Abstract
Because of the complex cascade of molecular events that can occur in the brain of an Alzheimer’s disease (AD) patient, the therapy of this neurodegenerative disease seems more likely to be achieved by multifunctional drugs. Herein, a new series of dual-targeting ligands have been developed and in vitro bioevaluated. Their architecture is based on conjugating the acetylcholinesterase inhibition and anti-oxidant properties in one molecular entity. Specifically, a series of naturally occurring phenolic acids with recognized anti-oxidant properties (derivatives of caffeic acid, rosmarinic acid, and trolox) have been conjugated with choline to account for the recognition by acetylcholinesterase (AChE). The synthesized hybrid compounds evidenced AChE inhibitory capacity of micromolar range (rationalized by molecular modeling studies) and good antioxidant properties. Their effects on human neuroblastoma cells, previously treated with beta-amyloid peptides and 1-methyl-4-phenylpyridinium ion neurotoxins (to simulate AD and Parkinson’s disease, respectively), also demonstrated a considerable capacity for protection against the cytotoxicity of these stressors.
| Abbreviations | ||
| AD | = | Alzheimer’s disease |
| AChE | = | acetylcholinesterase |
| AChEI | = | acetylcholinesterase inhibitors or inhibition |
| PD | = | Parkinson’s disease |
| Abeta | = | β-amyloid plaques |
| MPP+ | = | 1-methyl-4-phenylpyridinium ion |
| Abeta1–42 | = | 42 residues-long β-amyloid peptides |
| DPPH | = | 2,2-diphenyl-1-picrylhydrazyl radical |
| BHT | = | 3,5-di-tert-butyl-4-hydroxytoluene |
| ROS | = | reactive oxygen species |
| NMDA | = | N-methyl D-aspartate |
Introduction
Alzheimer’s disease (AD) is the most common form of age-related neurodegenerative disorders, accounting for 50–70% of all cases.Citation1,Citation2 AD individuals exhibit progressive memory loss and subsequent deficits in cognitive abilities. AD is typified by pathological depositions of β-amyloid (Abeta) plaques and neurofibrillary tangles within specific regions of the brain.
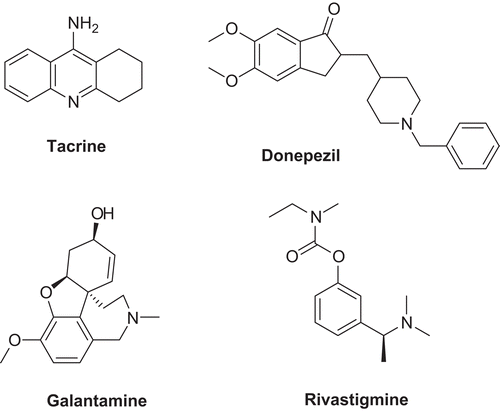
The complexity of the mechanisms underlying the progression of AD presents a current challenge for the development of new therapeutic agents. Despite the huge amount of research focused on tackling upstream or downstream pathways of the disease onset and plaque formation,Citation3 up to date the only Food and Drug Administration (FDA)-approved AD drugs consist of acetylcholinesterase inhibitors (AChEI; ) and a N-methyl D-aspartate (NMDA) receptor antagonist.Citation4 The AChEI-based therapies lead to the recovery of the cholinergic system to enhance synapses, but they may treat only soft to moderate severity cases, and do not reverse or heal the disease.Citation5,Citation6
Figure 1. Inhibitors of AChE currently approved by FDA as drugs against AD.
Complementary coadjutant therapeutics have also been proposed for AD treatments,Citation7 due to the brain vulnerability to oxidative damage. In fact, as a result of several mechanisms, radical reactive species can promote the formation of neurotoxic aggregates, such as the Abeta plaques. It is also known that AD brains display evidence of metal dyshomeostasis (namely of calcium, iron and copper), which in some cases may be related to an increase in the oxidative stress.Citation3,Citation8 Anti-oxidant compounds and metal chelators are able, respectively, to scavenge free radical species and modulate the metal excess. Thus, oxidative stress, protein aggregation, and redox active metal ions are all considered to be promising pharmacological targets.Citation9–11
However, the complexity of combined therapy protocols leads to the recent search for alternative therapeutic strategies based on multifunctional compounds.Citation12–16 Thus, following the same strategy, we have decided to develop new bifunctional compounds as drug candidates for anti-AD therapy, by conjugating, in the same compound, molecular fragments for both neurorestorative and neuroprotective roles, such as the inhibition of acetylcholinesterase (AChE) and the antioxidation.
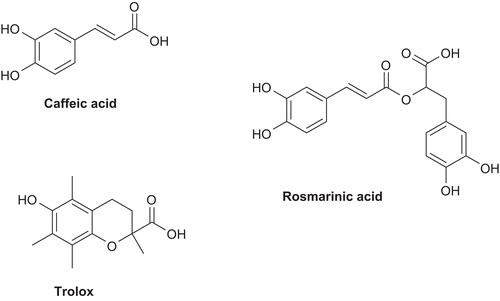
Concerning the antioxidant approach, we have selected molecular fragments containing a phenol (or catechol) group with an α,β-unsaturated carboxylic acid chain, namely caffeic and rosmarinic acids, as well as trolox (). These are well-known naturally occurring potent antioxidants with high radical scavenging capacity and also some metal chelating ability.Citation1,Citation17 Noteworthy is the fact that, besides having antioxidant properties, trolox is also an analogue of vitamin E, rosmarinic acid has proven antiinflammatory activity,Citation18 and caffeic acid has anti-(5-lipoxygenage) activity.Citation19
Figure 2. Caffeic acid, rosmarinic acid, and trolox, all nontoxic natural antioxidants.
