Abstract
A series of novel L-isoserine derivatives were synthesised and evaluated for their ability to inhibit aminopeptidase N (APN)/CD13. In our preliminary biological results, some of these compounds possessed a potent inhibitory activity against the APN. Within this series, compound 14b not only showed similar enzyme inhibition (IC50 of 12.2 μM) compared with the positive control bestatin (half maximal inhibitory concentration (IC50) of 7.3 μM), but also had a potent antiproliferative activity against human cancer cell lines cells.
Introduction
Aminopeptidase N (APN EC 3.4.11.2), also known as CD13, is a zinc-dependent membrane metalloprotease which belongs to the M1 family of ectoenzymes [Citation1]. It usually forms a noncovalently bound homodimer on the cell membrane of monocytes, myeloid, epithelial cells of the intestine and kidney, fibroblasts and tumour cells [Citation2–3]. This enzyme is able to cleave the N-terminal residues from polypeptide chains, as well as play an important role in modulating bioactive peptides or protein responses such as modification, activation, and degradation. APN is also involved in various physiological processes, such as cell proliferation, secretion, invasion and angiogenesis [Citation4–5]. For example, APN is over-expressed on the tumour cell surface for regulating ECM degradation and the invasion of tumour cells [Citation6]. Therefore, inhibition of APN/CD13 could prevent the spread and metastasis of cancer cells since several APN inhibitors have been well studied clinically over the last two decades. Among the APN inhibitors, bestatin is a representative molecule for the treatment of adult acute nonlymphocytic leukemia [Citation7].
So far, X-ray crystal structures for APN for both the holo- and apo-forms [Citation8] have been resolved along with a number of different ligands, such as bestatin [Citation9–10], arginine and lysine [Citation11]. According to the literature for binding modes, our group has developed various APN inhibitors, including L-lysine derivates [Citation12] and AHP A(α-Amino-β-Hydroxyl-phenylbutanoic acid) derivates, [Citation13] which both showed potent APN inhibitory activities. In our recent screening studies, L-isoserine showed the ability to inhibit APN at an half maximal inhibitory concentration (IC50 ) value of 563 μM, which could serve as a new lead compound for further chemical modification and optimisation.
It has been reported that L-isoserine-L-leucine dipeptide shows moderate aminopeptidase B inhibitory activity (IC50 of 140 μM) [Citation14]. This evidence indicates that the incorporation of an amino acid to L-isoserine may contribute to its inhibitory activity against APN. Therefore in our current work, different amino acids have been coupled with L-isoserine by forming dipeptides or tripeptides. This article will describe the synthesis, preliminary biological evaluation and structure-activity relationship (SAR) study of such a novel series of L-isoserine derivatives as potent APN inhibitors.
Materials and methods
Chemistry
The starting material L-Isoserine (7), is a white solid, (mp 199-201°C, [α]25D = -32.5° (c 1, H2O)), was purchased from JiaDing District, Shanghai, China. Unless otherwise specified, all reagents and buffer salts were purchased from commercial vendors and used without further purification. The solvents were distilled prior to use and flash chromatography was performed using silica gel (60 Å, 200 ± 300 mesh). Melting points were determined on an electrothermal melting point apparatus in an uncorrected form. Proton nuclear magnetic resonance (1H NMR) spectra were determined on a Brucker Avance 600 spectrometer using TMS as an internal standard in DMSO-d6 solutions. Chemical shifts were reported in delta (δ) units, parts per million (ppm) downfield from trimethylsilane. High-resolution mass spectral (HRMS) data are reported as m/e (relative intensity). The optical rotation was determined on a GYROMAT-HP Digital Automatic Polarimeter. All reported yields correspond to purified products.
General procedure for the synthesis of 2a, 2b, 4a and 4b
The title compound L-leucine methyl ester hydrochloride (2a) and L-phenylalanine methyl ester hydrochloride (2b) were prepared from L-leucine and L-phenylalanine according to the methods described in the literature [Citation15]. Using the same materials, the title compound L-leucine benzyl ester hydrochloride (4a) and L-phenylalanine benzyl ester hydrochloride (4b) were prepared as described by Kim et al. [Citation16].
General procedure for the synthesis of 6a, 6b, 6c and 6d
To a 200 mL solution of compound 3a (0.92 g, 4 mmol), 2b (0.88 g, 4.4 mmol), and hydroxybenzotriazole (HOBt) (0.65 g, 4.8 mmol), 4-Dimethylaminopyridine (DMAP) (0.09 g, 0.8 mmol) in dry DCM, was added TEA (1.33 g, 13.2 mmol). The reaction mixture was gently cooled to 0°C in an ice bath. To the reaction mixture was added dropwise a solution of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDCI) (1.53 g, 8 mmol) in DCM for 1 h. After removal of the ice bath, the reaction mixture was stirred at room temperature for 12h and then filtered to remove the precipitate. The filtrate was washed with a 1 mol/L citric acid solution, saturated NaHCO3 and brine, dried over Na2SO4, and evaporated in vacuo. The residue was purified by flash column chromatography (EtOAc/PE, 1:3(V/V)) to give the desired compound 5a as a white solid (1.48 g), yield: 94.2%, mp 70–73°C. 1H-NMR: (DMSO-d6) δ 7.919 (d, J = 7.8 Hz, 1H, -CONH-), 7.287–7.214 (m, 5H, Ar-H), 5.807 (d, J = 5.4 Hz, 1H, -CONH-), 4.474–4.459 (m, 1H, CH), 4.346–4.311 (m, 1H, CH), 3.568 (s, 3H, -OCH3), 3.102–3.089 (m, 1H, CH2), 2.787–2.75 (m, 1H, CH2), 1.526–1.501 (m, 1H, CH), 1.435 (s, 9H, CH3), 1.475–1.328 (m, 2H, CH2), 0.863 (d, J = 6.6 Hz, 3H, CH3), 0.828 (d, J = 6.6 Hz, 3H, CH3); ESI-MS m/z: 393.2 [M+H]+.
