpy of ligand binding, at T0; T0 is the temperature of ligand binding (usually 37°C); ![]() is the entropy of ligand binding, at T0; ΔbCP is the heat capacity of ligand binding, assumed to be temperature-independent over the temperature range studied.
is the entropy of ligand binding, at T0; ΔbCP is the heat capacity of ligand binding, assumed to be temperature-independent over the temperature range studied.
The binding constant at the physiological temperature T0 is determined by Equation 3:
Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) measurements were performed using the Nano-III ITC (Calorimetry Sciences, Lindon) or the VP-ITC calorimeter (MicroCal, Northampton,) at pH 7 in 50 mM phosphate buffer containing 50 mM NaCl. Titrations were carried out at 37°C. A typical titration consisted of 25 injections of each tested compound (10 µL per injection), at 200–240 s intervals, into the sample cell containing the protein sample.
Crystallisation
Crystals of hCA II were obtained by the sitting drop method. The protein in 20 mM sodium HEPES, pH 7.8 and 50 mM NaCl was concentrated by ultrafiltration to a concentration of 20 mg/mL. For crystallisation, equal volumes of the protein solution and crystallisation buffer were mixed. Crystallisation buffer was 0.1 M sodium Bicine, 0.2 M ammonium sulphate and 2 M sodium malonate prepared by diluting and mixing 1 M sodium Bicine, pH 9, 3.5 M ammonium sulphate, and 3.4 M sodium malonate, pH 7. Crystals appeared the next day and were soaked for 7–10 days with a 1 mM solution of inhibitor in reservoir buffer.
Data collection and structure determination
Data from the protein-inhibitor complex crystals were collected at the EMBL BW7B beamline at the DORIS storage ring, Deutsches Elektronen-Synchrotron (DESY), Hamburg. Images have been processed with MOSFLM [Citation23] and SCALA [Citation24]. Initial phases were obtained by molecular replacement using 2NNS [Citation25] PDB entry as an initial model (only the protein chain was used; all water molecules and ligands were removed from the model). The electron density of Zn and the inhibitor molecule at the active centre of the protein were clearly visible on the difference electron density map after initial refinement of the model by REFMAC [Citation26] and CNS [Citation27]. A file with the atomic coordinates of the compound 1d was generated by the DSVisualizer 1.7 (Accelrys, San Diego). The topology and parameters of the molecule used in refinement of the structure were generated by XPLO2D [Citation28] and LIBREFMAC [Citation29]. The data collection and refinement statistics are given in . The coordinates and structure factors were deposited under the Protein Data Bank (PDB) with ID 3HLJ.
Table 1. Data collection and refinement statistics for the hCA II – 1d co-crystal structure.
Results
Chemical structures of the compounds designed to inhibit CAs are shown in . The synthesis of benzimidazo[1,2-c][1,2,3]thiadiazole-7-sulphonamides has been described previously [Citation13]. These compounds are good binders and inhibitors of several CAs. To determine compound specificity towards three CA enzymes (hCA I, hCA II, and hCA IX) with precision, the compound binding was studied using three techniques: a thermal shift assay, isothermal titration calorimetry, and enzymatic inhibition methods. The binding and inhibition constants of the series of compounds on the above mentioned CA isozymes are listed in .
Figure 1. Chemical structures of the compounds used in the study.
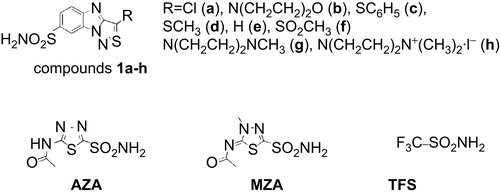
Table 2. Dissociation and inhibition constants (µM) of ligand binding to hCA I, hCA II, and hCA IX (hCA IX-M represents the monomeric protein form, and hCA IX-D the dimeric) as determined by thermal shift analysis (TSA), isothermal titration calorimetry (ITC) and the stopped-flow CO2 hydration assay. Average standard deviations for each method and enzyme are shown in percentuals.
Several typical isothermal titration calorimetry curves for an inhibitor binding to CA are shown in . The stoichiometries of ITC curves were slightly lower than or equal to one, indicating that the protein purity was good. This is a good test of the efficiency of the recombinant protein production and the purity of recombinant CAs, followed by their capability to bind inhibitors (70–100%). The binding constants obtained by ITC are listed in . These binding constants represent the binding strength at isothermal conditions, usually 37°C or room temperature. The protein remains in its native conformation and is not unfolded during the course of ITC measurement.
