Abstract
Despite being an ancient disease, tuberculosis (TB) remains the leading single-agent infectious disease killer in the world. The emerging serious problem of TB control and clinical management prompted us to synthesize a novel series of heterocyclic substituted diphenyl ether derivatives and determine their activity against the H37Rv strain of Mycobacterium. All ten compounds inhibited the growth of the H37Rv strain of Mycobacterium at concentrations of 1 μg/mL. This activity was found to be comparable to the reference drugs rifampicin and isoniazid at the same concentration. While the antimicrobial activity of other diphenyl ether analogues, such as triclosan, is associated with the inhibition of enoyl-ACP reductase (ENR), the synthesised substituted diphenyl ether derivatives did not affect this enzyme activity in spite of their structural similarity with triclosan. Therefore, these compounds appear to have a novel mechanism of action against M. tuberculosis, and their structural features should be studied further for their potential as new antitubercular drugs.
Introduction
Tuberculosis (TB), an ancient infectious disease of global influence, has re-emerged with multi-drug resistant strains (MDR-TB), extensively drug resistant strains (XDR-TB) and acquired immune defiency syndrome (AIDS). According to the World Health Organisation (WHO), one third of the world’s population is infected with Mycobacterium tuberculosis (MTB) and about 1 billion more people will be infected with TB by 2020 [Citation1,Citation2]. The estimated 8.8 million new cases per year correspond to 52,000 deaths per week or more than 7,000 each day [Citation3,Citation4]. These numbers however, are only a partial depiction of the global TB threat. More than 80% of TB patients are in the economically productive age of 15 to 49 years, which results in tremendous economical and social problems.
About 15% of the projected one billion group (150 million) will exhibit symptoms of the disease, and about 3.6% (36 million) will die from TB if new disease prevention and treatment measures are not developed [Citation5]. The dramatic increase in TB observed in the recent years is a result of two major factors. Firstly, the increased susceptibility of people infected with acquired immunodeficiency syndrome (AIDS) to TB, which augments the risk of developing the disease 100 fold [Citation6]. Secondly, the increase in resistant strains of the disease [Citation7] with some showing cross-resistance to as many as nine drugs.
Although one possible long term solution to the problem is a better vaccine, in the short term, the major reliance will be on chemotherapy requiring the development of novel, effective, non-toxic antitubercular agents [Citation8–10]. The identification of novel target sites will also be needed to circumvent the problems associated with the increasing occurrence of multi-drug resistant strains. To do this, biochemical pathways specific to the mycobacteria and related organism disease cycle must be better understood. Many unique metabolic processes occur during the biosynthesis of mycobacterial cell wall components [Citation11]. One attractive target for the rational design of new antitubercular agents are the mycolic acids, the major components of the cell wall of M. tuberculosis.
Mycolic acids are high molecular weight C74 to C90 α-alkyl, β-hydroxy fatty acids covalently linked to arabino-galactan [Citation12–14]. They represent the major lipid components of the mycobacterial cell wall and are unique to mycobacteria and its related species [Citation15]. Enzymes that comprise the fatty acid synthetase (FAS) complex responsible for fatty acid biosynthesis are considered ideal targets for designing new antibacterial agents. Enoyl-ACP reductase (ENR) is a key regulatory step in fatty acid elongation, and catalyses the NADH-dependent stereospecific reduction of α,β-unsaturated fatty acids bound to acyl carrier protein [Citation16–18]. Two studies have shown that 5-chloro-pyrazinamide (5-Cl-PZA) [Citation19,Citation20] and pyrazinamide (PZA) inhibit the M. tuberculosis FAS I, indicating that FAS I is also a good drug target.
The diphenyl ether triclosan 5-chloro-2-(2,4-dichloro-phenoxy) phenol ether is a broad spectrum biocide that has been used for over 30 years, mainly as a component of antimicrobial wash in consumer products such as toothpaste, mouthwashes, deodorant soaps, lotions, children toys and cutting boards [Citation21].