To a solution of compound 5a (0.78 g, 2 mmol) in dry EtOAc at 0°C was added dropwise a solution of EtOAc (10 mL) saturated by dry HCl gas. The reaction solution was stirred at 0°C for 2 h, and then the temperature was raised to room temperature and the reaction proceeded for 5 h before being concentrated in vacuo. The residue was recrystallised with MeOH and ether to give 6a (0.56 g) as a white crystals, yield: 85.3%, mp 190–193°C, [α]25D = +14.2° (c 1, MeOH). 1H-NMR: (DMSO-d6) δ 8.403 (d, J = 7.8 Hz, 1H, -CONH-), 8.054 (s, 3H, NH3), 7.285–7.202 (m, 5H, Ar-H), 4.482–4.472 (m, 1H, CH), 4.351–4.318 (m, 1H, CH), 3.573 (s, 3H, -OCH3), 3.11–3.092 (m, 1H, CH2), 2.791–2.751 (m, 1H, CH2), 1.527–1.495 (m, 1H, CH), 1.486–1.351 (m, 2H, CH2), 0.861 (d, J = 6.6 Hz, 3H, CH3), 0.831 (d, J = 6.6 Hz, 3H, CH3); ESI-MS m/z: 293.1 [M+H]+.
Compounds 6b, 6c and 6d were prepared following the general procedure as described above.
L-phenylalanyl-L-leucine methyl ester hydrochloride (6b)
White crystals, yield: 79.4%, mp 141–143°C; [α]25D = +17.5° (c 1, MeOH); 1H-NMR: (DMSO-d6) δ 8.578 (d, J = 7.8 Hz, 1H, -CONH-), 8.108 (s, 3H, NH3), 7.28–7.182 (m, 5H, Ar-H), 4.622–4.587 (m, 1H, CH), 4.325–4.198 (m, 1H, CH), 3.628 (s, 3H, -OCH3), 2.793–2.605 (m, 1H, CH2), 2.787–2.52 (m, 1H, CH2), 1.652–1.604 (m, 1H, CH), 1.541–1.479 (m, 2H, CH2), 0.893 (d, J = 6.6 Hz, 3H, CH3), 0.842 (d, J = 6.6 Hz, 3H, CH3); ESI-MS m/z: 293.2 [M+H]+.
L-leucyl-L-phenylalanine benzyl ester hydrochloride (6c)
A white solid, yield: 83.9%, mp 160–162°C; [α]25D = +6.3° (c 1, H2O); 1H-NMR: (DMSO-d6) δ 9.203 (d, J = 7.8 Hz, 1H, -CONH-), 8.345 (s, 3H, NH3), 7.363–7.226 (m, 10H, Ar-H), 5.09–5.04 (m, 2H, Ar-CH2-O-), 4.612–4.576 (m, 1H, CH), 3.805–3.781 (m, 1H, CH), 3.102–3.032 (m, 2H, Ar-CH2-),1.671–1.627 (m, 1H, CH), 1.522–1.499 (m, 2H, CH2), 0.85 (d, J = 6.6 Hz, 3H, CH3), 0.829 (d, J = 6.6 Hz, 3H, CH3); ESI-MS m/z: 369.3 [M+H]+.
L-phenylalanyl-L-leucine benzyl ester hydrochloride (6d)
A white solid, yield: 81.8%, mp 161–163°C; [α]25D = +5.6° (c 1, H2O); 1H-NMR: (DMSO-d6) δ 9.187 (d, J = 7.8, 1H, -CONH-), 8.32 (s, 3H, NH3), 7.389–7.24 (m, 10H, Ar-H), 5.168–5.105 (m, 2H, Ar-CH2-O-), 4.398–4.36 (m, 1H, CH), 4.133–4.111 (m, 1H, CH), 3.162–3.13 (m, 1H, Ar-CH2-), 2.953–2.917 (m, 1H, Ar-CH2-), 1.72–1.674 (m, 1H, CH), 1.678–1.526 (m, 2H, CH2), 0.896 (d, J = 6.6 Hz, 3H, CH3), 0.848 (d, J = 6.6 Hz, 3H, CH3); ESI-MS m/z: 369.2 [M+H]+.
Synthesis of (S)-3-(tert-butoxycarbonylamino)-2-hydroxypropanoic acid (8)
Compound 8 was synthesised from L-Isoserine following the general procedure as described above (preparation of 3a). a colourless solid, yield: 93.5%, mp 85–88°C; [α]25D = +6.7° (c 1, MeOH); 1H-NMR: (DMSO-d6) δ 7.934 (d, J = 7.8 Hz, 1H, -CONH-), 6.54 (s, 1H, OH), 4.674–4.651 (m, 1H, CH), 3.457 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H, CH2), 3.241 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H, CH2), 1.435 (s, 9H, CH3); ESI-MS m/z: 206.1 [M+H]+.