Concerning the molecular fragment for the AChE inhibition, we aimed at getting some emulation of donepezil (Dnp), one of the most potent AChEIs (IC50 value of 33 nM) still in use.Citation20 However, our main design strategy was to take profit of hybrid drugs, even with individual moderate effects, instead of very potent mono-targeting drug with eventual severe side effects (e.g., tacrine).Citation6 In the past, derivatives of cinnamoylcholine have been described as quite potent inhibitors of AChE, with inhibitory constants (KI) between sub-micromolar and sub-millimolar range.Citation21 On the other hand, α,β-dehydrophenylalanine choline esters have recently been found to inhibit that enzyme at micromolar level concentration.Citation22 Thus, we have decided to study the choline esters of these phenolic carboxylic acids, to conjugate, in each molecular entity, their antioxidant properties with the AChE inhibitory activity.
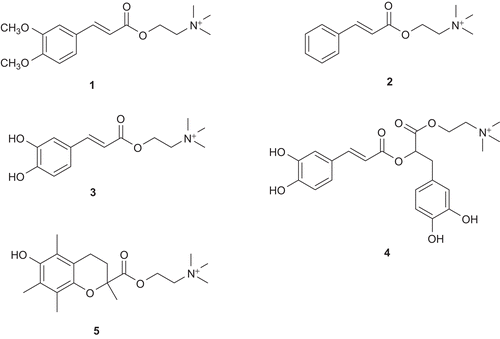
Herein, we describe the preparation of the bifunctional choline esters of three caffeic acid derivatives, rosmarinic acid, trolox (), as well as the resulting biological (neuroprotective) properties. These include their AChE inhibitory and antioxidant activities, and the neuroprotective efficacy in 1-methyl-4-phenylpyridinium ion (MPP+) and 42 residues-long β-amyloid peptides (Abeta1–42) induced cell death, as mimics of Parkinson’s disease (PD) and AD, respectively.Citation16
Figure 3. The synthesized choline ester derivatives of: 3,4-dimethoxycinnamic acid (1), cinnamic acid (2), caffeic acid (3), rosmarinic acid (4), and trolox (5).
Methods
Chemicals and equipment
Analytical grade reagents from Aldrich, Fluka, and Acros, were purchased from commercial suppliers (in Lisbon), and they were used as supplied. AChE type VI-S, from electric eel 349 U/mg solid, 411 U/mg protein, 5,5′-dithiobis(2-nitrobenzoic) acid] (DTNB), tris(hydroxymethyl)-aminomethane (Tris buffer), acetylthiocholine iodide (AChI), caffeic acid, and rosmarinic acid were obtained from Sigma. Solvents were dried according to standard methods.Citation23 The chemical reactions were monitored by thin-layer chromatography (TLC) using aluminium plates coated with silica gel 60 F254 (Merck). TLC detection methods were used: UV 254 nm, staining with I2 (general), ninhydrin (for amino groups), and dragendorff (for quaternary ammonium salts). The common mobile phases were used: C1 (nBuOH–HCOOH–H2O 75:14:12); C2 (CHCl3–MeOH 9:1); C3 (acetonitrile (ACN)-H2O 7:1 0.1% trifluoroacetic acid (TFA)); and C4 (ACN-H2O 5:1). Column flash chromatography separations were performed on silica gel Merck 230–400 mesh. The reverse-phase high-performance liquid chromatography (RP-HPLC) analysis was carried out in a Liquid Chromatograph Finnigan Surveyor Plus Modular LC System equipped with a Purospher STAR RP-18 column, from Merck and Xcalibur software. The compounds were analyzed by injecting a 0.5 mg/ml solution (25 μL) and using a gradient composed of solution A (0.05% trifluoroacetic acid), solution B (ACN), and solution C (methanol) as follows: 0 min 90% A, 10% B and 40 min 22% A, 66% B, 12% C. For choline caffeate, a better resolution was obtained when using a gradient composed of solution A (0.05% trifluoroacetic acid) and solution B (methanol) as follows: 0 min 70% A, 30% B; 20 min 10% A, 90% B; and 25 min 10% A, 90% B. The detection was carried out at wavelength between 200 and 600 nm with a diode array detector. To further purify the compounds, the HPLC system referred previously was also used, the peaks were collected separately, and this process was repeated several times. Melting points were measured with a Leica Galen III hot stage apparatus and are uncorrected. Compensating anions were determined by Ionex HPLC using Dionex ICS-1500 instrument equipped with Dionex IonPac AS14A 7 μm (4 × 250 mm) anion exchange column, anion self-regenerating suppressor (ASRS Ultra II), Dionex DS6 conductivity detector, and a 25 μL inject loop. Isocratic elution was carried out with 1.1 ml/min flow of carbonate buffer (16 mM Na2CO3, 1 mM NaHCO3) for 15 min. RT (Cl−) 3.7 ± 0.3 min. RT (CF3COO−) 9.7 ± 0.9 min. Chromatograms were collected by PC with Chromeleon software. The mass spectra of the compounds were normally performed in a Varian 500-MS LC Ion Trap instrument equipped with an electrospray ionisation (ESI) ion source. The high-resolution mass spectra (HRMS) were performed in a Bruker MicrOTOF ESI–TOF apparatus. The NMR spectra (for 1H, 13C, attached proton test (APT), and 2D experiments) were measured on Bruker AVANCE III 300 MHz and Bruker AVANCE III 400 MHz spectrometers. The structural assignment of the hydrogen and carbon chemical shifts was achieved using homonuclear two-dimensional 1H–1H correlation spectroscopy, 2D 1H–13C heteronuclear multiple quantum coherence, and 1H–13C heteronuclear multibond correlation experiments. The UV measurements were carried out in a Camspec M 350 Double beam UV–vis spectrophotometer.