Until recently, it was thought that triclosan, being a relatively small hydrophobic molecule, was absorbed via diffusion into the bacterial cell wall and that its antibacterial activity was the result of a nonspecific disruption of the organism’s cell wall [Citation22,Citation23]. However, the first evidence that this diphenyl ether inhibits fatty acid biosynthesis came when a genetic analysis of an Escherichia coli strain resistant to triclosan linked the resistance to the FabI gene which encodes for ENR [Citation24]. Subsequently extensive biochemical and structural studies have been performed to confirm that triclosan is a specific inhibitor of the E. coli ENR [Citation25–29]. Triclosan also directly inhibits the ENRs from Staphylococcus aureus [Citation30], Haemophilus influenzae [Citation31], M. tuberculosis and M. smegmatis (encoded by InhA) [Citation32–35], Plasmodium falciparum, the malarial parasite [Citation36–38], and in plants [Citation39]. The common theme in the inhibition of ENRs by triclosan is the requirement of the NAD+ cofactor. The interaction of triclosan with ENR is stabilised by the π-π stacking interaction between the hydroxychlorophenyl ring (ring A in ) and the nicotinamide ring of the NAD cofactor. The hydroxyl group of the NAD+ ribose ring and the hydroxyl group of a tyrosine form hydrogen bonding interactions with the hydroxyl group of triclosan. Ring B of triclosan makes several hydrophobic contacts with ENR. The ether oxygen of triclosan may also be critical in the formation of the stable ENR-triclosan-NAD+ complex, since the replacement of the group by a sulphur atom abolishes the inhibitory activity [Citation25].
Figure 1. Structure of triclosan.
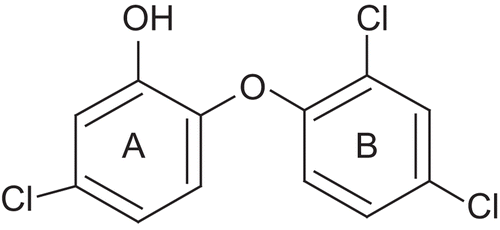
Since diphenyl ethers (including triclosan) are well known for their antitubercular activities [Citation40] and their mode of action has been studied [Citation41], our work has focused on the synthesis of compounds based on the diphenyl ether skeleton and tested them for their in vitro antitubercular activity. In vitro testing of their activity on over expressed E. coli ENR was also performed to determine whether their antitubercular activity was associated with inhibition of this target site.
Materials and methods
Reagents
All reagents were purchased from Sigma Chemicals (Bangalore, India) and were used without further purification.
TLC analysis
This was carried out on aluminum foil precoated with silica gel 60 F254 (Sigma- Aldrich, Bangalore). The solvent systems for TLC were chloroform:methanol (8:2 v/v), chloroform:ethyl acetate (1:1 v/v) and benzene:ethylacetate (1:2 v/v).
Equipment
Melting points were determined on a Toshniwal apparatus (Toshniwal Company, Bangalore, India) and are uncorrected. IR spectra were taken on Shimadzu FTIR 8300 (MCOPS, Manipal, India). 1H-NMR spectra were recorded on an Brucker AMX-400 NMR spectrometer (IISc, Bangalore, India) and were referenced to TMS with all chemical shifts reported as δ (ppm) values. Compounds were also analysed by GC-MS, (QP 5010, Shimadzu Corporation, Japan), LC-MS, FAB-MS (Joel SX-102, CDRI, Lucknow, India) and EI-MS (Autospec-5, IICT, Hyderabad, India).
Synthesis of oxadiazolethione (5a, 5b, 5c) ()
Phenoxy benzaldehyde 1 was oxidised to phenoxy benzoic acid 2 using an alkaline KMnO4 solution as per the procedure given in Vogel [Citation43]. Phenoxy benzoic acid was converted to its respective ester 3 using methanol and concentrated H2SO4. Phenoxy benzoic acid ester was converted to hydrazide 4 using hydrazine hydrate as per the procedure given in Udupi [Citation44].
Scheme 1. Synthesis of the oxadiazole thiones, triazoles, and pyrazoline derivatives of diphenyl ethers.
Ar, ortho-, meta-, or para- phenoxy phenyl (a, b, c) in case of oxadiazolethione (5a, 5b, and 5c) and triazole derivatives (7a, 7b, and 7c). Ar, meta-phenoxy phenyl for (9).
