Abstract
A series of 3-alkyl/aryl-8-(furan-2-yl)thiazolo[5,4-e][1,2,4]triazolo[1,5-c]pyrimidine-2(3H)-thiones (3a–3f) were synthesised in good yield and evaluated for their anti-Parkinsonian and neuroprotective potential. The structures of the synthesised compounds were confirmed on the basis of their spectral data and elemental analysis. All of the compounds were found to be active in haloperidol-induced catalepsy and oxidative stress in mice. The most active compound carried a propyl group at the 3-position of the thiazolotriazolopyrimidine nucleus while substitution with a phenyl ring produced the least active compound among the series. A computational study was carried out for the prediction of pharmacokinetic properties and none of the compounds violated Lipinski’s rule of five, making them potentially promising agents for the treatment of Parkinson’s disease.
Introduction
Parkinson’s disease (PD) is the prevalent neurodegenerative disorder of adults. Neuropathological hallmarks of PD involving molecular events include loss of dopaminergic neurons in the substantia nigra pars compacta, as well as the presence of Lewy bodies in the degenerating neurons [Citation1,Citation2]. Lewy bodies consist of large aggregates of oxidised, nitrated, ubiquitinated proteins. Over the past 50 years, extensive research has been done to elucidate the underlying molecular events leading to neuronal death and Lewy body formation, yet the cause and individual steps in the progression of the disease are still under debate [Citation3]. At present, the generally accepted hypothesis is that oxidative stress, via the generation of reactive oxygen species (ROS)/reactive nitrogen species (RNS), is in some way involved in causing protein oxidation and destruction of dopaminergic neurons [Citation4,Citation5].
Currently, the therapy for PD is largely focused on dopamine replacement strategies with the dopamine precursor levodopa and dopamine agonist drugs [Citation6,Citation7]. Although these strategies are highly effective in controlling the early stages of the disease, long-term treatment is associated with drug-related complications such as a loss of drug efficacy, the onset of dyskinesias and the occurrence of psychosis and depression [Citation8].
Based on the overwhelming genetic and pathophysiological evidence supporting ROS/RNS as the culprit molecules in PD, the major approaches for developing therapeutics to slow, stop, or reverse PD progression have attempted to target ROS/RNS by employing free radical scavengers [Citation9]. In the past decade, a large number of antioxidants such as Vitamin E, glutathione (GSH), melatonin, carotenoids, flavonoids, lipoic acid and coenzyme Q10 have been investigated as potential therapeutic agents to control oxidative stress in neurodegenerative disorders [Citation9]. Although such compounds have shown neuroprotection when used in animal models or in small clinical studies, their use is still relatively limited mainly because of poor bioavailability and/or inadequate antioxidant property under physiological conditions [Citation10].
In recent times, we have been investigating various thiazole derivatives as potential agents for the treatment of neurological disorders [Citation11–14]. Among these, 3-phenyl/ethyl-2-thioxo-2,3-dihydrothiazolo[4,5-d]pyrimidines () showed better activity than the standard drug levodopa in an animal model of PD at one instance, whereas at another instance, these agents proved their efficacy in reducing lipid peroxidation (LPO) as well as restoring GSH content and antioxidant enzyme activities in the brain homogenate of mice with Parkinsonian symptoms [Citation14]. We have also observed that the tricyclic thiazole moiety has a pronounced protective effect on haloperidol-induced catalepsy and oxidative stress in mice [Citation13]. Reports from other laboratories have also realised the importance of thiazolopyrimidines in the therapeutics of neurodegenerative disorders. Very recently, few thiazolo[4,5-d]pyrimidine derivatives () have been patented for their use in neurodegenerative disorders [15]. In addition, thiazolo[4,5-d]pyrimidines have been demonstrated to be associated with potent immunomodulating properties [Citation16]. Also, the recently demonstrated adenosine A2A receptor antagonistic activities of certain thiazoles [Citation17] and thiazolopyrimidines () for the development of a suitable approach to the treatment of PD may be the starting point for a future drug design [Citation18].
Figure 1. Design of the title compounds based on our previous report on thiazolo[4,5-d]pyrimidines and other known compounds.
![Figure 1. Design of the title compounds based on our previous report on thiazolo[4,5-d]pyrimidines and other known compounds.](/cms/asset/ff0c6683-ab92-4e48-8069-a3354ebe52b1/ienz_a_467627_f0001_b.gif)
Coupled with these observations and to extend our stride toward the development of potential therapeutic agents for neurological disorders [Citation11–14], we have modified our previously reported bicyclic molecule [Citation14] to a tricyclic moiety () because our recent review on antiparkinsonian agents [Citation19] reflects that tricyclic molecules are novel and potent drugs with fewer adverse effects than existing drugs for the treatment of PD [Citation20].
Materials and methods
Chemistry
Synthetic starting material, reagents and solvents were of analytical reagent grade or of the highest quality commercially available and were purchased from Aldrich Chemical Co. Inc., WI, Merck Chemical (Darmstadt, Germany) and were dried when necessary. The progress of the reactions was monitored by thin layer chromatography with F254 silica-gel precoated sheets (Merck) using chloroform/methanol 95/5 as eluent; UV light and iodine vapours were used for detection. IR spectra were recorded, as KBr pellets, on a Schimadzu 8201 PC FT-IR spectrophotometer (Shimadzu Corporation, Japan) and wave numbers are given in cm−1. The mass spectra were recorded on Jeol (USA Inc.) SX-102 FAB. 1H NMR and 13C NMR spectra, in CDCl3 solution, were recorded on a Bruker DRX-300 instrument (Germany) at 298 K. Chemical shifts are reported as ppm relative to TMS as internal standard. Melting points (°C) were determined with an open glass capillary tube and are uncorrected. Elemental analyses were performed on Elementar Vario EL III instrument (Germany).