Synthesis of the compounds
Chlorocholine chloride (6)
Choline chloride (2.89 g, 20.1 mmol) was dried in high vacuum at 200°C for 2 h. Freshly distilled SOCl2 (20 ml) was added to the white solid.Citation24 The solution was heated under reflux for 1 h. After cooling, solvent was evaporated to dryness. Solid was redissolved in MeOH (3 × 20 ml), and re-evaporated to dryness. White crystalline solid was dried in high vacuum at 80°C until constant mass. Yield 2.58 g (16.3 mmol, 81%) of white extremely hygroscopic solid 3. 1H-NMR (300 MHz, D2O), δ: 4.04 (m, 2H, Cl–CH2), 3.80 (t, 2H, 7 Hz, N–CH2), 3.24 (s, 9H, 3 × CH3). 13C-APT-NMR (400 MHz, D2O), δ: 67.29 (CH2N), 67.26 (CH2N), 67.23 (CH2N), 54.59 (CH3), 54.55 (CH3), 54.51 (CH3), 36.59 (CH2–Cl).
Methyl 3,4-dimethoxycinnamate (7)
This compound was prepared according to the procedure of Jung et al.Citation25 To a solution of caffeic acid (1 g, 5.6 mmol) and K2CO3 (2.49 g, 18 mmol) in THF (20 ml), dimethylsulfate (1.58 ml, 16.6 mmol) was added drop-wise. The mixture was heated under reflux over night. Because the reaction was not completed, new portions of dimethylsulfate (0.5 ml) and K2CO3 (1 g) were added. After one more day of reflux, there was no progress. Reaction mixture was evaporated to dryness and separated by flash column chromatography (using CH2Cl2 as eluent). After solvent evaporation, the residue was dried on vacuum line, affording compound 7 as a white solid (0.291 g, 24%); m.p. 66–70°C. TLC Rf 0.93 in C2 mixture. TLC Rf 0.52 in CH2Cl2. 1H-NMR (300 MHz, CDCl3), δ: 7.64 (d, 1H, 16 Hz, Ph–CH=), 7.11 (dd, 1H, 8 Hz, 2 Hz, HPh-6), 7.05 (d, 1H, 2 Hz, HPh-2), 6.86 (d, 1H, 8 Hz, HPh-5), 6.32 (d, 1H, 16 Hz, =CH–CO), 3.91 (s, 6H, 2 × CH3O–Ph), 3.79 (s, 3H, COOCH3). 13C-APT-NMR (400 MHz, CDCl3), δ: 167.82 (COO), 151.29 (CPh-4), 149.36 (CPh-3), 144.94 (Ph–CH=), 127.52 (CPh-1), 122.75 (CPh-6), 115.64 (=CH–CO), 111.18 (CPh-5), 109.77 (CPh-2), 56.12 (Ph–OCH3), 56.04 (Ph–OCH3), 51.77 (COOCH3).
3,4-Dimethoxycinnamic acid (8)
Procedure was adopted according to Bisogno et al.Citation26 Compound 7 (235 mg, 1.1 mmol) was dissolved in dioxane (8 ml) and water (5 ml) mixture. The pH was set up to 12 with 2 M NaOH. After 2 h stirring, pH was again raised to 12 with several drops of 2M NaOH. After the next 2 h, dioxane was evaporated and reaction mixture was remained at pH 12–14. Reaction mixture was washed with ether (2 × 6 ml). The pH was set up to 1–2, and the compound was taken to EtOAc (3 × 6 ml). It was dried over Na2SO4, filtered off, and the solvent was evaporated to dryness. Yield 186 mg (0.9 mmol, 81%) of compound 8. m.p. 182–190°C. TLC Rf 0.53 in C2 mixture. 1H-NMR (300 MHz, CDCl3), δ: 7.74 (d, 1H, 16 Hz, Ph–CH=), 7.14 (dd, 1H, 8 Hz, 2 Hz, HPh-6), 7.08 (d, 1H, 2 Hz, HPh-2), 6.88 (d, 1H, 8 Hz, HPh-5), 6.32 (d, 1H, 16 Hz, =CH–CO), 3.93 (s, 6H, 2 × CH3O–Ph). 13C-APT-NMR (400 MHz, CDCl3), δ: 172.62 (COO), 151.64 (CPh-4), 149.38 (CPh-3), 147.09 (Ph–CH=), 127.16 (CPh-1), 123.26 (CPh-6), 115.02 (=CH–CO), 111.15 (CPh-5), 109.89 (CPh-2), 56.11 (Ph–OCH3), 56.03 (Ph–OCH3).