General procedure for the synthesis of 11a–11d and 13a–13d
Compound 9a was synthesised from N protected L-Isoserine (8) and L-phenylalanine methyl ester hydrochloride (2b) following the general procedure as described above (preparation of 5a), yield: 84.5%, mp 95–97°C; [α]25D = -9.72° (c 1, MeOH); 1H-NMR (DMSO-d6) δ 7.921 (d, J = 7.8 Hz, 1H, -CONH-), 7.287–7.362 (m, 2H, Ar-H), 7.177–7.166 (m, 3H, NH3), 6.526 (s, 1H, OH), 5.801 (d, J = 5.4 Hz, 1H, -CONH-), 4.604–4.567 (m, 1H, CH), 3.889–3.869 (m, 1H, CH), 3.627 (s, 3H, -OCH3), 3.173–3.135 (m, 1H, Ar-CH2- ), 3.075 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H, CH2), 3.032 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H, CH2), 2.817–2.77 (m, 1H, Ar-CH2-),1.435 (s, 9m, CH3); HRMS (FAB) calcd for C18H26N2O6 389.1683; found: 389.167 [M+Na]+.
Compound 11a was obtained through the deprotection of compound 9a in the saturated HCl/EtOAc solution as the preparation of 6a, yield: 77.7%, mp 138–140°C; [α]25D = -19.8° (c 1, H2O). 1H-NMR (DMSO-d6) δ 8.25 (d, J = 7.8 Hz, 1H, -CONH-), 8.094 (s, 3H, NH3), 7.294–7.27 (m, 2H, Ar-H), 7.23–7.197 (m, 3H, Ar-H), 6.541 (s, 1H, OH), 4.6–4.563 (m, 1H, CH), 4.203–4.173 (m, 1H, CH), 3.107 (s, 3H, -OCH3), 3.101 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H, CH2), 3.05 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H, CH2), 2.927 (dd, J = 13.2 Hz, J = 9 Hz, 1H, Ar-CH2-),2.569 (dd, J = 13.2 Hz, J = 9 Hz, 1H, Ar-CH2-); HRMS (FAB) calcd for C13H18N2O4 267.1339; found: 267.1335 [M+H]+.
Compounds 9c–9d were prepared from N protected L-Isoserine and amino acid methyl/benzyl ester hydrochlorides (2a,4a and 4b) and compounds 10a–10d were prepared from N protected L-Isoserine and dipeptide methyl/benzyl ester hydrochlorides(6a, 6b, 6c and 6d). Compounds 11c–11d and 13a–13d were obtained through the deprotection of compounds 9c–9d and 10a–10d.
L-Isoseryl-L-leucine methyl ester hydrochloride (11b)
A white solid, yield: 63.1%, mp 120–123°C; [α]25D = -36.1° (c 1, H2O); 1H-NMR (DMSO-d6) δ 8.054 (d, J = 7.8 Hz, 1H, -CONH-), 7.903 (s, 3H, NH3), 6.427 (s, 1H, OH), 4.607–4.593 (m, 1H, CH), 4.186–4.152 (m, 1H, CH), 3.218 (s, 3H, -OCH3), 3.106 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H, CH2), 3.023 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H, CH2), 1.523–1.704 (m, 3H, CH2CH), 0.926–0.947 (m, 6H, CH3); HRMS (FAB) calcd for C10H20N2O4 233.1496; found: 233.1493 [M+H]+.
L-Isoseryl-L- phenylalanine benzyl ester hydrochloride (11c)
A white solid, yield: 76.6%, mp 177–179°C; [α]25D = -23.3° (c 1, H2O); 1H-NMR (DMSO-d6) δ 8.285 (d, J = 7.8 Hz, 1H, -CONH-), 8.032 (s, 3H, NH3), 7.391–7.303 (m, 5H, Ar-H), 7.274–7.177 (m, 5H, Ar-H), 6.511 (s, 1H, OH), 5.14–5.097 (m, 2H, Ar-CH2-O-), 4.644–4.607 (m, 1H, CH), 4.195–4.164 (m, 1H, CH), 3.126 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H, CH2), 3.028 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H, CH2), 2.95–2.93 (m, 1H, Ar-CH2-), 2.601–2.566 (m, 1H, Ar-CH2-); HRMS (FAB) calcd for C19H22N2O4 343.1652; found: 343.1650 (M+H)+.
L-Isoseryl-L-leucine benzyl ester hydrochloride (11d)
A white solid, yield: 84.6%, mp 98–103°C; [α]25D = -25.7° (c 1, H2O); 1H-NMR (DMSO-d6) δ 8.387 (d, J = 7.8 Hz, 1H, -CONH-), 8.189 (s, 3H, NH3), 7.405–7.324 (m, 5H, Ar-H), 6.48 (s, 1H, OH), 5.187–5.13 (m, 2H, Ar-CH2-O-), 4.372–4.335 (m, 1H, CH), 4.287–4.256 (m, 1H, CH), 3.088 (dd, J = 12.6 Hz, J = 8.4 Hz, 1H, CH), 2.778 (dd, J = 12.6 Hz, J = 8.4 Hz, 1H, CH), 1.7–1.594 (m, 2H, CH2), 1.567–1.522 (m, 1H, CH), 0.882 (d, J = 6.6 Hz, 3H, CH3), 0.839 (d, J = 6.6 Hz, 3H, CH3); HRMS (FAB) calcd for C16H24N2O4 309.1809; found: 309.1807 (M+H)+.