General method for the synthesis of 4-amino-3-alkyl/aryl-2-thioxo-2,3-dihydrothiazole-5-carbonitriles (1a–1f)
To appropriate alkyl/aryl isothiocyanate (0.05 mol) in 50 mL dimethylformamide (DMF) was added finely divided sulphur (0.05 mol) and malononitrile (0.05 mol). Triethylamine (0.1 mol) was added drop wise with constant stirring to the mixture at room temperature and the stirring was continued for 3 h. The thick solution so obtained was poured into a mixture of 90 mL ice cold water and 10 mL methanol. The solid so obtained was filtered, washed with water, dried and recrystallised from methanol.
4-amino-3-ethyl-2-thioxo-2,3-dihydrothiazole-5-carbonitrile (1a)
Yield 42%; mp 170°C (Lit 168°C)
4-amino-3-propyl-2-thioxo-2,3-dihydrothiazole-5-carbonitrile (1b)
IR (KBr) υmax 3332, 3228, 2204, 1207 cm−1; 1H NMR (CDCl3) δ 4.78 (s, 2H, NH2), 3.92 (t, 2H, J = 6.9 Hz), 1.81–1.84 (m, 2H, CH2), 1.12 (t, 3H, J = 6.9 Hz); MS (FAB) m/z 200 (M+1)+.
4-amino-3-butyl-2-thioxo-2,3-dihydrothiazole-5-carbonitrile (1c)
IR (KBr) υmax 3320, 3232, 3201, 2202, 1210 cm−1; 1H NMR (CDCl3) δ 6.44 (s, 2H, NH2), 3.96 (t, 2H, J = 7.1 Hz), 1.68–1.78 (m, 2H, CH2), 1.28–1.46 (m, 2H, CH2), 0.92 (t, 3H, J = 7.2 Hz); MS (FAB) m/z 214 (M+1)+.
4-amino-3-phenyl-2-thioxo-2,3-dihydrothiazole-5-carbonitrile (1d)
Yield 48%; mp 262°C (Lit 263°C)
4-amino-3-(4-fluorophenyl)-2-thioxo-2,3-dihydrothiazole-5-carbonitrile (1e)
IR (KBr) υmax 3374, 3302, 3205, 3152, 3060, 2202, 1274, 872 cm−1; 1H NMR (CDCl3) δ 7.02-7.58 (m, 4H, ArH), 6.17 (s, 2H, NH2); MS (FAB) m/z 252 (M+1)+.
4-amino-3-(4-chlorophenyl)-2-thioxo-2,3-dihydrothiazole-5-carbonitrile (1f)
IR (KBr) υmax 3362, 3334, 3184, 3062, 2204, 1242, 764 cm−1; 1H NMR (CDCl3) δ 7.21–7.67 (m, 4H, ArH), 5.87 (s, 2H, NH2); MS (FAB) m/z 268 (M+1)+.
General method for the synthesis of ethyl N-(5-cyano-3-alkyl/aryl-2-thioxo-2,3-dihydrothiazol-4-yl)formimidates (2a–2f)
Compounds 1a–1f (0.005 mol), triethylorthoformate (0.01 mol) and a catalytic amount of p-toluenesulphonic acid were added in 25 mL toluene. The mixture was refluxed with stirring for 7–8 h and allowed to stand overnight in an ice bath. The reaction mixture was evaporated, and the residue was washed with water to give 2a–2f.
Ethyl N-(5-cyano-3-ethyl-2-thioxo-2,3-dihydrothiazol-4-yl)formimidate (2a)
IR (KBr) υmax 3011, 2952, 2901, 2205, 1585, 1227, 872 cm−1; 1H NMR (CDCl3) δ 8.14 (s, 1H, N = CH), 4.12 (t, 2H, J = 6.9 Hz), 3.72 (q, 2H, J = 7.5 Hz), 0.92 (t, 6H, J = 7.5 Hz); MS (FAB) m/z 241 (M+1)+.
Ethyl N-(5-cyano-3-propyl-2-thioxo-2,3-dihydrothiazol-4-yl)formimidate (2b)
IR (KBr) υmax 2999, 2956, 2876, 2206, 1599, 1267, 881 cm−1;1H NMR (CDCl3) δ 8.31 (s, 1H, N = CH), 4.43 (q, 2H, J = 7.2 Hz), 3.87 (m, 2H, CH2), 2.01–2.23 (m, 2H, CH2), 1.14 (t, 3H, J = 7 Hz), 0.93 (t, 3H, J = 7); MS (FAB) m/z 255 (M+1)+.
Ethyl N-(3-butyl-5-cyano-2-thioxo-2,3-dihydrothiazol-4-yl)formimidate (2c)
IR (KBr) υmax 2985, 2961, 2865, 2208, 1602, 1270, 1205 cm−1; 1H NMR (CDCl3) δ 8.43 (s, 1H, N = CH), 4.11 (m, 4H, CH2), 1.61 (m, 2H, CH2), 1.39 (m, 2H, CH2), 1.17 (t, 3H, J = 7.1 Hz), 0.95 (t, 3H, J = 6.9 Hz); MS (FAB) m/z 269 (M+1)+.
Ethyl N-(5-cyano-3-phenyl-2-thioxo-2,3-dihydrothiazol-4-yl)formimidate (2d)
IR (KBr) υmax 3001, 2954, 2844, 2208, 1612, 1247, 1008 cm−1; 1H NMR (CDCl3) δ 8.56 (s, 1H, N = CH), 6.84–7.39 (m, 3H, ArH), 6.68 (d, 2H, J = 7.2 Hz), 4.32 (q, 2H, J = 6.9 Hz), 1.12 (t, 3H, J = 6.8 Hz); MS (FAB) m/z 289 (M+1)+.