3,4-Dimethoxycinnamoylcholine chloride (1)
The procedure was modified according to the method of Alemany et al.Citation21 for the syntheses of choline esters of various cinnamates. To a solution of compound 8 (186 mg, 0.89 mmol) in a dioxane–water (3:2) mixture (5 ml), a solution of K2CO3 (61.7 mg, 0.45 mmol) in water (1 ml) was added. After 10 min stirring, it was evaporated to dryness. To the remaining solid, compound 6 (167 mg, 1.05 mmol) was added. The combined solids were dried in high vacuum at 70°C for 1 h. Dry DMSO was added, the mixture was heated up to 60°C, and it was left stirring at that temperature for 44 h. DMSO was distilled off and the residue was dried under vacuum for 1 h. It was redissolved in CHCl3 and loaded on 1-mm TLC plates from Merck. Chromatographic separation was performed using an ACN–H2O mixture (3:1) as eluent. Yield 107 mg (0.32 mmol, 36%) of white hygroscopic solid 1. TLC Rf 0.26 in C1 mixture. RP-HPLC RT 15.4 min (84%). 1H-NMR (300 MHz, CDCl3), δ: 7.64 (d, 1H, 16 Hz, Ph–CH=), 7.36 (d, 1H, 2 Hz, HPh-2), 7.27 (dd, 1H, 8 Hz, 2 Hz, HPh-6), 7.00 (d, 1H, 8Hz, HPh-5), 6.58 (d, 1H, 16 Hz, =CH–CO), 4.58 (m, 2H, COO–CH2), 3.81 (s, 3H, 3-CH3O–Ph), 3.80 (s, 3H, 4-CH3O–Ph), 3.73 (m, 2H, N–CH2), 3.18 (s, 9H, 3 × CH3). 13C-APT-NMR (400 MHz, DMSO-d6), δ: 165.8 (COO), 151.2 (CPh-4), 149.0 (CPh-3), 145.5 (Ph–CH=), 126.6 (CPh-1), 123.1 (CPh-6), 114.7 (=CH–CO), 111.5 (CPh-5), 110.5 (CPh-2), 63.9 (N–CH2), 57.7 (COO–CH2), 55.7 (O–CH3), 55.6 (O–CH3), 52.9 (N–CH3). HRMS (ESI–TOF), for C16H24NO4+: calculated 294.1705; found 294.1707.
Cinnamoylcholine chloride (2)
It was prepared from cinnamic acid and the chloride 6, following the same procedure used for compound 1. Yield 167 mg (0.62 mmol, 45%) of white hygroscopic solid 2. TLC Rf 0.12 in C1 mixture. TLC Rf 0.36 in C4 mixture. RP-HPLC RT 18.5 min (97%). 1H-NMR (300 MHz, DMSO-d6), δ: 7.73 (m, 3H, Ph–CH=, HPh-2), 7.45 (m, 3H, HPh-3, HPh-4), 6.69 (d, 1H, 16 Hz, =CH–CO), 4.60 (m, 2H, COO–CH2), 3.75 (m, 2H, N–CH2), 3.18 (s, 9H, 3 × CH3). 13C-NMR (400 MHz, DMSO-d6), δ:165.6 (COO), 145.4 (Ph–CH=), 133.9 (CPh-1), 130.8 (CPh-4), 129.0 (CPh-3), 128.5 (CPh-2), 117.6 (=CH–CO), 63.9 (N–CH2), 58.0 (COO–CH2), 53.0 (N–CH3). HRMS (ESI–TOF), for C14H20NO2+: calculated 234.1494; found 234.1487.
Caffeoylcholine trifluoroacetate (3)
Prepared as compound 1, but starting from caffeic acid and compound 6. Separation was performed by column chromatography (RP-18 silica gel, gradient from 1 to 88% ACN (with 0.1% TFA)). Yield 100 mg (0.26 mmol, 19%) of white hygroscopic solid 3. TLC Rf 0.19 in C1 mixture. TLC Rf 0.33 in C4 mixture. RP-HPLC RT 11.8 min (97%). 1H-NMR (300 MHz, D2O), δ: 7.64 (d, 1H, 16 Hz, Ph–CH=), 7.19 (d, 1H, 2 Hz, HPh-2), 7.12 (dd, 1H, 8 Hz, 2 Hz, HPh-6), 6.94 (d, 1H, 8 Hz, HPh-5), 6.40 (d, 1H, 16 Hz, =CH–CO), 4.67 (m, 2H, COO–CH2), 3.80 (m, 2H, N–CH2), 3.25 (s, 9H, 3 × CH3). 13C-APT-NMR (300 MHz, D2O), δ: 168.1 (COO), 147.2 (CPh-4), 146.5 (Ph–CH=), 144.2 (CPh-3), 126.7 (CPh-1), 122.8 (CPh-6), 116.0 (CPh-5), 115.0 (CPh-2), 113.3 (=CH–CO), 64.6 (N–CH2), 58.1 (COO–CH2), 53.7 (CH3). HRMS (ESI–TOF), for C14H20NO4+: calculated 266.1392; found 266.1395.