Figure 2. Representative isothermal titration calorimetry data for 1d binding to hCA II (Panels A and B) and hCA IX (Panels C and D). Panels A and C show the raw isothermal titration calorimetry data, panels B and D show integrated ITC data fitted with a single binding site model. Binding constants are listed in .
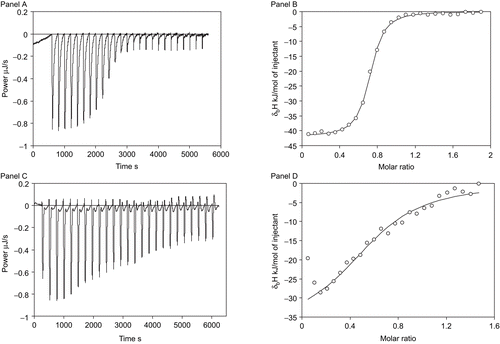
A very different biophysical method, is the thermal shift assay (ThermoFluor®, Johnson&Johnson, Exton) which measures the effect of protein stabilisation by ligands. Proteins are unfolded by increasing the temperature over the course of the experiment. Enzyme unfolding is tracked using the fluorescence of tryptophan or a solvatochromic probe such as 1-anilino-8-naphthalene sulphonate. shows typical thermal shift assay data for an inhibitor binding to carbonic anhydrase. Curves showing the dependence of fluorescence on temperature are shown in panel A. There is a significant increase in fluorescence at the unfolding temperature. The midpoint of the melting transition, the melting temperature Tm, is around 55°C without the addition of ligand, and the Tm shifts to 60°C or higher when the added ligand binds and stabilises the protein. Stronger binders shifted protein melting temperatures to a greater extent than the weak binders, as illustrated in panel B, where the Tm was higher for the same concentration of ligand indicating stronger binding.
Figure 3. Representative thermal shift assay data. Panel A shows fluorescence curves upon thermal denaturing of hCA II in the presence of various concentrations of compound 1d: Δ- 0 µM, ▴ - 3.5 µM, □ - 12 µM, • - 40 µM, and ○ - 130 µM. Datapoints are experimental values and the lines are fitted according to Equation 1. Panel B shows the dependence of Tm on the concentration of the added ligand for three inhibitors bound to hCA II: ○ - compound 1g, Δ - compound 1a, and □ - compound 1f. Datapoints are values from curves as in Panel A and the lines are fitted to the model according to Equation 2.
There was relatively good agreement between the three methods for determining binding and inhibition. Comparison of binding constants determined by the two methods and inhibition constants showed that sometimes there are small discrepancies between the three methods. These differences are usually within a factor of two to three.
Several novel inhibitors, such as 1e and 1f, bind and inhibit hCA IX with medium potency, on the order of 200 to 500 nM. All the inhibitors tested bound more strongly to the dimeric form of hCA IX than to the monomeric form. Only the thermal shift assay could distinguish between the binding to monomeric and dimeric forms of hCA IX because the forms melt at significantly different temperatures. Therefore, stabilisation of both forms could be observed in a single experiment, enabling determination of the binding constants to both CA forms. ITC and inhibition methods could not distinguish the binding to both forms of the protein without prior separation of the forms by gel filtration. Since some of the dimeric form separate towards the monomeric form, the results obtained by the thermal shift assay are considered more reliable, and therefore the ITC data is not listed. It is likely that there is a cooperative effect when the inhibitor binds to the dimeric state that increases binding strength.
The influence of the His6-tag on ligand binding to CA was studied by comparing the binding constants for two hCA I constructs, one containing an N-terminal His6-tag and the other without the tag. Comparison of many binding constants did not yield any statistically significant difference (data not shown). Similarly, since hCA II contains a significant number of natural histidines, it was possible to purify the enzyme by the same Sepharose-IDA-Ni+2 affinity column without attaching the tag.