Ethyl N-(5-cyano-3-(4-fluorophenyl)-2-thioxo-2,3-dihydrothiazol-4-yl)formimidate (2e)
IR (KBr) υmax 2989, 2950, 2851, 2203, 1607, 1263 cm−1; 1H NMR (CDCl3) δ 8.12 (s, 1H, N = CH), 7.19–7.53 (m, 4H, ArH), 4.27 (q, 2H, J = 7.1 Hz), 1.11 (t, 3H, J = 7.2 Hz); MS (FAB) m/z 307 (M+1)+.
Ethyl N-(3-(4-chlorophenyl)-5-cyano-2-thioxo-2,3-dihydrothiazol-4-yl)formimidate (2f)
IR (KBr) υmax 3002, 2923, 2873, 2205, 1617, 1261 cm−1; 1H NMR (CDCl3) δ 7.98 (s, 1H, N = CH), 6.79–7.42 (m, 4H, ArH), 4.35 (q, 2H, J = 6.9 Hz), 0.99 (t, 3H, J = 7.1 Hz); MS (FAB) m/z 324 (M+1)+.
General method for the synthesis of 3-alkyl/aryl-8-(furan-2-yl)thiazolo[5,4-e][1,2,4]triazolo[1,5-c]pyrimidine-2(3H)-thiones (3a–3f)
Equimolar quantities of compounds 2a–2f, 2-furoic hydrazide and isobutyric acid in freshly dried toluene were refluxed for 1–3 h using a Dean-Stark assembly. Completion of reaction was monitored by TLC. The reaction mixture was evaporated, and the residue was washed with 70% ethanol to afford 3a–3f.
3-ethyl-8-(furan-2-yl)thiazolo[5,4-e][1,2,4]triazolo[1,5-c]pyrimidine-2(3H)-thione (3a)
IR (KBr) υmax 3062, 2996, 2853, 2808, 1268, 1050 cm−1; 1H NMR (CDCl3) δ 8.53 (s, 1H, N = CH), 7.91 (m, 1H, furyl), 7.43 (m, 1H, furyl), 7.16 (d, 1H, J = 3.2 Hz), 4.54 (q, 2H, J = 7.2 Hz), 1.31 (t, 3H, J = 7.2 Hz); 13C NMR (CDCl3) δ 12.6 (CH3), 49.9 (CH2), 112.3 (CH), 113.2 (CH), 116.8 (CH), 121.6 (CH), 135.1 (C), 142.6 (C), 153.5 (C), 158.7 (C), 160.1 (C), 190.2 (C = S); MS (FAB) m/z 304 (M+1)+.
8-(furan-2-yl)-3-propylthiazolo[5,4-e][1,2,4]triazolo[1,5-c]pyrimidine-2(3H)-thione (3b)
IR (KBr) υmax 3012, 2956, 2842, 1236, 1001 cm−1; 1H NMR (CDCl3) δ 8.48 (s, 1H, N = CH), 7.62 (d, 1H, J = 3.3 Hz), 7.17 (m, 1H, furyl), 6.99 (m, 1H, furyl), 4.41 (q, 2H, J = 7.1), 2.01–2.24 (m, 2H), 0.97 (t, 3H, J = 7.1); 13C NMR (CDCl3) δ 12.1 (CH3), 22.3 (CH2), 49.2 (CH2), 112.1 (CH), 112.9 (CH), 114.1 (CH), 128.2 (CH), 136.3 (C), 142.8 (C), 152.2 (C), 159.5 (C), 161.2 (C), 188.9 (C = S); MS (FAB) m/z 318 (M+1)+.
3-butyl-8-(furan-2-yl)thiazolo[5,4-e][1,2,4]triazolo[1,5-c]pyrimidine-2(3H)-thione (3c)
IR (KBr) υmax 3011, 2987, 2932, 2865, 1277, 1186, 982 cm−1; 1H NMR (CDCl3) δ 9.12 (s, 1H N = CH), 7.55 (m, 1H, furyl), 7.32 (m, 1H, furyl), 6.87 (d, 1H, J = 3.2 Hz), 4.34 (t, 2H, J = 7.1), 1.67 (m, 2H), 1.45 (m, 2H), 0.94 (t, 3H, J = 7.3 Hz); 13C NMR (CDCl3) δ 14.2 (CH3), 20.6 (CH2), 29.4 (CH2), 45.8 (CH2), 112.7 (CH), 113.5 (CH), 116.3 (CH), 127.1 (CH), 133.5 (C), 144.4 (C), 149.1 (C), 157.5 (C), 159.1 (C), 189.1 (C = S); MS (FAB) m/z 332 (M+1)+.
8-(furan-2-yl)-3-phenylthiazolo[5,4-e][1,2,4]triazolo[1,5-c]pyrimidine-2(3H)-thione (3d)
IR (KBr) υmax 3041, 2987, 2866, 1234, 1012 cm−1; 1H NMR (CDCl3) δ 8.88 (s, 1H, N = CH), 8.34 (m, 1H, furyl), 7.26–8.01 (m, 6H, ArH), 6.89 (m, 1H, furyl); 13C NMR (CDCl3) δ 112.6 (CH), 113.5 (CH), 113.8 (CH), 115.2 (CH), 118.3 (CH), 122.6 (CH), 124.3 (CH), 125.8 (CH), 127.2 (CH), 139.5 (C), 143.9 (C), 147.2 (C), 156.4 (C), 160.1 (C), 161.3 (C), 191.3 (C = S); MS (FAB) m/z 352 (M+1)+.