Rosmarinylcholine trifluoroacetate (4)
Prepared as compound 1, but starting from rosmarinic acid and compound 6. Yield 254 mg (0.45 mmol, 45%) of yellow hygroscopic oil 4. TLC Rf 0.33 in C1 mixture. TLC Rf 0.37 in C4 mixture. RP-HPLC RT 18.7 min (84%). 1H-NMR (300 MHz, DMSO-d6), δ: 9.79 (s, 1H, cin. Ph–OH), 9.23 (s, 1H, cin. Ph–OH), 8.92 (s, 1H, lact. Ph–OH), 8.89 (s, 1H, lact. Ph–OH), 7.49 (d, 1H, 16 Hz, Ph–CH=), 7.08 (d, 1H, 2 Hz, cin. HPh-2), 7.02 (dd, 1H, 8 Hz, 2 Hz, cin. HPh-6), 6.78 (d, 1H, 8 Hz, cin. HPh-5), 6.68 (m, 2H, lact. HPh-2, lact. HPh-5), 6.55 (dd, 8 Hz, 2 Hz, lact. HPh-6), 6.28 (d, 1H, 16 Hz, =CH–CO), 5.18 (t, 1H, 7 Hz, lact. α-CH), 4.50 (m, 2H, COO–CH2), 3.61 (m, 2H, N–CH2), 3.05 (m, 11H, 3 × N–CH3, lact. β-CH2). 13C-APT-NMR (400 MHz, DMSO-d6), δ: 169.1 (lact. COO), 166.0 (cin. COO), 148.9 (cin. CPh–OH), 146.7 (Ph–CH=), 145.7 (cin. CPh–OH), 145.2 (lact. CPh–OH), 144.4 (lact. CPh–OH), 126.5 (lact. ipso-CPh), 125.3 (cin. ipso-CPh), 121.8 (lact. CPh-2), 120.2 (cin. CPh-6), 116.8 (lact. CPh-6), 115.8 (lact. CPh-5), 115.6 (cin. CPh-5), 115.0 (cin. CPh-2), 112.7 (=CH–CO), 72.7 (lact. δ-CH), 63.6 (N–CH2), 58.6 (COO–CH2–CH2–N), 52.9 (N–CH3), 36.1 (lact. δ-CH2). HRMS (ESI–TOF), for C23H28NO8+: calculated 446.1805; found 446.1815.
2-(6-Hydroxy-2,5,7,8-tetramethylchroman-2-carbonyloxy)-N,N,N-trimethylethanaminium chloride (5)
It was prepared as compound 1, starting from trolox and compound 6. Yield 43 mg (0.11 mmol, 11%) of hygroscopic oil 5. TLC Rf 0.15 in C1 mixture. TLC Rf 0.41 in C4 mixture. TLC Rf 0.43 in C3. RP-HPLC RT 19.2 min (83%). 1H-NMR (300 MHz, DMSO-d6), δ: 7.57 (br, 1H, OH), δ 4.47 (m, 2H, COO–CH2), 3.63 (m, 2H, N–CH2), 3.01 (s, 9H, 3 × CH3), 2.58 (m, 1H, H-4), 2.45 (m, 1H, H-4), 2.32 (m, 1H, H-3), 2.06 (s, 3H, 7-CH3), 2.03 (s, 3H, 8-CH3), 1.99 (s, 3H, 5-CH3), 1.82 (m, 1H, H-3), 1.54 (s, 3H, 2-CH3). 13C-APT-NMR (300 MHz, DMSO-d6), δ: 172.510 (COO), 145.9 (C-6), 144.5 (C-8a), 122.8 (C-8), 120.8 (C-7), 120.3 (C-5), 116.4 (C-4a), 76.4 (C-2), 63.7 (N–CH2), 58.8 (COO–CH2), 52.7 (N–CH3), 30.2 (C-3), 25.0 (2-CH3), 20.3 (C-4), 12.8 (7-CH3), 11.8 (5-CH3, 8-CH3). HRMS (ESI–TOF), for C19H30NO4+: calculated 336.2175; found 336.2172.
Bioassays of AChE inhibition
AChE enzymatic activity was measured using an adaptation of the method previously describedCitation24; 98 μl of 50 mM Tris–HCl buffer (pH 8), 30 μl of a solution sample of the inhibitor, at different concentrations in methanol, and 7.5 μl of AChE solution containing 0.26 U/ml were mixed in a microplate and left to incubate for 15 min. Subsequently, 22.5 μl of 0.023 mg/ml AChI and 142 μl of 3 mM DTNB were added. The initial rate of the enzymatic reaction was followed by reading the absorbance at 405 nm during the first 5 min of reaction. Samples were prepared in a range of concentrations of the compounds in water (choline caffeate, choline trolox, choline cinnamate) or in an aqueous solution of 50% methanol (choline 3,4-dimethoxicinnamate, choline rosmarinate). A control reaction was carried out using the sample solvent instead of sample and it was considered 100% activity.
where Asample is the absorbance of the compound containing reaction, and Acontrol is the absorbance of the control reaction, which were recorded on a Camspec M350 double-beam scanning UV-vis spectrophotometer. Tests were carried out in triplicate, and a blank with Tris–HCl buffer instead of the enzyme solution was used.