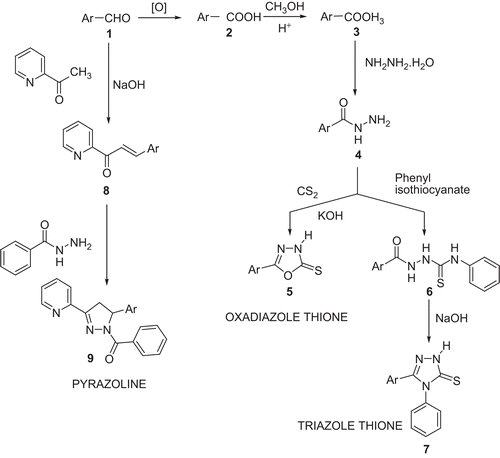
Mixtures of equimolar quantities of phenoxy benzoic acid hydrazide, CS2 and KOH were taken in absolute ethanol in a round bottom flask. The solution was refluxed until the evolution of H2S had nearly stopped (about 18 h). The solvent was removed under reduced pressure, the residue dissolved in water and the solution was acidified with dilute HCl (10%). The resulting product 5 was collected, washed with water, dried and recrytallised from aqueous ethanol (80%). The purity of the compound was ascertained by a single spot on the TLC plate. The physical and molecular data are given in . The solvent system for TLC was chloroform:methanol (8:2 v/v).
Table 1. Structures of the compounds synthesised and some of their physical properties.
5-(3-phenoxyphenyl)-1,3,4-oxadiazole-2(3H)-thione (5b)
FTIR Spectral Data: KBr pellet
N-H (3456.2), C-O-C stretch (1693.4), C=N (1583.4), C-O-C (oxadiazole) (1336.6), C-O-C (diphenyl ether) (1232.4), C=S (1180.4), Ar stretch (688.5).
1H-NMR (DMSO)
6.9–7.9 δ (m, 9H,Ar), 8.3 δ(s, 1H, NH of oxadiazole).
Mass
Calculated for C14H10N2O2S 270, found 270.
Synthesis of triazole thione (7a, 7b, 7c) ()
Equimolar quantities of acid hydrazide (4) and phenyl isothiocynate were dissolved in absolute ethanol in a round bottom flask. The solution was refluxed for 3 h on a water bath. The solution was concentrated under reduced pressure, the solid separated was collected and recrystallised from appropriate solvent to get the isothiocyanate derivative 6. This was further used to prepare the triazole whereby it was suspended in 4% NaOH and refluxed for 1 h. The resulting solution was filtered and the filtrate was cooled and acidified carefully with diluted acetic acid. The precipitate formed was filtered, washed with water, and the product 7 crystallised from ethanol. The purity of the compounds were established by a single spot on the TLC plate. The physical and molecular data are given in . The solvent system used consisted of chloroform:ethyl acetate (1:1 v/v).
5-(2-phenoxyphenyl)-4-phenyl-2,4-dihydro-3H-1,2,4-Triazole-3-thione (7a)
FTIR Spectral Data: KBr pellet
N-H (3436.9), C-N stretch (1612.4), C=N (1577.7), C-N-C (1394.4), C-O-C (diphenyl ether) (1242.1), C=S (1157.2), Ar stretch (692.4).
1H-NMR (DMSO)
3.2δ(s, 1H, NH of triazole), 6.7–7.7δ (m, 14 H, Ar).
Mass
Calculated for C20H15N3OS 345.14 and molecular ion (m+1) peak from LCMS was obtained at 346.14.