3-(4-fluorophenyl)-8-(furan-2-yl)thiazolo[5,4-e][1,2,4]triazolo[1,5-c]pyrimidine-2(3H)-thione (3e)
IR (KBr) υmax 3016, 2998, 2865, 1265, 1112, 998 cm−1; 1H NMR (CDCl3) δ 8.76 (s, 1H, N = CH), 6.74–7.71 (m, 7H, ArH); 13C NMR (CDCl3) δ 110.3 (CH), 112.4 (CH), 114.1 (CH), 115.9 (CH), 119.5 (CH), 123.2 (CH), 125.6 (CH), 126.5 (CH), 142.8 (C), 146.3 (C), 148.7 (C), 154.2 (C), 156.7 (C), 160.8 (C), 162.3 (C), 191.4 (C = S); MS (FAB) m/z 370 (M+1)+.
3-(4-chlorophenyl)-8-(furan-2-yl)thiazolo[5,4-e][1,2,4]triazolo[1,5-c]pyrimidine-2(3H)-thione (3f)
IR (KBr) υmax 3034, 2989, 2854, 1273, 1117, 1014 cm−1; 1H NMR (CDCl3) δ 9.33 (s, 1H, N = CH), 8.07 (m, 1H, furyl), 7.85 (d, 1H, J = 3.1 Hz), 7.51 (m, 1H, furyl), 6.89–7.24 (m, 4H, ArH); 13C NMR (CDCl3) δ 112.4 (CH), 113.1 (CH), 114.6 (CH), 116.2 (CH), 120.1 (CH), 121.3 (CH), 122.4 (CH), 124.9 (CH), 136.5 (C), 141.6 (C), 146.8 (C), 152.1 (C), 155.4 (C), 157.1 (C), 158.6 (C), 189.9 (C = S); MS (FAB) m/z 387 (M+1)+.
Pharmacological evaluation
Compounds 3a–3f were screened for antiparkinsonian activity in haloperidol-induced catalepsy test in mice. The results obtained are summarised in .
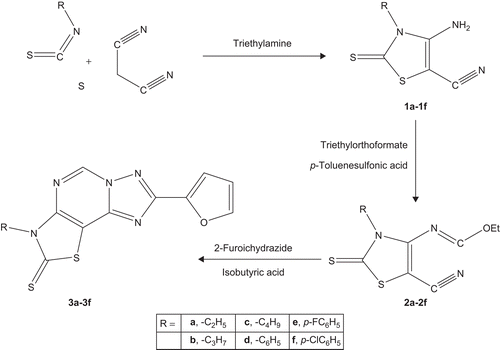
Scheme 1. Synthetic protocol of the title compounds.
Animals and drugs
Adult male pathogen-free Swiss albino mice weighing 18–25 g, were used. All animal experimentation was conducted in accordance with the Animal Ethics Committee of the Institute. The procedures adhered to the NIH Guidelines for the Care and Use of Laboratory Animals. Levodopa (Sigma-Aldrich) was injected ip at a dose of 100 mg/kg. Haloperidol (Sigma-Aldrich) was administered in dose of 5 mg/kg ip. The synthesised compounds 3a–3f were administered at 100 mg/kg ip. All the drugs and synthesised compounds were suspended in 0.5% gum acacia in redistilled water and administered at a volume of 0.1 mL/100 g.
Haloperidol-induced catalepsy test
Haloperidol-induced catalepsy was measured with the standard bar test [21], in a wooden chamber (length, 23 cm; width, 10.5 cm; height, 9 cm) with a horizontal metal bar (diameter, 0.4 cm; length, 10.5 cm) fixed at 9 cm above the floor, and at 4 cm from the back of the box. All experiments were carried out between 8:00 and 15:30 h in a room with controlled temperature (23 ± 1°C), and light intensity of 20 lux. Five mice were taken in each group. Control animals received 0.5% gum acacia in redistilled water. Synthesised compounds and levodopa were injected intraperitoneally at 100 mg/kg, 30 minutes prior to haloperidol injection. Animals were used only once. Catalepsy was measured every 30 min during the whole session that lasted 3 h after haloperidol injection. To assess whether the repeated handling of animals could have any influence on catalepsy intensity over time [Citation21], the bar test was performed in groups of mice that were injected only with the vehicle in which haloperidol was dissolved.
To measure catalepsy, the mouse was gently lifted until its forepaws firmly grasped the metal bar. Then, the mouse body was released and simultaneously a stopwatch was started. The time elapsed until the animal released both forepaws from the bar, up to a maximum of 300 s, was defined as the descent time. The sum of the descent time values measured every 30 min during the 3 h after haloperidol or vehicle was defined as the cumulative descent time (CDT[3h]). The mean CDTs measured in animals treated by the vehicle in which haloperidol was dissolved were subtracted from the mean CDTs recorded in mice treated with haloperidol. This difference was taken as 100% of catalepsy, and served as a reference value to calculate the percent inhibition of drugs on catalepsy intensity.
Biochemical evaluation
Mice were sacrificed by decapitation 3 h after the last injection. The brains were quickly removed and were washed twice with ice-cold saline solution, placed into glass bottles, labelled, and stored in a deep freeze (-30°C) until processing (maximum 10 h). Tissues were homogenised in four volumes of ice-cold Tris-HCl buffer (50 mM, pH 7.4) using a glass Teflon homogeniser (Ultra Turrax IKA T18 Basic, IKA Works, USA) after cutting up the brain into small pieces with scissors (for 2 min at 5000 rpm). Malondialdehyde (MDA) and protein levels were carried out at this stage. The homogenate was then centrifuged at 10500 × g for 20 min to remove nuclear debris. Clear supernatant fluid was taken and glutathione peroxidase (GSH-Px) activity was carried out in this stage. The supernatant solution was then extracted with an equal volume of an ethanol/chloroform mixture (5:3, v/v). After centrifugation at 5000 × g for 30 min, the clear upper layer (the ethanol phase) was taken and used in the superoxide dismutase (SOD) activity. All preparation procedures were performed at +4°C.