Antioxidant activity
Antioxidant activity was measured by the 2,2-diphenyl-1-picrylhydrazyl radical (DPPH) method, as described by Tepe et al.Citation27 To a 2.5 ml solution of DPPH (0.002% in methanol), 25 μl of compound solution was added. The mixture was incubated for 30 min at room temperature. The absorbance was measured at 517 nm against the corresponding blank. The antioxidant activity was calculated as:
where AA is the antioxidant activity, ADPPH is the absorption of the DPPH solution against the blank, Asample is the absorption of the sample compound against the blank. The tests were carried out in triplicate and the compound concentration providing 50% of antioxidant activity (IC50) was obtained by plotting the antioxidant activity against the compound concentration.
Docking of ligands
All X-ray structures of AChE complexed with different inhibitors were taken from the RCSB Protein Data Bank (PDB).Citation28 To define the best docking procedure, the crystal structures with PDB entry codes 1EVE (containing Dnp) and 1ACL [containing trimethyl-(10-trimethylazaniumyldecyl) azanium, a bi-quaternary ligand; see Figure S1, Supplementary Material] were used. Solvent, co-crystallization molecules, and counterions were removed from the original structure whenever they were present, and hydrogen atoms were added by means of Maestro 7.5.Citation29 The ligands were extracted from the complexes and were submitted to a conformational search (CS) of 1000 cycles, using a water environment model (generalized-Born/surface-area model) by means of Macromodel.Citation30 The algorithm used was based on the Monte-Carlo method with the molecular Merck force field (MMFFs) and a distance-dependent dielectric constant of 1.0. Each minimized ligand was then docked to the respective AChE structure, by means of the Gold program,Citation31 where the region of interest was defined to contain the residues within 15 Å from the original position of the ligand. The Gold default parameters were used, and the ligands were submitted to 100 genetic algorithm runs, applying the three fitness functions available with Gold (GoldScore, ChemScore, and astex statistical potential (ASP)), at a first stage without any other constraints. The best docked conformation for each scoring function was then compared with the experimental conformation of the ligands in the crystal structure, and the root-mean-square deviation (rmsd) between the positions of the heavy atoms was calculated; this parameter being considered as a measure of the docking accuracy. The rmsd values for Dnp and the quaternary ligand were 1.199 and 1.101 Å for ASP, 1.211 and 2.828 Å for ChemScore, and 10.165 and 2.456 Å for GoldScore fitness function, respectively, resulting in ASP as the best scoring function to dock these ligands into their original AChE structures with program Gold.
The choline conjugates were built using program Maestro 7.5 and underwent CS and structure optimization with Macromodel, using the same procedures described earlier. To perform the docking calculations of these ligands into AChE, the protein structure of 1ACL complex was used as receptor, with the same docking procedure mentioned earlier, and ASP as scoring function. Because of some difficulties in correctly placing the trimethylammonium moiety in the active site of the gorge, a scaffold match constraint was added. Here, the N+(CH3)3 moiety of the quaternary ligand (from structure 1ACL, see Figure S1 on Supplementary Material), in its original position, was saved and used by Gold as a scaffold be matched; the scaffold match constraint weight, a parameter determining how closely the ligand atoms should fit onto the scaffold, was set to 5.0.
Prediction of pharmacokinetic properties
For the compounds in study, a brief in silico calculation was performed to enable a comparative prediction of some relevant properties in drug delivery, namely, octanol/water partition coefficient (log P); brain/blood partition coefficient (log BB); polar surface activity (PSA); % human oral absorption, and the central nervous system activity.
The compounds were built and minimized as previously mentioned for the docking calculations but, in this case, a chloride ion was used as counter-ion for all the compounds (except for the neutral Dnp). The final structures were then submitted to the calculation of some relevant pharmacokinetic properties and descriptors, using the QikProp, version 2.5.Citation32 These predictions are for orally delivered drugs and assume nonactive transport.Citation33
Cell culture and treatments
Human neuroblastoma SH-SY5Y cells (ATCC number CRL-2266) were grown in Dulbecco’s modified Eagle’s medium with nutrient mixture F-12 (DEMEM/F12) with 10% heat-inactivated fetal calf serum, containing 50 U/ml penicillin and 50 µg/ml streptomycin, under a humidified atmosphere of 95% air–5% CO2 at 37°C. Cells were plated at 0.15 × 106 cells/ml for cell viability assays.
Medium was changed after 24 h and immediately before treatments. MPP+ was prepared as 200 mM stocks (in H2O) immediately before use and added to the medium to 1 mM final concentration. Abeta1–42 (1 μM) peptides were added from a 221.5 μM stock phosphate buffered saline (PBS) solution. Previously, the peptides were dissolved in sterile distilled water at a concentration of 6 mg/ml and diluted to 1 mg/ml (221.5 μM) with PBS and then incubated for 5–7 days to induce fibril formation. The tested phenolic-choline conjugates (1−3) were freshly prepared on each experiment day. First, they were dissolved in H2O at a concentration of 1 mM and then added to the medium to 5 μM final concentration.