The co-crystal structure of one of the compounds (1d) bound to the active site of hCA II is shown in . The sulphonamide moiety of 1d is coordinated by the Zn(II) ion in the active centre of the enzyme. The planar heterocyclic ring system is restrained from one side by the hydrophobic residues Phe131 and Leu198 as well as van der Waals contact with the Gln92 side chain from another side. Mobility of the molecule in the lateral direction is sterically fixed from one side by the hydrophobic residues Val121 and Ile91 (from the N5-N14 side of the ring). From the opposite side, the aromatic ring is restrained by Ser200 and the carbonyl oxygen of Pro201. The only hydrogen bond is between the nitrogen of the sulphonamide group and the hydroxyl of the side chain of Thr199. Thus, hydrophobic interactions ensure tight binding and inhibition of enzymatic activity by this compound. The buried surface area, as calculated by the Naccess program [Citation30], corresponding to the contact between the protein and the inhibitor is 520 Å2, which is more than half of the surface of the compound. Thus, one side of the aromatic ring is completely enclosed by protein side chains and the other side of the ring has contact with the solvent.
Figure 4. A view of inhibitor 1d located in the active centre of hCA II. Residues which are in hydrophobic or van der Waals contact with the inhibitor molecule, are also shown. The electron density map is contoured at 1.2σ. The picture has been generated using MOLSCRIPT [Citation33], Raster3D [Citation34], and BOBSCRIPT [Citation35].
![Figure 4. A view of inhibitor 1d located in the active centre of hCA II. Residues which are in hydrophobic or van der Waals contact with the inhibitor molecule, are also shown. The electron density map is contoured at 1.2σ. The picture has been generated using MOLSCRIPT [Citation33], Raster3D [Citation34], and BOBSCRIPT [Citation35].](/cms/asset/af86ce0c-15f1-4f3b-a34d-6eae5c660a73/ienz_a_457664_f0004_b.gif)
Discussion
Several biophysical and enzymatic techniques for ligand binding and inhibition were compared for inhibitor potency for several CA isozymes, including hCA I, hCA II, and hCA IX. Isozyme hCA IX is an important anticancer target. Despite the fact that none of the inhibitors studied appear to be specific for hCA IX, there are several inhibitors that bind the enzyme with submicromolar potency and could be developed further into more potent, and possibly more specific compounds.
Acetazolamide (AZA) is an example of a relatively specific inhibitor of hCA II; there is a concentration of AZA at which only hCA II is inhibited. All three methods show that the binding and inhibition constants are about 10-20 nM for hCA II, but around 100 nM for hCA I and hCA IX. Despite repeated experiments, there is still uncertainty in the dissociation constant of AZA to hCA I. It appears to be approximately 1 µM, while the Ki is about 0.25 µM. There is similar uncertainty for hCA IX. This apparent discrepancy may mean that results determined using disparate techniques will not match exactly. Even Kis determined by the esterase method do not match the results obtained by the hydratase method [Citation31]. This is because different processes are being monitored. Therefore, it is realistic that different methods will yield slightly different values. However, the rankings of compound affinities and inhibitor constants should be consistent across different methods.
Determining compound binding by a single method could be misleading. For example, the measurement of hCA II inhibition by compound 1b yielded a Ki of 9 nM. However, testing of the compound binding by titration calorimetry and the thermal shift assay indicates that the dissociation constant is around 100 nM. It is not clear which value is correct. Although it requires more work, including two or three methods helps to avoid mistakes in determining structure-activity relationships. However, when all three methods yield exactly the same results, this consistency helps establish the certainty of the ranking of compound affinities and potencies. For example, compound 1d binding to hCA II was determined to be about 50 nM (Ki = 57 nM, Kd(TSA) = 50 nM, and Kd(ITC) = 54 nM). Similarly, AZA binding to hCA II yielded essentially the same results with all three methods.
The dimeric form of the hCA IX bound all the inhibitors more strongly than the monomeric form of the enzyme. The native enzyme forms dimeric or possibly trimeric forms and thus may be able to exhibit a cooperative effect on binding. The binding constants are about 5–10 times greater for the dimeric form than for the monomeric form of hCA IX. This was most clearly visible using the thermal shift assay since it uses a mixture of both enzyme forms. However, the enthalpy of unfolding of the dimeric form of the protein has not been previously determined. Therefore, the precision of the determinations is lower than for other measurements.
The analysis of standard deviations in indicates that the inhibition assay is the most precise of the three methods. The thermal shift assay has biggest uncertainty of the data. However, the TSA measurements do not require expensive stop-flow equipment and can be carried out with a high throughput. The error may be reduced by determining the enthalpy of protein unfolding by an alternative method such as differential scanning calorimetry.