L-Isoseryl-L-leucyl-L-phenylalanine methyl ester hydrochloride (13a)
A white solid, yield: 64.3%, mp 65–68°C; [α]25D = -33.2° (c 1, H2O); 1H-NMR (DMSO-d6) δ 8.588 (d, J = 7.8 Hz, 1H, -CONH-), 8.12 (s, 3H, NH3), 7.843 (d, J = 7.8 Hz, 1H, -CONH-), 7.298–7.2 (m, 5H, Ar-H), 6.485 (s, 1H, OH), 4.486–4.499 (m, 1H, CH), 4.368–4.329 (m, 1H, CH), 4.12–4.199 (m, 1H, CH), 3.579 (s, 3H, -OCH3), 3.102–3.093 (m, 1H, Ar-CH2-), 3.055 (dd, J = 13.80 Hz, J = 8.4 Hz, 1H, CH2), 2.977 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H, CH2), 2.798–2.752 (m, 1H, Ar-CH2-), 1.528–1.494 (m, 1H, CH), 1.483–1.359 (m, 2H, CH2), 0.861 (d, J = 6.6 Hz, 3H, CH3), 0.831 (d, J = 6.6 Hz, 3H, CH3); HRMS (FAB) calcd for C19H29N3O5 380.2180; found: 380.2174 (M+H)+.
L-Isoseryl-L-phenylalanyl-L-leucine methyl ester hydrochloride (13b)
A white solid, yield: 50.9%, mp 77–80°C; [α]25D = -26.7° (c 1, H2O); 1H-NMR (DMSO-d6) δ 8.631(d, J = 7.8 Hz, 1H, -CONH-), 8.1 (s, 3H, NH3), 7.862 (d, J = 7.8 Hz, 1H, -CONH-), 7.298–7.195 (m, 5H, Ar-H), 6.512 (s, 1H, OH), 4.633–4.596 (m, 1H, CH), 4.33–4.269 (m, 1H, CH), 4.166–4.145 (m, 1H, CH), 3.639 (s, 3H, -OCH3), 3.077 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H, CH2), 2.918 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H, CH2), 2.895–2.55 (m, 1H, Ar-CH2-), 2.568–2.521 (m, 1H, Ar-CH2-), 1.656–1.597 (m, 2H, CH2), 1.544–1.497 (m, 1H, CH), 0.894 (d, J = 6.6 Hz, 3H, CH3), 0.847 (d, J = 6.6 Hz, 3H, CH3); HRMS (FAB) calcd for C19H29N3O5 380.2180; found: 380.2179 (M+H)+.
L-Isoseryl-L-leucyl-L-phenylalanine benzyl ester hydrochloride (13c)
A white solid, yield: 52.2%, mp 135–137°C; [α]25D = -30.5° (c 1, H2O); 1H-NMR (DMSO-d6) δ 8.631(d, J = 7.8 Hz, 1H, -CONH-), 8.134 (s, 3H, NH3), 7.827 (d, J = 7.8 Hz, 1H, -CONH-), 7.368–7.311 (m, 3H, Ar-H), 7.27–7.202 (m, 7H, Ar-H), 6.493 (s, 1H, -OH), 5.088–5.033 (m, 2H, Ar-CH2-O-), 4.547–4.51 (m, 1H, CH), 4.381–4.341 (m, 1H, CH), 4.216–4.201 (m, 1H, CH), 3.084–3.074 (m, 1H, Ar-CH2-), 3.067 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H, CH2), 2.993 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H, CH2), 2.782–2.63 (m, 1H, Ar-CH2-),1.512-1.35 (m, 3H, CH2CH), 0.844 (d, J = 6.6 Hz, 3H, CH3), 0.797 (d, J = 6.6 Hz, 3H, CH3); HRMS (FAB) calcd for C25H33N3O5 456.2493; found: 456.2490 (M+H)+.
L-Isoseryl-L-phenylalanyl-L-leucine benzyl ester hydrochloride (13d)
A white solid, yield: 58.1%, mp 140–142°C; [α]25D = -28.6° (c 1, H2O); 1H-NMR (DMSO-d6) δ 8.644 (d, J = 7.8 Hz, 1H, -CONH-), 8.037 (s, 3H, NH3), 7.852 (d, J = 7.8 Hz, 1H, -CONH-), 7.377–7.319 (m, 3H, Ar-H), 7.242–7.171 (m, 5H, Ar-H), 6.494 (s, 1H, OH), 5.166–5.121 (m, 2H, Ar-CH2-O-), 4.64–4.603 (m, 1H, CH), 4.385–4.348 (m, 1H, CH), 4.142–4.126 (m, 1H, CH), 3.028 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H, CH2), 2.912–2.892 (m, 1H, Ar-CH2-), 2.851 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H, CH2), 2.54–2.504 (m, 1H, Ar-CH2-),1.657–1.529 (m, 3H, CH2CH), 0.906 (d, J = 6.6 Hz, 3H, CH3), 0.846 (d, J = 6.6 Hz, 3H, CH3); HRMS (FAB) calcd for C25H33N3O5 456.2493; found: 456.2496 (M+H)+.