For combinatorial treatments, both simultaneous and sequential treatment approaches were tested and the selected compound’s concentrations shown to exert the maximal protective action and had no toxic effects to cells.
For all conditions tested, control experiments were performed in which the compounds were tested or the stress agents was not added.
Cell reduction ability assay
Cell reduction ability as a surrogate of cell viability was measured by using a quantitative colorimetric assay with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), according to the method of Mosmann.Citation34 In brief, cells were incubated with MTT solution (in Krebs medium) for 3 h at 37°C. The medium was then discarded, the stained cells were dissolved with isopropanol/HCl, and thereafter the absorbance at 570 nm was measured using a Spectramax Plus 384 spectrophotometer (Molecular Devices). MTT reduction ability was expressed as a percentage of the control value obtained for untreated cells.
Results and discussion
Chemistry
For the preparation of the new compounds, the first step consisted of the conversion of choline into chlorocholine (6) with thionyl chloride (). The synthesis of the cinnamoylcholine esters was based on a reported method for alkylation of the respective silver carboxylates with chlorocholine,Citation21 but the silver salts were substituted with cheaper potassium salts. The preparation of 2,6-dimethoxycinnamoylcholine analogue, 1, was slightly more complex, because the caffeic acid was used as the starting material, which was methylated using dimethylsulfate, in the presence of K2CO3, to afford 7 (). The ester 7 was then hydrolyzed to the corresponding carboxylic acid, 8, under aqueous basic conditions (NaOH).
Figure 4. Synthesis of compound 1. Reagents and conditions: (i) SOCl2, reflux; (ii) (MeO)2SO2, K2CO3, THF, reflux; (iii) NaOH, dioxane/H2O, pH 12–14, r.t.; (iv) K2CO3, dioxane/H2O, r.t.; and (v) DMSO, chlorocholine chloride (6), 60°C.
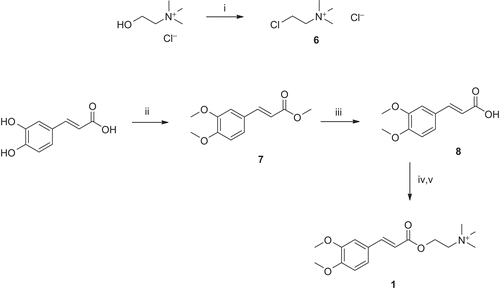
The carboxylic acid (8, cinnamic, caffeic, rosmarinic, or trolox) was converted to the corresponding potassium salt, using K2CO3. It was then alkylated with the chlorocholine derivative, 6, to afford the respective target compounds (1–5, respectively). Although compounds 1, 2, and 5 were obtained as the chloride salts, compounds 3 and 4 were obtained as trifluoroacetates, due to the use of TFA in ion-pairing reverse-phase chromatography for purification purpose.
The stability of the new compounds in aqueous solution was evaluated by 1H-NMR in neutral D2O. The 1H-NMR spectra of compounds 1 and 3 were recorded for a long time, and no changes were observed in their spectra in a 48-h timeframe, thus meaning that the compounds are stable in aqueous medium, at least along that time window.
Inhibition of AChE
Bioassays
The AChE inhibitory activities of the new phenolic-choline esters, and their parent compounds ( and ), were evaluated and the results are summarized in . All the carboxylic homologues displayed low AChE inhibitory activities, but the insertion of the choline moiety resulted, for all cases, in a drastic increase in those inhibitory properties. The choline-conjugate compounds proved to inhibit AChE, with IC50 values in micromolar range. Although, the new compounds are in general weaker AChEI than the standard AChEI drugs, some of them present activities with the same order of magnitude (low micromolar range). However, direct comparison between the new and the reference inhibitors renders difficult, because the corresponding inhibitor–enzyme interactions are quite different, except Dnp, which presents some structural analogies with these compounds, and is the strongest standard inhibitor (IC50 value of 30 nM). Among these new compounds, 3,4-dimethoxycinnamic choline ester (compound 1) appeared as the strongest inhibitor, with IC50 values (7.3 μM) close to those reported for some reference inhibitors, such as galantamine (1.07 μM),Citation35,Citation36 rivastigmine (1.5 μM),Citation37 or a choline ester derivative of α,β-dehydrophenylalanine (37 μM).Citation22 Within this series of caffeic analogues (1–3), compound 3 displayed the lowest activity, with IC50 value of 91 μM. The activity decreases from compounds with 3,4-dimethoxy-substituted aromatic ring (1) to the non-substituted (2), and then to the 3,4-dihydroxy-substituted (3) aromatic system; thus, it is suggested that the methoxy and hydroxyl groups can establish favorable and unfavorable interactions, respectively, with the active site of AChE, with concomitant effects on the stability of the corresponding protein-inhibitor adduct. This trend also suggests that those moieties should interact with the hydrophobic residues at the active site of AChE.