LPO assay
The extent of LPO in brain homogenate was determined by measuring the release of thiobarbituric acid reactive substance (TBARS) in terms of MDA equivalent using a molar extinction coefficient of 1.56 × 105/min/cm as described by Ohkawa et al [Citation22]. Briefly, the homogenate was centrifuged at 3000 × g for 15 min and the supernatant was used for the assay. Samples of 0.1 mL homogenate was mixed with 0.2 mL of 8.1% SDS, 1.5 mL 20% glacial acetic acid and 1.5 mL of 0.8% thiobarbituric acid (TBA). Following these additions, the tubes were mixed and heated at 95°C for 1 h on a water bath and cooled under tap water before mixing 1 mL of distilled water and a 5 mL mixture of n-butanol and pyridine (15:1). The mixture was centrifuged at 2200 × g for 10 min. The amount of MDA formed was measured by the absorbance of the upper organic layer at a wavelength of 532 nm in a Perkin Elmer (USA) spectrophotometer using appropriate controls. The results were expressed as nmol MDA/mg protein.
GSH determination
The amount of GSH in brain was measured according to the method of Sedlak and Lindsay [Citation23]. Briefly, brain tissue was deproteinised with an equal volume of 10% TCA and was allowed to stand at 4°C for 2 h. The contents were centrifuged at 2000 × g for 15 min. The supernatant was added to 2 mL of 0.4 M Tris buffer (pH 8.9) containing 0.02 M EDTA (pH 8.9) followed by the addition of 0.01 M DTNB {5, 5′-dithiobis(2-nitrobenzoic acid)}. Finally, the mixture was diluted with 0.5 mL of distilled water, to make the total mixture to 3 mL and the absorbance was read in a spectrophotometer at 412 nm and results expressed as µg GSH/gm Tissue.
SOD activity determination
Cu, Zn-SOD activity was determined according to the method of Sun et al [Citation24]. In this method, a xanthine–xanthine oxidase complex produces superoxide radicals, which react with nitrobluetetrazolium (NBT) to form the farmazan compound. In brief, a reactive was prepared with 0.1 mM of xanthine, 0.1 mM of EDTA, 50 mg of bovine serum albumin, 25 μM of NBT and 40 mM of Na2CO3 (pH 10.2). To 2.45 mL of reactive was added 0.5 mL of an ethanol/chloroform (5:3, v/v) extract, previously prepared from brain homogenate. Subsequently, 50 µl of 9.9 nM of xanthine oxidase solution was added, the mixture was kept in a water bath of 25°C for 20 min, and the reaction was terminated using 1 mL of CuCl2. The absorbance of the samples was read at 560 nm. In the control sample the amount of the ethanol supernatant was replaced by an equivalent volume of PBS buffer. One unit SOD activity was defined as the amount of enzyme causing 50% inhibition of NBT reduction to formazan. SOD activity was expressed as U/mg protein.
GSH-Px activity determination
The GSH-Px activity was measured by the method of Paglia and Valentine [Citation25]. The enzymatic reaction was conducted in 3 mL quartz cuvettes of 1 cm path length in a Perkin-Elmer spectrophotometer. Each 3 mL assay volume contained 50 mM potassium phosphate buffer (pH 7), 1 mM EDTA, 1 mM sodium azide, 0.2 mM NADPH, 1 U glutathione reductase and 1 mM reduced GSH. The sample (0.2 mL of the tissue homogenate), after its addition, was allowed to equilibrate for 5 min at 25°C. The reaction was initiated by adding 0.1 mL of 2.5 mM H2O2. Changes in absorbance were recorded at 340 nm for 5 min. Values were expressed as units of NADPH oxidised to NADP by using the extinction coefficient of 6.22 mM−1 cm−1 at 340 nm. All samples were assayed in duplicate. GSH-Px activity was expressed as units per gram protein.
Total protein determinations
Total protein concentration of brain homogenates was determined by the folin–phenol reaction as described by Lowry et al [Citation26] with bovine serum albumin (BSA) was used as a standard.
Calculation of physicochemical parameters
Absorption (%ABS) was calculated by: %ABS = 109–[0.345 × Topological polar surface area (TPSA)] according to the method of Zhao et al [Citation27]. TPSA [Citation28], miLogP, number of rotable bonds, and violations of Lipinski’s rule of five [Citation29] were calculated using Molinspiration online property calculation toolkit [Citation30].
Statistical analysis
Data were expressed as the mean ± standard error (SE) of the means. For statistical analysis of the data, group means were compared by one-way analysis of variance (ANOVA) with post hoc analysis. The post hoc Bonferroni multiple comparison test was applied to identify significance among groups where a p < 0.05 was considered to be statistically significant.
Results and discussion
Synthesis
In the present study, 3-alkyl/aryl-8-(furan-2-yl)thiazolo[5,4-e][1,2,4]triazolo[1,5-c]pyrimidine-2(3H)-thiones (3a–3f) were synthesised as presented in Scheme 1. 4-amino-3-alkyl/aryl-2-thioxo-2,3-dihydrothiazole-5-carbonitriles (1a–1f) were prepared when appropriate alkyl/aryl isothiocyanate was treated with sulphur, malononitrile and triethylamine in DMF. Compounds 1a–1f were refluxed with excess of triethylorthoformate in toluene using catalytic amount of p-toluenesulphonic acid to afford ethyl N-(5-cyano-3-alkyl/aryl-2-thioxo-2,3-dihydrothiazol-4-yl)formimidates (2a–2f). The final tricyclic compounds (3a–3f) were prepared by reaction of 2a–2f with 2-furoic hydrazide and isobutyric acid in freshly dried toluene with azeotropic removal of water molecules. All compounds had IR, 1H NMR/13C NMR and mass spectra in accord with their anticipated structures. The physical characterisation data of the compounds are given in .
Table 1. Physical characterisation data of the 3-alkyl/aryl-8-(furan-2-yl)thiazolo[5,4-e][1,2,4]triazolo[1,5-c]pyrimidine-2(3H)-thiones (3a–3f).