General procedure for the synthesis of 12a–12b and 14a–14b
Compound 9c (1.77 g, 4 mmol) was hydrogenated in absolute MeOH (30 mL) with a catalytic quantity of 10% Pd/C (0.17 g). The mixture was stirred for 10 h under normal pressure then filtered. The filtrate was evaporated to give a white solid. The solid was dissolved in dry EtOAc (5 mL) at 0°C and added dropwise to a solution of EtOAc (10 mL) saturated by dry HCl gas. The reaction solution was stirred at 0°C for 12 h to provide a white solid. The solution was removed by filtration, and the remaining solid was recrystallised with MeOH and Et2O to give 12a white crystals (0.91 g), yield: 78.9%, mp 187–189°C; [α]25D = -13.4° (c 1, H2O); 1H-NMR (DMSO-d6) δ 12.964 (s, 1H, COOH), 8.081 (s, 3H, NH3), 8.07 (d, J = 8.4 Hz, 1H, -CONH-), 7.275 (t, J = 7.8 Hz, 2H, Ar-H), 7.223–7201 (m, 3H, Ar-H), 6.527 (s, 1H, OH), 4.543–4.507 (m, 1H, CH), 4.183–4.169 (m, 1H, CH), 3.113 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H, CH2), 3.031 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H, CH2), 2.957–2.938 (m, 1H, Ar-CH2-), 2.62–2.561 (m, 1H, Ar-CH2-); HRMS (FAB) calcd for C12H16N2O4 253.1183; found: 253.1180 (M+H)+.
L-Isoseryl-L-leucine hydrochloride (12b)
A white solid, yield: 88.3%, mp 213–215°C; [α]25D = -44.5° (c 1, H2O); 1H-NMR (DMSO-d6) δ 12.713 (s, 1H, COOH), 8.102 (s, 3H, NH3), 8.088 (d, J = 8.4 Hz, 1H, -CONH-), 6.441 (s, 1H, OH), 4.236–4.229 (m, 1H, CH), 4.274–4.251 (m, 1H, CH), 3.086 (dd, J = 12.6 Hz, J = 7.2 Hz, 1H, CH2), 2.784 (dd, J = 12.6 Hz, J = 7.2 Hz, 1H, CH2), 1.681–1.585 (m, 2H, CH2), 1.556–1.513 (m, 1H, CH), 0.893 (d, J = 6.6 Hz, 3H, CH3), 0.854 (d, J = 6.6 Hz, 3H, CH3); HRMS (FAB) calcd for C9H18N2O4 219.1339; found: 219.1340 (M+H)+.
L-Isoseryl-L-leucyl-L-phenylalanine hydrochloride (14a)
A white solid, yield: 56.9%, mp 90–92°C; [α]25D = -34.7° (c 1, H2O); 1H-NMR (DMSO-d6) δ 12.69 (s, 1H, COOH), 8.468 (d, J = 7.8 Hz, 1H, -CONH-), 8.119 (s, 3H, NH3), 7.836 (d, J = 7.8 Hz, 1H, -CONH-), 7.245–7.168 (m, 5H, Ar-H), 6.52 (s, 1H, OH), 4.652–4.573 (m, 1H, CH), 4.263–4.225 (m, 1H, CH), 4.176–4.084 (m, 1H, CH), 3.986 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H, CH2), 2.887 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H, CH2), 2.812–2.798 (m, 1H, Ar-CH2), 2.51–2.439 (m, 1H, Ar-CH2), 1.502–1.414 (m, 3H, CH2CH), 0.862 (d, J = 6.6 Hz, 3H, CH3), 0.813 (d, J = 6.6 Hz, 3H, CH3); HRMS (FAB) calcd for C18H27N3O5 366.2023; found: 366.2026 (M+H)+.
L-Isoseryl-L-phenylalanyl-L-leucine hydrochloride (14b)
A white solid, yield: 53.6%, mp 87–89°C; [α]25D = -35.1° (c 1, H2O); 1H-NMR (DMSO-d6) δ 12.647 (s, 1H, COOH), 8.475 (d, J = 7.8 Hz, 1H, -CONH-), 8.109 (s, 3H, NH3), 7.847 (d, J = 7.8 Hz, 1H, -CONH-), 7.265–7.176 (m, 5H, Ar-H), 6.524 (s, 1H, OH), 4.637–4.6 (m, 1H, CH), 4.274–4.236 (m, 1H, CH), 4.159–4.095 (m, 1H, CH), 3.09 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H, CH2), 2.905 (dd, J = 13.8 Hz, J = 8.4 Hz, 1H, CH2), 2.896–2.882 (m, 1H, Ar-CH2), 2.539–2.505 (m, 1H, Ar-CH2), 1.72–1.526 (m, 3H, CH2CH), 0.907 (d, J = 6.6 Hz, 3H, CH3), 0.853 (d, J = 6.6 Hz, 3H, CH3); HRMS (FAB) calcd for C18H27N3O5 366.2023; found: 366.2027 (M+H)+.