The enthalpies of inhibitor binding to the three isozymes are not listed because these enthalpies are so called “observed enthalpies”. These enthalpy values include the contribution of the enthalpy that come from processes such as buffer titration, inhibitor deprotonisation, and the protonisation of the hydroxide ion bound to the active site of carbonic anhydrase, as previously discussed [Citation2]. All these linked protonation effects are highly dependent on pH. Since the emphasis of the manuscript is to determine binding constants, the enthalpies are not discussed.
Crystallographic analysis of the compound-enzyme co-crystal structure shows the contacts of inhibitor-enzyme binding (). A full crystallographic characterisation of the system requires successful crystallisation of hCA IX. However, the active site of all three enzymes is similar and it is expected that some prediction of the binding potency could be made by modelling. Such analysis could lead to the design of more potent and selective inhibitors of carbonic anhydrases.
Table 3. Contacts between hCA II and compound 1d in the crystal structure.
Conclusion
The benzimidazo[1,2-c][1,2,3]thiadiazole-7-sulphonamide series bind stoichiometrically and inhibit the carbonic anhydrases I, II, and IX. Comparing results obtained using three different inhibition and binding methods increases the accuracy of compound affinity ranking and the ability to determine compound inhibitory specificity towards a particular carbonic anhydrase isoform. Thermal shift assay is a convenient method to measure compound binding constant.
Acknowledgements
We thank our local contacts at the EMBL beamlines, Santosh Panjikar, Matthew Groves and Paul Tucker for help with beamline operation. We thank Fernando Ridoutt for help with the beamline cryosystems.
Declaration of interest
The project was supported in part by the Lithuanian Science and Studies Foundation (N-06/09), EEA and Norway Grants 2004-LT0019-IP-1EEE, and by an EU grant of FP6 (DeZnIT project). Crystallographic data were collected at the EMBL/DESY, Hamburg. Access to the measurement facilities were funded by the European Community - Research Infrastructure Action under the FP6, structuring the European Research Area Programme contract number RII3/CT/2004/5060008.
References
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–181.
- Krishnamurthy VM, Kaufman GK, Urbach AR, Gitlin I, Gudiksen KL, Weibel DB, Whitesides GM. Carbonic anhydrase as a model for biophysical and physical-organic studies of proteins and protein-ligand binding. Chem Rev 2008;108:946–1051.
- Velazquez-Campoy A, Ohtaka H, Nezami A, Muzammil S, Freire E. Isothermal titration calorimetry. Curr Protoc Cell Biol 2004;Chapter 17:Unit 17 8.
- Matulis D, Kranz JK, Salemme FR, Todd MJ. Thermodynamic stability of carbonic anhydrase: measurements of binding affinity and stoichiometry using ThermoFluor. Biochemistry 2005;44:5258–5266.
- Supuran CT. Carbonic anhydrases–an overview. Curr Pharm Des 2008;14:603–14.
- Supuran CT, Scozzafava A. Carbonic anhydrases as targets for medicinal chemistry. Bioorg Med Chem 2007;15:4336–4350.
- Lindskog S. Structure and mechanism of carbonic anhydrase. Pharmacol Ther 1997;74:1–20.
- Hilvo M, Baranauskiene L, Salzano AM, Scaloni A, Matulis D, Innocenti A, Scozzafava A, Monti SM, Di Fiore A, De Simone G, Lindfors M, Janis J, Valjakka J, Pastorekova S, Pastorek J, Kulomaa MS, Nordlund HR, Supuran CT, Parkkila S. Biochemical characterization of CA IX, one of the most active carbonic anhydrase isozymes. J Biol Chem 2008;283:27799–27809.
- Dorai T, Sawczuk IS, Pastorek J, Wiernik PH, Dutcher JP. The role of carbonic anhydrase IX overexpression in kidney cancer. Eur J Cancer 2005;41:2935–2947.
- Chen J, Rocken C, Hoffmann J, Kruger S, Lendeckel U, Rocco A, Pastorekova S, Malfertheiner P, Ebert MP. Expression of carbonic anhydrase 9 at the invasion front of gastric cancers. Gut 2005;54:920–927.
- Kon-no H, Ishii G, Nagai K, Yoshida J, Nishimura M, Nara M, Fujii T, Murata Y, Miyamoto H, Ochiai A. Carbonic anhydrase IX expression is associated with tumor progression and a poor prognosis of lung adenocarcinoma. Lung Cancer 2006;54:409–418.