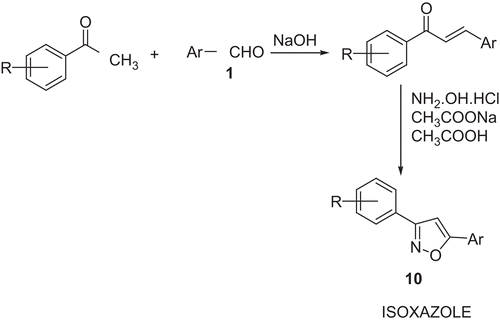
Synthesis of isoxazole derivatives (10a, 10b, 10c) ()
Preparation of chalcone: m-phenoxy benzaldehyde 1 (0.01 mole) in ethanol (95%, 15 mL) was added to the mixture of acetophenone (0.01mole), ethanol (95%, 20 mL) and NaOH (40%, 8 mL) and stirred for 24 h. The content was poured into crushed ice, isolated by acidification and crystallised from ethanol (95%). Anhydrous sodium acetate (0.01 mol) dissolved in a minimum amount of hot acetic acid was added to the solution of hydroxylamine hydrochloride (0.01 mol) in ethanol (15 mL). This solution was added to a solution of chalcone (0.01 mol) in ethanol (20 mL). The mixture was refluxed on an oil-bath for 8 h, concentrated and neutralised with a NaOH solution (0.1%). The product 10 was crystallised from ethanol (95%). The physical data are presented in .
Scheme 2. Synthesis of isoxazoles
Ar, meta-phenoxy phenyl; R = H (10a), 4-chloro (10b), and 4-methoxy (10c).
3-(4-methoxyphenyl)-5-(3-phenoxyphenyl) isoxazole (10c)
FTIR Spectral Data: KBr pellet
C=N (1579.6), C-O-C stretch (1614.3.6), C=C (isoxazole) (1515.9), CH3 (1461.9), C-O-C (diphenyl ether) (1245.9), Ar stretch (692.4).
1H-NMR (DMSO)
2.3δ (s, 1H, CH of isoxazole), 3.8δ (s, 1H,CH3), 6.9–8.0δ (m, 13H, Ar).
Mass
Calculated for C22H17NO3 is 343.37 and molecular ion peak was obtained at 343 indicating that the compound was formed.
Synthesis of pyrazoline derivatives (9) ()
The m phenoxy benzaldehyde (0.01 mol) was dissolved in 15mL of ethanol and 2-Methylpyridyl ketone dissolved in 15 mL of ethanol was added. This mixture was kept on a magnetic stirrer and slowly a stream of NaOH solution (40%, 10 mL) was added and stirred overnight. The mixture was then acidified to obtain the chalcone. The precipitate was filtered, dried and crystallised from ethanol. The hydrazide of the benzoic acid was synthesised by the usual method.
The above prepared chalcone (0.01 mol) was dissolved in ethanol (15 mL) and 0.01 mol of the hydrazide of the benzoic acid was dissolved in ethanol (20 mL) and added. To this mixture a few drops of glacial acetic acid was added and the mixture was refluxed for 12 hrs. The solvent was then removed under reduced pressure and the product poured into ice cold water. The precipitate of pyrazoline (9) was obtained which was filtered and dried. The product was crystallised from ethanol. The physical and molecular data are given in The solvent system for TLC was benzene:ethylacetate (1:2 v/v).
2-[5-(3-phenoxyphenyl)-4,5-dihydro-1(benzoyl)-pyrazol-3-yl] pyridine (9)
FTIR Spectral Data: KBr pellet
C=O (1685.7), C=N (1442.7), C-O-C (diphenyl ether) (1242.1), Ar stretch (694.3, 3055).
1H-NMR (DMSO)
3.7 δ (7.08 Hz and 18.0 Hz) (d, 2H,CH2 of pyrazoline) & 4. 4δ (7.62 Hz) (d, 1H,CH pyrazoline), 6.9-8.5 δ(m, 17H, Ar).
Mass
Calculated for C27H22 N3O3 is 435.23 and the molecular ion (m+1) peak was obtained at m/z 436.23. The other fragment peaks were obtained at m/z 316.2, 269.15, and 121.05.
Antitubercular activity
Antitubercular activity of the set of diphenyl ether derivatives was tested using the Lowenstein-Jensen medium (LJ medium) method [Citation45]. Briefly, eggs were broken aseptically to obtain 200 mL of egg solution. The solution was filtered through a sterile muslin cloth into a sterile conical flask containing glass beads. Sterilised mineral salt solution (120 mL) (consisting of 4 g potassium phosphate (anhydrous), 0.4 g of magnesium sulphate, 1.6 g magnesium citrate, 6 g of asparagine, 20 mL of glycerol, distilled water to fill to 1L) and 4 mL of sterilised malachite green solution (2%) were added to the 200 ml of egg solution. The contents were mixed well to form a uniform medium.