In vitro APN inhibition assay
The IC50 values against APN were determined using L-Leu-p-nitroanilide as the substrate and microsomal aminopeptidase from porcine kidney microsomes (Sigma) in 50 mM PBS, pH 7.2, at 37°C. The hydrolysis of the substrate was monitored by following the change in the absorbance measured at 405 nm with a UV-Vis spectrophotometer Pharmacia LKB, Biochrom 4060. All solutions of inhibitors were prepared in the assay buffer, and the pH was neutralised to 7.5 by the addition of either 0.1 M HCl or 0.1 M NaOH.
The samples and positive controls were serial diluted to various concentrations: 1280 μg/mL, 320 μg/mL, 80 μg/mL, and 20 μg/mL, 5 μg/mL and 1.25 μg/mL. All compounds were pre-incubated with APN on a 96-well plate for 30 min. The assay mixture, including the compound solution, the enzyme solution (5 μg/mL final concentration) and the assay buffer, was adjusted to give a total volume of 200 μL. Fifteen minutes later, the absorbance (OD405) of the wells was recorded on a microplate reader. The percentage inhibition was calculated according to the absorbance of the assay wells relative to those of the control wells. Finally, the IC50 values were determined using a linear regression analysis of the concentration/inhibition data.
In vitro MMP-2 inhibition assay
Gelatinase A (MMP-2) and picrylsulphonic acid (TNBS) were purchased from Sigma. Succinylated gelatin was synthesised as described by Baragi et al. [Citation17]. The compound samples were assayed for inhibitory activity against MMP-2 in 96-well microplates using succinylated gelatin as the substrate. The compounds and gelatinase were dissolved in sodium borate buffer (pH 8.5, 50 mM), and incubated at 37°C for 30 min. The substrate was added and incubated at 37°C for another 60 min. Then 0.03% picrylsulphonic acid solution was added and incubated at room temperature for an additional 20 min. The resulting solutions were detected under 450 nm wavelength to gain absorbance (OD450). MMP-2 inhibition activity was measured by the following formula [Citation17]: Inhibition ration (%) = [OD450(100%)- OD450(compound)]/[OD450(100%)- OD450(blank)]×100%. The IC50 values were obtained from the above inhibitory rates using the OriginPro 7.5 software.
In vitro HL-60 and SKOV3 cells viability assay
The HL-60 and SKOV3 Cells were grown in RPMI1640 medium with 10% fetal bovine serum at 37°C in a 5% CO2 humidified incubator. Cell proliferation was determined by the MTT (3-[4, 5- dimethyl-2-thiazolyl]-2.5-diphenyl-2H-tetrazolium bromide) assay. Briefly, 10,000 HL-60 cells (or 5,000 SKOV3 cells) per well were plated in a 96-well plate, cultured for 4 h in complete growth medium, then treated with 400 μg/mL, 200 μg/mL, 100 μg/mL, 50 μg/mL and 25 μg/mL of compounds for 48 h, respectively. An aliquot of 0.5% MTT (10 μL/well) was then added to each well. After further incubation for 4h, the formazan formed from the MTT was extracted by adding DMSO and mixing for 10 min. The optical absorbance was read with an micoplate reader at 550 nm, and the IC50 values were calculated according to a regression analysis of the concentration/inhibition data.
Results and discussion
Chemistry
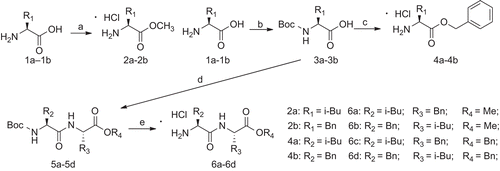
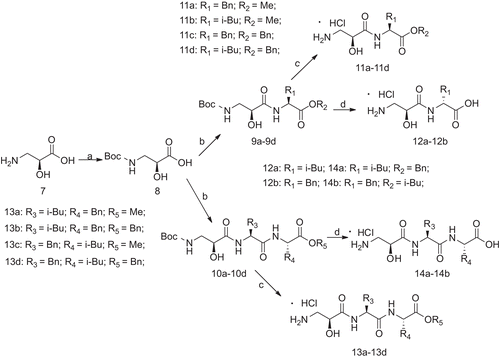
L-leucine (1a) and L-phenylalanine (1b) were used as the starting materials to yield their methyl esters 2a and 2b, and then protected by (Boc)2O to obtain 3a and 3b. The carboxylic acids of 3a–3b were protected using benzyl chloride, and then the Boc group was deprotected with 3 N HCl in ethyl acetate to give 4a and 4b. N-Boc-protected amino acids (3a and 3b) were coupled with intermediates (2a, 2b, 4a and 4b) by a classical EDCI/HOBt method, and then converted into 6a–6d by deprotecting the Boc group (). Using the same procedure, Boc-protected L-isoserine 8 was coupled with the intermediate (2a, 2b, 4a and 4b) and 6a–6d to yield the dipeptide 9a–9d and tripeptide 10a–10d containing protecting group. Finally, target compounds 11a–11d and 12a–12d were obtained by easily cleaving Boc, while the target compounds 13a–13b and 14a–14b were obtained from 9c–9d and 10c–10d by first reacting in a hydrogen atmosphere with 15% Pd/C and then deprotecting Boc ().
Scheme 1. Reagents and conditions: a. CH3OH, HCl; b. (Boc)2O, CH3OH, NaOH; c. i) K2CO3, KI, TBAI, benzyl chloride, CH3COCH3; ii) HCl/anhydrous EtOAc; d. EDCI, HOBt/anhydrous, DCM, 2a∼2b,4a∼4b, 0°C to room temperature; e. HCl/anhydrous EtOAc.