- Pastorekova S, Zatovicova M, Pastorek J. Cancer-associated carbonic anhydrases and their inhibition. Curr Pharm Des 2008;14:685–698.
- Dudutiene V, Baranauskiene L, Matulis D. Benzimidazo[1,2-c][1,2,3]thiadiazole-7-sulfonamides as inhibitors of carbonic anhydrase. Bioorg Med Chem Lett 2007;17:3335–3338.
- Pocker Y, Stone JT. The catalytic versatility of erythrocyte carbonic anhydrase. 3. Kinetic studies of the enzyme-catalyzed hydrolysis of p-nitrophenyl acetate. Biochemistry 1967;6: 668–678.
- Cimmperman P, Baranauskiene L, Jachimoviciute S, Jachno J, Torresan J, Michailoviene V, Matuliene J, Sereikaite J, Bumelis V, Matulis D. A quantitative model of thermal stabilization and destabilization of proteins by ligands. Biophys J 2008;95:3222–3231.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–2573.
- Nishimori I, Minakuchi T, Onishi S, Vullo D, Cecchi A, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: cloning, characterization, and inhibition studies of the cytosolic isozyme III with sulfonamides. Bioorg Med Chem 2007;15:7229–7236.
- Vullo D, Voipio J, Innocenti A, Rivera C, Ranki H, Scozzafava A, Kaila K, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of the human cytosolic isozyme VII with aromatic and heterocyclic sulfonamides. Bioorg Med Chem Lett 2005;15:971–976.
- Nishimori I, Vullo D, Innocenti A, Scozzafava A, Mastrolorenzo A, Supuran CT. Carbonic anhydrase inhibitors: inhibition of the transmembrane isozyme XIV with sulfonamides. Bioorg Med Chem Lett 2005;15:3828–3833.
- Vullo D, Innocenti A, Nishimori I, Pastorek J, Scozzafava A, Pastorekova S, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of the transmembrane isozyme XII with sulfonamides-a new target for the design of antitumor and antiglaucoma drugs? Bioorg Med Chem Lett 2005;15:963–969.
- Pantoliano MW, Petrella EC, Kwasnoski JD, Lobanov VS, Myslik J, Graf E, Carver T, Asel E, Springer BA, Lane P, Salemme FR. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J Biomol Screen 2001;6:429–440.
- Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc 2007;2:2212–2221.
- Leslie AGW. The integration of macromolecular diffraction data. Acta Crystallogr D Biol Crystallogr 2006;62:48–57.
- Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr 2006;62:72–82.
- Srivastava DK, Jude KM, Banerjee AL, Haldar M, Manokaran S, Kooren J, Mallik S, Christianson DW. Structural analysis of charge discrimination in the binding of inhibitors to human carbonic anhydrases I and II. J Am Chem Soc 2007;129:5528–5537.
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 1997;53:240–255.
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr 1998;54:905–921.
- Kleywegt GJ, Zou JY, Kjeldgaard M, Jones TA. Crystallography of biological macromolecules. In: Rossmann MG, Arnold E, Eds. International Tables for Crystallography. Vol F. The Netherlands, Kluwer Academic Publishers 2001:L353–367.
- Collaborative Computational Project (CCP). The CCP4 suite: programs for protein crystallography. Acta Cryst D 1994;50:760–763.
- Hubbard SJ, Thornton JM. Department of Biochemistry and molecular biology, UCL. ‘NACCESS’, Computer Program. 1993.
- Briganti F, Pierattelli R, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Part 37. Novel classes of isozyme I and II inhibitors and their mechanism of action. Kinetic and spectroscopic investigations on native and cobalt substituted enzymes. Eur J Med Chem 1996;31:1001–1010.
- Hilvo M, Salzano AM, Innocenti A, Kulomaa MS, Scozzafava A, Scaloni A, Parkkila S, Supuran CT. Cloning, expression, post-translational modifications and inhibition studies on the latest mammalian carbonic anhydrase isoform, CA XV. J Med Chem 2009;52:646–54.
- Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Cryst 1991;24:946–950.
- Merritt EA, Bacon DJ. Raster3D: Photorealistic molecular graphics. Methods Enzymol 1997;277:505–524.
- Esnouf RM. Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr D Biol Crystallogr 1999;55:938–940.