Compounds (10 mg) were dissolved in 10 mL of DMSO to obtain a concentration of 1000 µg/mL. The samples were diluted further with DMSO to make 100 µg/mL and 10 µg/mL stock solutions. A 0.8 mL aliquot of each concentration was transferred into different McCartney bottles. To this, 7.2 mL of LJ medium was added and mixed well.
INH and rifampicin were chosen as the standard drug for the comparison of antitubercular activity. The drug was dissolved in DMSO and diluted and tested as described above. The bottles were incubated at 75–80°C for 3 days for solidification and sterilisation.
Procedure for inoculation
A sweep from H37Rv strain of M. tuberculosis culture was discharged with the help of 22 SWG nichrome wire loop of 3 mm external diameter into a sterile bijou bottle containing six 3 mm glass beads and 4 mL of sterile distilled water. Each loop of culture delivered approximately 4 mg of bacilli cells. The bottle was shaken with the help of a mechanical shaker for 2 min to constitute the suspension. The suspension was inoculated on the surface of each LJ medium containing the test compounds using 27 SWG nichrome wire loop of 3 mm external diameter. LJ medium containing INH and rifampin as well as the medium containing DMSO (control) were inoculated with the test organism for positive and negative controls. Medium without any test compound/DMSO was also inoculated with the test organism to check whether the media supports the growth of the tubercle bacilli or not. The inoculated bottles were incubated at 37°C for 6 weeks, at the end of which readings were taken.
Cloning and purification of E. coli ENR reductase
Genomic DNA was isolated from E. coli (DH5α) according to the method of Sambrook et al. [Citation46]. Briefly, pelleted cells from 1.5 ml of saturated culture were resuspended in 1 mL of buffer (20 mM Tris-HCl, pH 8, 1 mM EDTA, 50 mM NaCl, 8% sucrose and 1 mg/mL lysozyme) by vortexing for 30 s. After 10 min incubation at room temperature, 50 mL of 20% SDS was added. The mixture was extracted with an equal volume of phenol/chloroform/isoamyl alcohol and precipitated with one volume of isopropanol. The pellet was washed with 70% ethanol and dissolved in 0.4 mL TE (pH 8). The coding region of fabI was amplified from the isolated genomic DNA by using polymerase chain reaction (PCR). The PCR reaction was performed using PfuTurbo DNA polymerase (Stratagene, La Jolla, CA) according to the manufacturer’s instruction with upstream primer 5′-gccATGGGTTTTCTTTCCGGTAAG-3′ and downstream primer 5′-gctcgagTTTCAGTTCGAGTTCGTTCATT-3′. The PCR product was digested with NcoI and XhoI that was introduced into the primers at the 5′ and 3′ ends, respectively, and cloned into similarly prepared pET28b (Novagen, Madison, WI). The clone was sequenced to confirm without errors. This plasmid was designated pETFab and transformed into BL21 (DE3) cells for expression. ENR was purified by metal affinity chromatography using a HisTrap HP column (GE Healthcare Bio-Sciences Corp, Piscataway, NJ) as described previously.
Enzymatic assay of ENR
ENR activity was determined spectrophotometrically by monitoring oxidation of NADH at A340 according to Ward et al. [Citation26]. The assay buffer consisted of 10 mM sodium phosphate, pH 7.2. The compounds were first tested at 100 μM to determine which diphenyl ether analogue was active against ENR activity. All inhibitors were dissolved in acetone and control samples received equivalent amount of acetone (1% v/v final concentration). ENR (24 nM or 1 ng/mL) was incubated with the compounds in the presence of 100 μM NAD+ for 30 minutes at room temperature prior to assay. Crotonyl-CoA (100 μM) was added to the mixture and the reaction was initiated by the addition of (100 μM) NADH. The oxidation of NADH was monitored for 60 sec by measuring the change in A340 in a spectrophotometer (UV3101PC, Shimadzu Corporation, Japan) with the cell thermostabilised at 25°C.