Scheme 2. Reagents and conditions: a. (Boc)2O, CH3OH, NaOH; b. EDCI, HOBt/anhydrous, DCM, 2a∼2b,4a∼4b,6a∼6d, 0°C to room temperature; c. HCl/anhydrous EtOAc; d. i) 15% Pd/C, CH3OH. ii) HCl/anhydrous EtOAc.
In vitro APN and MMP assays
All the target compounds were tested for their inhibitory potential against APN and MMP-2 and the results are shown in . The matrix metalloproteinase-2 (MMP-2) also belongs to the zinc-dependent metalloproteinase family and is associated with tumour invasion and metastasis. These inhibitory studies were performed on both APN and MMP-2 so as to identify the selectivity of the target compounds.
Table 1. The structures and IC50 values of target compounds against APN and MMP-2.
Table
From the results of the enzyme assay, compound 14b (IC50 of 12.2 μM) is the most potent APN inhibitor of all the target compounds, and showed an inhibitory activity similar to that of bestatin (IC50 of 7.3 μM). In addition, the dipeptides derivatives (11a, 11b, 11c, 11d, 12a and 12b) showed less inhibition than their tripeptide analogues (13a, 13b, 13c, 13d, 14a and 14b). This phenomenon suggests that the third amino acid residue helps to increase the interaction with the APN binding site. One the other hand, the tripeptide compounds (13c, 13d and 14b) with a scaffold of isoser-L-Phe-L-Leu exhibited a better inhibitory activity than that of the compounds (13a, 13b and 14a) with an isoser-L-Leu-L-Phe scaffold. As for the compounds (13c, 13d and 14b) with a substituted R5, the hydrogen seems to be the best substitution for this position. A possible reason may be that the free terminal carboxylate group could enhance the hydrogen bond interaction with the active sites of APN.
In our previous studies, most of the compounds tested showed better inhibition on MMP-2 than APN [Citation18]. However, most of these L-isoserine dipeptides and tripeptides derivatives exhibited a better inhibitory activity on APN than MMP-2. This result may be due to the differences between the 3D structure of the active sites of APN and MMP-2. According to the binding site of APN (PDB code: 1HS6) and MMP-2 (PDB code: 1HOV, 1CK7, 1QIB), the active site of APN was much deeper than the active site of MMP-2 [Citation17].The scaffold of our L-isoserine derivatives are linear structures, and therefore there is easier access to the APN active site.
Antiproliferative activity assays on HL-60 and SKOV3 cells
The human promyelocytic leukaemia cell line (HL-60) was used to evaluate antiproliferative activity with the MTT assay due to APN being over-expressed. In addition, the human ovarian carcinoma cell line (SKOV3) was chosen as an additional model for MTT assays. According to the results (), all the test compounds showed cytostatic activity except for compound 13d. It should also be pointed out that compound 14b not only displays the most potent inhibition on APN, but also has a higher antiproliferative activity than bestatin.
Table 2. In vitro antiproliferative inhibiting potency of compounds 11c, 13c, 13d and 14b.
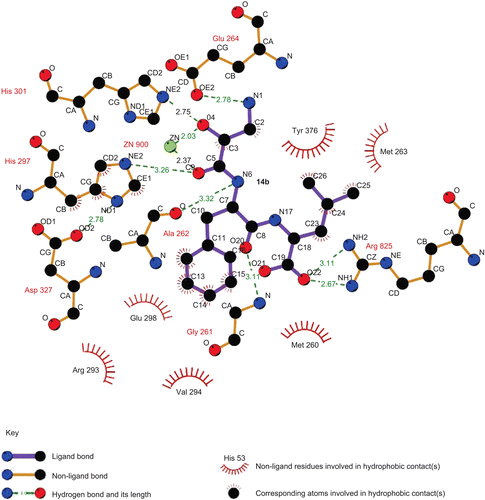
In order to determine the interaction between our target compounds with APN, the most potent compound, 14b, was constructed with a Sybyl/Sketch module and optimised using the Tripos force field. The docking study performed with the Sybyl/FlexX module was based on the active site of the APN co-crystal structure with bestatin (PDB code: 2DQM). The docking results showed that the carbonyl group and hydroxyl group of compound 14b could chelate with the zinc ion in APN ( and ). The terminal amino group of 14b could form a hydrogen bond with Glu264 and the carboxylic group of 14b is able to interact with Arg825. In addition, the phenylalanine residue of 14b can insert an S1′ pocket which is surrounded by Gly261, Ala262 and Glu298, while the leucine residue in 14b interacts with the S2′ pocket containing Met263 and Tyr376
Figure 1. The docking result for 14b with the active site of APN (PDB: 2DQM).
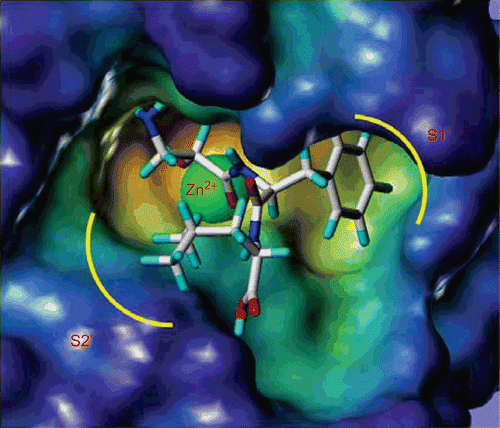
Figure 2. The docking results for 14b with APN showed by LIGPLOT.