Haloperidol-induced catalepsy
PD is a motor deficit based on the degeneration of the nigrostriatal dopaminergic neurons. When haloperidol, a dopamine D2 receptor antagonist is administered, catalepsy is induced by the block of postsynaptic D2 receptor [31]. This haloperidol-induced catalepsy is known as a classic model in which symptoms of PD are produced by drug administration [Citation32]. Neuroleptic-induced catalepsy has long been used as an animal model for screening drugs for Parkinsonism [Citation12–14,Citation21,Citation33]. This is a behavioral condition in which the animal is unable to correct an externally imposed posture. Catalepsy-free mice, i.e., those without haloperidol, should be able to come down from the horizontal bar within a certain time period. On the other hand, cataleptic mice, when placed in this awkward position, were unable to come down from the bar over a period of 300 s or more.
Haloperidol (5 mg/kg) produced a profound increase in catalepsy as shown by a progressive increase in the latency to step down from the rod over time as compared with controls (p<0.001). Efficacious compounds were defined as those that allowed the mice to come down from the bar within 300 s. The result obtained from acute administration of haloperidol is in agreement with previous reports from other laboratories [Citation34] and our research group [Citation12–14]. Intraperitoneal administration of the compounds (3a–3f) at a dose of 100 mg/kg significantly antagonised haloperidol-induced catalepsy as evident from the decrease in mean descent time in the treated animals when compared to the mice injected with haloperidol alone. All of the tested compounds except 3a and 3d exhibited better activity than the standard drug levodopa. The effect of 3-alkyl/aryl-8-(furan-2-yl)thiazolo[5,4-e][1,2,4]triazolo[1,5-c]pyrimidine-2(3H)-thiones and standard drug in terms of mean descent time observed at different time intervals, mean CDT[3h] and % catalepsy are presented in .
Table 2. Mean descent time observed at different time intervals.
Structure-activity analysis revealed that a propyl group at position 3 (compound 3b) was better than any other alkyl or aromatic moiety in terms of both antiparkinsonian and antioxidant activities. Either increasing (compound 3c) or decreasing (compound 3a) the methylene bridge by one carbon resulted in a decrease in antiparkinsonian activity. When the 3-propyl group of compound 3b was replaced with phenyl ring (compound 3d), a drop in anticataleptic activity was observed. However, when a p-fluoro or a p-chloro group was substituted in the phenyl ring, the anticataleptic activity was improved but was still was less than the compounds with aliphatic substituents at position 3. In our previous studies, compounds substituted with a halogen in the C4 position of phenyl ring had better anticataleptic as well as neuroprotective activities [Citation12,Citation14].
Haloperidol-induced oxidative stress
In this study, we investigated the possible role of compounds 3b and 3c in resisting oxidative stress based on their role in alleviating catalepsy induced by haloperidol in mice. Cellular redox imbalance has been attributed to the development of several neurodegenerative diseases such as Alzheimer’s, Parkinson’s, and Huntington’s disease. The brain is especially prone to oxidative stress due to its high oxygen consumption, abundance of polyunsaturated fatty acids, transition metals, and relatively low levels of antioxidant enzymes [Citation35]. The importance of the antioxidant system in PD is supported by alterations in the levels of antioxidant enzymes and LPO in PD patients as well as in animal models. Chronic administration of neuroleptics enhance free radical generation leading to oxidative stress [Citation36,Citation37], which has been implicated in the pathophysiology of PD [Citation38]. Our results reveal that haloperidol increases LPO whereas it decreases the GSH level as well as antioxidant enzyme (SOD and GSH-Px) activities in brain homogenate. A reduction in GSH may impair H2O2 clearance and promote OH• formation, thus increasing the free radical load, which triggers oxidative stress and consequently disrupts homeostasis. GSH comprises one of the major non-enzymatic antioxidants serving as a cofactor of several detoxifying enzymes against oxidative stress, e.g. GSH-Px, glutathione transferase and others. GSH scavenges OH• and O2•− directly, detoxifying H2O2 and lipid peroxides by the catalytic action of GSH-Px [Citation39]. In addition, GSH is able to regenerate the most important antioxidants, vitamins C and E, back to their active forms; GSH can reduce the tocopherol radical of vitamin E directly, or indirectly, via reduction of semidehydroascorbate to ascorbate [Citation40,Citation41]. GSH-Px plays a predominant role in removing excess free radicals and hydroperoxides and is a major defence system against oxidative stress in the brain [Citation42]. SOD, which is present in both cytosol and mitochondria, neutralises the deleterious effects of O2•− [Citation43].
Compounds 3b and 3c administered 30 min before haloperidol significantly suppressed oxidative stress, restoring enzyme activities as well as GSH and MDA contents to be almost similar to the controls (). The present data demonstrate that 3-alkyl/aryl-8-(furan-2-yl)thiazolo[5,4-e][1,2,4]triazolo[1,5-c]pyrimidine-2(3H)-thiones possess antioxidant effect on catalepsy model of PD, offering protection by enhancing GSH content and antioxidant enzymes (SOD and GSH-Px) activities as well as decreasing LPO in mice treated with haloperidol. Such regulation of oxidative stress markers and antioxidant enzymes by the title compounds in the present study may be well correlated with our previous reports, where thiazole derivatives and 3-phenyl/ethyl-2-thioxo-2,3-dihydrothiazolo[4,5-d]pyrimidine derivatives have been shown to restore the level of oxidative stress markers and antioxidative enzymes up to basal level against oxidative stress induced by pentylenetetrazole [Citation11] or haloperidol [Citation12–14] in mice brain homogenate.
Table 3. Biochemical estimation from brain homogenate.