Results and discussion
A series of cyclic azole substituted diphenyl ether derivatives were synthesised using ortho-, meta-, or para- phenoxy benzaldehydes with different reagents to form the desired heterocyclic diphenyl ether derivatives. Overall, the yields were in the range of 60–88% and the compounds crystallised readily. Their structures were confirmed by GC-MS, LC-MS, 1H-NMR and IR spectroscopy (given in the synthetic procedure).
The compounds listed in were evaluated for antitubercular activity. All ten diphenyl ether derivatives were highly active against the H37Rv strain of M. tuberculosis (). The compounds inhibited the growth of the human pathogen at concentrations as low as 1 µg/mL. A concentration of 1 µg/mL corresponds to 3.7 µM for the compounds in series 5, 2.9 µM for compounds in series 7, and 2.4, 3.2, 2.9 and 2.9 µM for compounds 9, 10a, 10b, and 10c, respectively. Such level of antitubercular activity is comparable to other standard drugs such as isoniazid and rifampicin, which have MICs at 0.01–1.25 and 0.06–0.25 µg/mL, respectively [Citation42]. The diphenyl ether triclosan has a MIC of 5 µg/mL (17.4 µM) for M. tuberculosis [Citation33].
Table 2. Biological activity of the diphenyl ether derivatives against H37Rv strain of M. tuberculosis and inhibitory activity against ENR.
The mechanism of action of the diphenyl ether derivatives synthesised for this study was investigated by testing the activity of these compounds on E. coli ENR. In spite of the very high antitubercular activity, these compounds had very low activity on ENR. Unlike triclosan, which completely inhibited E. coli ENR activity at 1 µM, none of the diphenyl ether derivatives from this study inhibited ENR to any great extent at 100 µM. This is in stark contrast to the high in vitro activity displayed by these compounds, suggesting that the cyclic azole substituted diphenyl ether derivatives used in this study have a different mode of action than triclosan.
There may be several factors contributing to the lack of activity of these compounds on ENR. One of them might be the 5-chloro group on triclosan that is known to be very important for tight-binding and for slow-binding inhibition, and removal of this group reduces the binding 450,000 fold [Citation40]. The low ENR inhibitory activity of the diphenyl series used in this study may also be the result of the absence of similar halogenated substitutions.
Acknowledgements
We would like to thank the Microbiology Department of Kasturba Medical College, Manipal for helping with the antitubercular assay, and to the Central Drug Research Institute, Lucknow; Indian Institute Science, Bangalore and Indian Institute Chemical Technology, Hyderabad for providing the elemental analysis, 1H-NMR and Mass spectra of the compounds. ENR assay was performed at USDA-ARS Natural Products Utilization Research Unit, University, MS, USA.
Declaration of Interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- World Health Organisation. http://www.who.int/tb. 2008 Tuberculosis Facts.
- Nunn P, Williams B, Floyd K, Dye C, Elzinga G, Raviglione M. Nat Rev Immunol 2005;5:819–826.
- World Health Organization. Bridging the gaps: the world health report, Geneva: The Organization. (1995).
- World Health Organization report on TB epidemic. Global TB programme, Geneva: The Organization. (1997).
- World Health Organization. Tuberculosis; Fact Sheet No.104 (2002). Site accessed: www.who.int/mediacentre/factsheets/who104/en/index.html.
- El Sayed KA, Bartyzel P, Shen XY, Perry TL, Zjawiony JK, Hamann MT. Marine natural products as antituberculosis agents. Tetrahedron 2000;56:949–953.
- Goldberg MJ. Antituberculosis agents. Med Clin North Am 1988;72:661–668.
- Maranetra KN. Quinolones and multidrug-resistant tuberculosis. Chemotherapy 1999;45:3–11.
- Berning SE. The role of fluoroquinolones in tuberculosis today. Drugs 2001;61:9–18.
- Reddy VM, Nadadhur G, Daneluzzi DD, Osullivan JF and Gangadharam PRJ. Antituberculosis activities of clofazimine and its new analogs B4154 and B4157. Antimicrob Agents Chemother 1996;40:633–636.