Conclusion
In summary, we have described the synthesis and performed a SAR study for a novel series of L-isoserine derivatives as inhibitors of APN. Most of these compounds possessed potent activity toward APN. Among them, compound 14b exhibited a potent inhibitory activity and a significant selectivity for APN over MMP-2. Furthermore, this compound also showed good cell-based inhibitory activity, and could be used as a potential lead compound for new anti-cancer agents in the future.
Declaration of interest
This work was supported by National Natural Foundation Research Grant (Grant No. 9071304; No 30772654) and National High Technology Research and Development Program of China (863 proJect; Grant No 2007AA02Z314) and Doctoral Foundation of Ministry of Education of the People’s Republic of China (Grant No 20060422029).
References
- Danziger RS. Aminopeptidase N in arterial hypertension. Heart Fail Rev 2008; 13:293–298.
- Dixon J, Kaklamanis L, Turley H, Hickson ID, Leek RD, Harris AL, Gatter KC. Expression of aminopeptidase-N (CD 13) in normal tissues and malignant neoplasms of epithelial and lymphoid origin. J Clin Pathol 1994;47:43–47.
- Zhang R, Hua G, Andacht TM, Adang MJ. A 106-kDa aminopeptidase is a putative receptor for Bacillus thuringiensis Cry11Ba toxin in the mosquito Anopheles gambiae. Biochemistry 2008;47:11263–11272.
- Uehara N, FuJita M, Shimizu T. Colorimetric assay of aminopeptidase N activity based on inhibition of the disassembly of gold nano-composites conjugated with a thermo-responsive copolymer. Anal Sci 2009;25:267–273.
- Bauvois B, Dauzonne D. Aminopeptidase-N/CD13 (EC 3.4.11.2) inhibitors: chemistry, biological evaluations and therapeutic prospects. Med Res Rev 2006;26:88–130.
- Fukasawa K, FuJii H, Saitoh Y, Koizumi K, Aozuka Y, Sekine K, Yamada M, Saiki I, Nishikawa K. Aminopeptidase N (APN/CD13) is selectively expressed in vascular endothelial cells and plays multiple roles in angiogenesis. Cancer Lett 2006;243:135–143.
- Xu W, Li Q. Progress in the development of aminopeptidase N (APN/CD13) inhibitors. Curr Med Chem AntiCancer Agents 2005;5:281–301.
- Onohara Y, NakaJima Y, Ito K, Xu Y, Nakashima K, Ito T, Yoshimoto T. Crystallization and preliminary X-ray characterization of aminopeptidase N from Escherichia coli. Acta Crystallogr Sect F Struct Biol Cryst Commun 2006;62:699–701.
- Ito K, NakaJima Y, Onohara Y, Takeo M, Nakashima K, Matsubara F, Ito T, Yoshimoto T. Crystal structure of aminopeptidase N (Proteobacteria alanyl aminopeptidase) from Escherichia coli and conformational change of methionine 260 involved in substrate recognition. J Biol Chem 2006;281:33664–33676.
- Addlagatta A, Gay L, Matthews BW. Structure of aminopeptidase N from Escherichia coli suggests a compartmentalized, gated active site. Proc Natl Acad Sci USA 2006;103:13339–13344.
- Addlagatta A, Gay L, Matthews BW. Structural basis for the unusual specificity of Escherichia coli aminopeptidase N. Biochemistry 2008;47:5303–5311.
- Mou J, Fang H, Jing F, Wang Q, Liu Y, Zhu H, Shang L, Wang X, Xu W. Design, synthesis and primary activity evaluation of L-arginine derivatives as amino-peptidase N/CD13 inhibitors. Bioorg Med Chem 2009;17:4666–4673.
- Yang K, Wang Q, Su L, Fang H, Wang X, Gong J, Wang B, Xu W. Design and synthesis of novel chloramphenicol amine derivatives as potent aminopeptidase N (APN/CD13) inhibitors. Bioorg Med Chem 2009;17:3810–3817.
- Nishizawa R, Saino T, Takita T, Suda H, Aoyagi T. Synthesis and Structure-Activity Relationships of Bestatin Analogues, Inhibitors of Aminopeptidase B. J Med Chem 1977;20:510–515.
- Jordis U, Sauter F, Siddiqi SM. Synthesis of (1S,4S)-2-thia-5-azabicyclo[2.2.1]heptane. Indian J Chem Sec B 1989;28:294–296.
- Kim S, Lee JI, Kim YC. A simple and mild esterification method for carboxylic acids using mixed carboxylic-carbonic anhydrides. J Org Chem 1985;50:560–565.
- Baragi VM, Shaw BJ, Renkiewicz RR, Kuipers PJ, Welgus HG, Mathrubutham M, Cohen JR, Rao SK. A versatile assay for gelatinases using succinylated gelatin. Matrix Biol 2000;19:267–273.
- Wang Q, Chen M, Zhu H, Zhang J, Fang H, Wang B, Xu W. Bioorg Med Chem 2008;16:5473–5481.