Prediction of pharmacokinetic properties
A computational study for the prediction of the pharmacokinetic properties of compounds 3a–3f was performed and is presented in . Poor solubility and poor permeability are among the main causes for failure during drug development [Citation44,Citation45]. It is therefore important to determine the physicochemical properties associated with a drug before synthetic work is undertaken. The estimated log P values (P is the partition coefficient of the molecule in the water/octanol system), which can be used as an indicator of passive diffusion across cell membranes and cellular uptake [Citation46–48], were determined for compounds 3a–3f and is presented in . The lipophilicity study showed that most of compounds possess the optimum lipophilicities (log P 1.89–3.46) required for oral absorption and biomembrane penetration, even for blood-brain barrier (BBB) penetration according to Lipinski’s drug-likeness ‘rules of five’ [Citation29]. This bioavailability feature makes it possible to use these compounds in the treatment of neurodegenerative diseases. Topological polar surface area (TPSA) ie the surface belonging to polar atoms, is a descriptor that was shown to correlate well with passive molecular transport through membranes and, therefore, allows prediction of the transport properties of drugs in the intestines and BBB crossing [Citation28]. TPSA was used to calculate the percentage of absorption (%ABS) according to the equation: %ABS = 109 − 0.345 × TPSA, as reported by Zhao et al [Citation27]. In addition, the number of rotatable bonds (n-ROTB) and Lipinski’s rule of five were also calculated [Citation29]. From all these parameters, it is evident that all 3-alkyl/aryl-8-(furan-2-yl)thiazolo[5,4-e][1,2,4]triazolo[1,5-c]pyrimidine-2(3H)-thione derivatives showed no obvious indications that they would not be active orally, as none of the compounds violated Lipinski’s parameters, making them potentially promising agents for the treatment of PD.
Table 4. Physicochemical parameters of 3-alkyl/aryl-8-(furan-2-yl)thiazolo[5,4-e][1,2,4]triazolo[1,5-c]pyrimidine-2(3H)-thiones (3a–3f)a.
Conclusion
We have innovatively extended our previously reported bicyclic thiazolo[4,5-d]pyrimidines to tricyclic 3-alkyl/aryl-8-(furan-2-yl)thiazolo[5,4-e][1,2,4]triazolo[1,5-c]pyrimidine-2(3H)-thiones as our efforts to discover novel antiParkinsonian agents with an improved pharmacological profile for haloperidol-induced catalepsy and oxidative stress in mice. Altogether, this study further outlines the importance of neuroprotective/antioxidant therapy for PD, supporting the notion that the oxidative stress may play a significant role in the pathophysiological mechanisms underlying PD.
Acknowlegdement
Mr Nitin is acknowledged for his kind help and support.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Jellinger KA. Formation and development of Lewy pathology: a critical update. J Neurol 2009;256:270–279.
- Olanow CW. Oxidation reactions in Parkinson’s disease. Neurology 1990;40:32–39.
- Markesbery WR, Jicha GA, Liu H, Schmitt FA. Lewy body pathology in normal elderly subjects. J Neuropathol Exp Neurol 2009;68:816–822.
- Halliwell B. The wanderings of a free radical. Free Radic Biol Med 2009;46:531–542.
- Beal MF. Mitochondria, oxidative damage, and inflammation in Parkinson’s disease. Ann N Y Acad Sci 2003;991:120–131.
- Allain H, Bentue-Ferrer D, Akwa Y. Disease modifying drugs and Parkinson’s disease. Prog Neurobiol 2008;84:25–39.
- Rascol O, Katzenschlager R. The treatment of early Parkinson’s disease. In: Hallett M, Poewe W ed. Therapeutics of Parkinson’s Disease and Other Movement Disorders. New York: John Wiley & Sons, 2008:49–70.
- Stacy M, Galbreath A. Optimizing long-term therapy for Parkinson disease: levodopa, dopamine agonists, and treatment-associated dyskinesia. Clin Neuropharmacol 2008;31:51–56.
- Azam F. Therapeutic Potential of Free Radical Scavengers in Neurological Disorders. In: Kozyrev D, Slutsky V, ed. Handbook of Free radicals: Formation, Types and Effects. New York: Nova Publishers, 2010; 57–97.
- Gilgun-Sherki Y, Rosenbaum Z, Melamed E, Offen D. Antioxidant therapy in acute central nervous system injury: current state. Pharmacol Rev 2002;54:271–284.
- Azam F, Alkskas IA, Khokra SL, Prakash O. Synthesis of some novel N4-(naphtha1,2-d]thiazol-2-yl)semicarbazides as potential anticonvulsants. Eur J Med Chem 2009;44:203–211.
- Azam F. Synthesis of some urea and thiourea derivatives of naphtha[1,2-d]thiazol-2-amine as anti-Parkinsonian agents that cause neuroprotection against haloperidol-induced oxidative stress in mice. Med Chem Res 2009;18:287–308.
- Azam F, Barodia SK, Anwer T, Alam MM. Neuroprotective effect of naphtha[1,2-d]thiazol-2-amine in an animal model of Parkinson’s disease. J Enzyme Inhib Med Chem 2009;24:808–817.
- Azam F, Alkskas IA, Ahmed MA. Synthesis of some urea and thiourea derivatives of 3-phenyl/ethyl-2-thioxo-2,3-dihydrothiazolo[4,5-d]pyrimidine and their antagonistic effects on haloperidol-induced catalepsy and oxidative stress in mice. Eur J Med Chem 2009;44:3889–3897.
- Nordvall, G; Ray, C; Rein, T; Sohn, D. Novel 5,7-disubstituted [1,3]thiazolo[4,5-d]Pyrimidin-2(3H)-one derivatives 794. US Patent 20090124637, May 14, 2009.