- Barry CE. New horizons in the treatment of tuberculosis. Biochem Pharmacol 1997;54:1165–1172.
- Pasquato KFM and Ferreira EI. An approach for the rational design of new antitubercular agents. Curr Drug Targets 2001;2:427–437.
- Brenan PJ, Nikaido H. The envelope of mycobacteria. Annu Rev Biochem 1995;64:29–63.
- Barry CE III, Lee RE, Mdluli K, Sampson AE, Schroeder BG, Slayden RA, Yaun Y. Mycolic acid structure, biosynthesis and physiological functions. Prog Lipid Res 1998;37:143–179.
- Kolattukudy PE, Fernandes ND, Azad AK, Fitzmaurice AM, Sirakova TD. Biochemistry and molecular genetics of cell-wall lipid biosynthesis in 16 mycobacteria. Mol Microbiol 1997;24:263–270.
- Bergler H, Fuchsbichler S, Hogenauer G, Turnowsky F. The enoyl-[acyl-carrier-protein] reductase (FabI) of Escherichia coli, which catalyzes a key regulatory step in fatty acid biosynthesis, accepts NADH and NADPH as cofactors and is inhibited by palmitoyl-CoA. Eur J Biochem 1996;242:689–694.
- Stewart MJ, Parikh S, Xiao G, Tonge PJ, Kisker C. Structural basis and mechanism of enoyl reductase inhibition by triclosan. J Mol Biol 1999;290:859–865.
- Rozwarski DA, Vilcheze C, Sugantino M, Bittman R, Sacchettini JC. Crystal structure of the Mycobacterium tuberculosis enoyl-ACP reductase, InhA, in complex with NAD+ and a C16 fatty substrate. J Biol Chem 1999;274:15582–15589.
- Boshoff HI, Mizrahi V, Barry CE III. Effects of pyrazinamide on fatty acid synthesis by whole mycobacterial cells and purified fatty acid synthase I. J Bacteriol 2002;184:2167–2172.
- Zimhony O, Cox JS, Welch JT, Vilcheze C, Jacobs WR Jr. Pyrazinamide inhibits the eukaryotic-like fatty acid synthetase I (FASI) of Mycobacterium tuberculosis. Nat Med 2000);6:1043–1047.
- Schweizer HP. Triclosan: A widely used biocide and its link to antibiotics. FEMS Microbiol Lett 2001;202:1–7.
- Regos J, Zak O, Solf R, Vischer WA, Weirich EG. Antimicrobial spectrum of triclosan, a broad-spectrum antimicrobial agent for topical application. II. Comparison with some other antimicrobial agents. Dermatologica 1979;1158:72–79.
- Vischer WA, Regos J. Antimicrobial spectrum of triclosan, a broad-spectrum antimicrobial agent for topical application. Zentralbl Bakteriol [Orig A] 1974;226:376–389.
- McMurry LM, Oethinger M, Levy SB. Triclosan targets lipid synthesis. Nature 1998;394:531–532.
- Heath RJ, Yu YT, Shapiro MA, Olson E, Rock CO. Broad spectrum antimicrobial biocides target the FabI component of fatty acid synthesis. J Biol Chem 1998;273:30316–30320.
- Ward WH, Holdgate GA, Rowsell S, McLean EG, Pauptit RA, Clayton E, Nichols W, Colls JG, Minshull CA, Jude DA, Mistry A, Timms D, Camble R, Hales NJ, Britton CJ, Taylor IW. Kinetic and structural characteristics of the inhibition of enoyl (acyl carrier protein) reductase by triclosan. Biochemistry 1999;38:12514–12525.
- Levy CW, Roujeinikova A, Sedelnikova S, Baker PJ, Stuitje AR. Molecular basis of triclosan activity. Nature 1999;398:383–384.
- Stewart MJ, Parikh S, Xiao G, Tonge PJ, Kisker C. Structural basis and mechanism of enoyl reductase inhibition by triclosan. J Mol Biol 1999;290:859–865.
- Roujeinikova A, Levy CW, Rowsell S, Sedelnikova S, Baker PJ, Minshull CA, Mistry A, Colls JG, Camble R, Stuitje AR, Slabas AR, Rafferty JB, Pauptit RA, Viner R, Rice DW. Crystallographic analysis of triclosan bound to enoyl reductase. J Mol Biol 1999;294:527–535.