- Nagahara K, Anderson JD, Kini GD, Dalley NK, Larson SB, Smee DF, Jin A, Sharma BS, Jolley WB. Thiazolo[4,5-d]pyrimidine nucleosides. The synthesis of certain 3-.beta.-D-ribofuranosylthiazolo[4,5-d]pyrimidines as potential immunotherapeutic agents. J Med Chem 1990;33:407–415.
- Slee D, Lanier M, Vong BG, Chen Y, Zhang X, Lin E, Moorjani M, Castro P, Laria JC. Substituted pyrimidines as adenosine receptor antagonists. US Patent 20080275064, November 06, 2008.
- Sugihara Y, Kawakita Y. Thiazolopyrimidine derivative. US Patent 20080269238, October 30, 2008.
- Azam F, Ibn-Rajab IA, Alruiad AA. Adenosine A2A receptor antagonists as novel antiParkinsonian agents: A review of structure-activity relationships. Pharmazie 2009;64:771–795.
- Wardas J, Konieczny J, Lorenc-Koci E. SCH 58261, an A2A adenosine receptor antagonist, counteracts parkinsonian-like muscle rigidity in rats. Synapse 2001;41:160–171.
- Sanberg PR, Giordano M, Bunsey MD, Norman AB. The Catalepsy Test: Its Ups and Downs. Behav Neurosci 1988;102:748–759.
- Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem 1979;95:351–358.
- Sedlak J, Lindsay RH. Estimation of total, protein-bound and non-protein sulfhydryl groups in tissue with Ellmann’s reagent. Anal Biochem 1968;25:192–205.
- Sun Y, Oberley LW, Li Y. A simple method for clinical assay of superoxide dismutase Clin Chem 1988;34:497–500.
- Paglia DE, Valentine WN. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J Lab Clin Med 1967;70:158–170.
- Lowry OH, Rosenbrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem 1951;193:265–275.
- Zhao Y, Abraham MH, Le J, Hersey A, Luscombe CN, Beck G, Sherborne B, Cooper I. Rate-limited steps of human oral absorption and QSAR studies. Pharm Res 2002;19:1446–1457.
- Ertl P, Rohde B, Selzer P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J Med Chem 2000;43:37:14–3717.
- Lipinski CA, Lombardo L, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Delivery Rev 2001;46:3–26.
- Molinspiration Cheminformatics, Bratislava, Slovak Republic, http://www.molinspiration.com/services/properties.html. Accessed on October 4, 2009.
- Sanberg PR. Haloperidol-induced catalepsy is mediated by postsynaptic dopamine receptors. Nature 1980;284:472–473.
- Ossowska K, Karcz M, Wardas J, Wolfarth S. Striatal and nucleus accumbens D1/D2 dopamine receptors in neuroleptic catalepsy. Eur J Pharmacol 1990;182:327–334.
- Frank ST, Schmidt WJ. Burst activity of spiny projection neurons in the striatum encodes superimposed muscle tetani in cataleptic rats. Exp Brain Res 2003;152:519–522.
- Correa M, Wisniecki A, Betz A, Dobson D.R, O’Neill M.F, O’Neill M.J, Salamone J.D. The adenosine A2A antagonist KF17837 reverses the locomotor suppression and tremulous jaw movements induced by haloperidol in rats: possible relevance to parkinsonism. Behav Brain Res 2004;148:47–54.
- Calabrese V, Bates TE, Stella AM. NO synthase and NO-dependent signal pathways in brain aging and neurodegenerative disorders: the role of oxidant/antioxidant balance. Neurochem Res 2000;25:1315–1341.
- Lohr JB, Kuczenski R, Niculescu AB. Oxidative mechanisms and tardive dyskinesia. CNS Drugs 2003;17:47–62.
- Sagara Y. Induction of reactive oxygen species in neurons by haloperidol. J Neurochem 1998;71:1002–1012
- Tsang AH, Chung KK. Oxidative and nitrosative stress in Parkinson’s disease. Biochim Biophys Acta 2009;1792:643–650.
- Valko M, Leibfritz D, Moncol J, Cronin MTD, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 2007;39:44–84.
- Masella R, Di Benedetto R, Vari R, Filesi C, Giovannini C. Novel mechanisms of natural antioxidant compounds in biological systems: Involvement of glutathione and glutathione related enzymes. J Nutr Biochem 2005;16:577–586.
- Martin HL, Teismann P. Glutathione—a review on its role and significance in Parkinson’s disease. FASEB J 2009;23:3263–3272.
- Imam SZ, Ali SF. Selenium, an antioxidant, attenuates methamphetamine-induced dopaminergic toxicity and peroxynitrite generation. Brain Res 2000;855:186–191.
- Pong K. Oxidative stress in neurodegenerative diseases: therapeutic implications for superoxide dismutase mimetics. Expert Opin Biol Ther 2003;3:127–139.
- Avdeef A. Physicochemical profiling (solubility, permeability, and charge state). Curr Top Med Chem 2001;1:277–351.
- Opera TI. Current trends in lead discovery: are we looking for the appropriate properties? J Comp Aided Mol Des. 2002;16:325–334.
- Grande F, Aiello F, Grazia OD, Brizzi A, Garofalo A, Neamati N. Synthesis and antitumor activities of a series of novel quinoxalinhydrazides. Bioorg Med Chem 2007;15:288–294.
- Yakaiah T, Lingaiah BP, Narsaiah B, Shireesha B, Ashok Kumar B, Gururaj S, Parthasarathy T, Sridhar B. Synthesis and structure-activity relationships of novel pyrimido1,2-b]indazoles as potential anticancer agents against A-549 cell lines. Bioorg Med Chem Lett 2007;17:3445–3453.
- Kamal A, Khan MN, Srinivasa Reddy K, Rohini K. Synthesis of a new class of 2-anilino substituted nicotinyl arylsulfonylhydrazides as potential anticancer and antibacterial agents. Bioorg Med Chem 2007;15:1004–1013.