- Heath RJ, Li J, Roland GE, Rock CO. Inhibition of the Staphylococcus aureus NADPH-dependent enoyl-acyl carrier protein reductase by triclosan and hexachlorophene. J Biol Chem 2001;275:4654–4659.
- Marcinkeviciene J, Jiang W, Kopcho LM, Locke G, Luo Y. Enoyl-ACP reductase (FabI) of Haemophilus influenzae: steady-state kinetic mechanism and inhibition by triclosan and hexachlorophene. Arch Biochem Biophys 2001;390:101–108.
- McMurry LM, McDermott PF, Levy SB. Genetic evidence that InhA of Mycobacterium smegmatis is a target for triclosan. Antimicrob Agents Chemother 1999;43:711–713.
- Parikh SL, Xiao G, Tonge, PJ. Inhibition of InhA, the enoyl reductase from Mycobacterium tuberculosis, by triclosan and isoniazid. Biochemistry 2000;39:7645–7650.
- Kuo MR, Morbidoni HR, Alland D, Sneddon S F, Gourlie BB. Targeting tuberculosis and malaria through inhibition of enoyl reductase: Compound activity and structural data. J Biol Chem 2003;278:20851–20859.
- Surolia N, Surolia A. Triclosan offers protection against blood stages of malaria by inhibiting enoyl-ACP reductase of Plasmodium falciparum. Nat Med 2001;7:167–173.
- Kapoor M, Dar MJ, Surolia N, Surolia A. Kinetic determinants of the interaction of enoyl-ACP reductase from Plasmodium falciparum with its substrates and inhibitors. Biochem Biophys Res Commun 2001;289:832–837.
- Perozzo R, Kuo M, bir Singh Sidhu A, Valiyaveettil JT, Bittman R, Jacobs WR Jr, Fidock DA, Sacchettini JC. Structural elucidation of the specificity of the antibacterial agent triclosan for malarial enoyl ACP reductase. J Biol Chem 2002;277:13106–13114.
- Vincent CB, Twomey D. Derivatives of diploicin. Proc Royal Irish Acad 1950;53:55–59.
- Dayan FE, Ferreira D, Wang Y-H Khan, IA, McInroy JA, Pan Z. A pathogenic fungi diphenyl ether phytotoxin targets plant enoyl (acyl carrier protein) reductase. Plant Physiol 2008;147:1062–1071.
- Sivarana S, Sullivan TJ, Johnson F, Novichenok P, Cui G, Simmerling C, Tonge PJ Inhibition of the bacterial Enoyl reductase FabI by triclosan: A structure-reactivity analysis of FabI inhibition by triclosan analogues. J Med Chem 2004;47:509–518.
- Kini SG, Bhat AR, Bryant B, Williamson JS, Dayan FE. Synthesis, antitubercular activity and docking study of novel cyclic azole substituted diphenyl ether derivatives. Eur J Med Chem 2009;44:492–500.
- Dosages and Pharmacokinetics of antituberculosis medications-a report in India. [13–06–2006] Site accessed: http://www.angelfire.com/indie/tbindia/attdrugs.html
- Furniss BS, Hannaford AJ, Rogers V, Smith PWG, Tatchell AR. Aromatic carboxylic acids, In: Vogel’s Textbook of Practical Organic Chemistry, Ed IV London: Longman Group, 1980:824.
- Udupi R.Ph.D thesis on “Studies on the synthesis of substituted Triazoles, Azetidinones, Quinazolinones and related compounds for possible antitubercular activity and other pharmacological profiles”;.
- Watt B, Rayner A, Harris G, Mackie, McCartney. Chapter 18. In: Colle JG, Fraser AG, Marmion BP, Simmons A. Eds Practical Medical Microbiology. New York: Churchill Livingstone, 1996:331–335.
- Sambrook J, Fritsch E, Maniatis T. In Molecular Cloning, A Laboratory Manual. 2nd edition, New York: Cold Spring Harbor Laboratory Press, 1989.