Table 1. Results of AChE inhibition and antioxidant activity (DPPH assay) measured for the compounds in study, some parent compounds and standard AChE inhibitors.
Regarding the other studied compounds, the rosmarinyl- and trolox-choline derivatives (4 and 5, respectively), and their AChE inhibitory activities (IC50 300 and 1190 μM) are much lower than those found for the caffeic analogues. This activity decrease may be due to the bulkiness of these compounds, which may hamper their fitting into the enzyme-active cavity and/or to their hydroxyl groups that may form particularly unfavorable interactions with the protein, as was observed for compound 3. This shall be further clarified below by the docking studies.
Globally, this new type of bifunctional antioxidant compounds revealed interesting properties as AChEI, with activities of the same order of magnitude as some reference inhibitors, but further efforts on structure optimization may enable the improvement of the AChE inhibitory capacity.
Molecular modeling
The AChE protein is a very efficient enzyme with quite interesting structural features.Citation38–40 The active site is buried in a narrow deep gorge, which is mostly surrounded by aromatic residues, and it includes two main subsites. The catalytic anionic subsite (CAS) is formed by Trp84, Phe330, and Glu199 (sequence numbering of Torpedo californica AChE, TcAChE)Citation38–40, and can establish cationic–π interactions with the positively charged choline, and help the stabilization of the transition state. The catalytic triad (Ser200, His440, and Glu327; and Figure S1 of Supplementary Material) binds covalently to the substrate (through Ser200) and, aided by a water molecule, leads to the hydrolysis of the acetylcholine, generating choline and acetic acid. There is also a peripheral anionic site (PAS), located at the entrance of the gorge and formed by Trp279, Tyr70, and Asp72, which may act as a “trap” for binding substrate molecules, before directing them into the active site gorge. The structure of this enzyme seems to be highly optimized, in terms of electrostatic field orientation and hydrophobic interactions, to rapidly guide the substrate into the bottom of the active site.
Figure 5. Docking of compound 1 (blue) into the active site of TcAChE (PDB entry 1ACL). The catalytic triad is formed by Ser200, His440, and Glu327, the CAS is formed by Trp84, Phe330, and Glu199, and the PAS located at the entrance of the gorge is formed by Trp279, Tyr70, and Asp72.
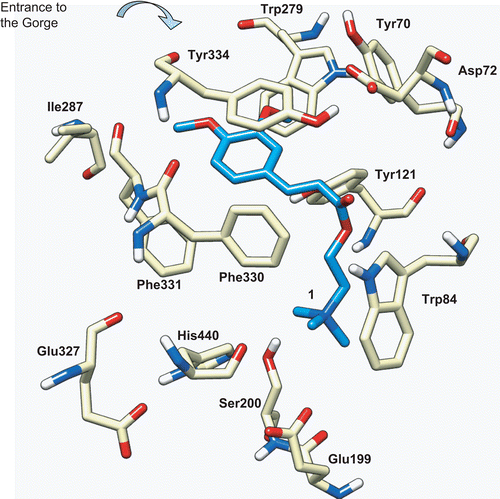
Aimed at getting some rationalization of the inhibitory activities of our new ligands toward AChE, a protein–ligand docking study was carried out using the program Gold, version 4.0.Citation31 To define the best docking procedure, the crystal structures of two complexes of TcAChE were taken from the RCSB PDB,Citation28 containing one of the strongest AChEI, the FDA-approved drug Dnp,Citation41 and another one with decamethonium ion, a biquaternary ligandCitation42 (PDB entries 1EVE and 1ACL, respectively) (see Figure S2, Supplementary Material). These two complexes were chosen because of some similarity between the corresponding inhibitors and our ligands. In the first case, Dnp has a dimethoxybenzene moiety, as compound 1; in the second case, the inhibitor is a quaternary ligand, inserted in the catalytic site but positioned along the gorge, rather than being close to the catalytic Ser200, as we expect to happen with our inhibitors, for sterical reasons. To validate the method, the ligands were redocked into the respective proteins, using the three scoring functions available with Gold (ASP, ChemScore and GoldScore). The rmsd of the best scored conformations was calculated for each case, with respect to the experimental, and ASP was found to give the best prediction for both ligands (rmsd values of 1.20 and 1.10 Å, for Dnp and the quaternary inhibitor, respectively), so it was used on the docking calculations with our inhibitors. Furthermore, 1EVE and 1ACL structures are very similar in their 3D conformations (the overall rmsd for the heteroatoms was calculated as 0.41 Å), but 1ACL structure was chosen for docking, for containing a quaternary ligand.
The first results were not always reliable. Either the choline moiety was pointing outward the gorge, unlike what is known for some ligands of this type, or it was in variable positions within the active site, which made no sense. Hence, we decided to apply a scaffold match constraint, allowing to define (constrain) the position that a certain fragment should occupy along the docking calculations. In this case, the N+(CH3)3 fragment from the original inhibitor was used as “scaffold” to dock the ligands into the active site of 1ACL structure, and the constraint weight was set to the value of 5.0.
The docking results for our compounds (1–5) with TcAChE showed the ligands well inserted into its active gorge, blocking the entrance of the substrate and water molecules, and therefore hindering the catalytic activity of this enzyme. For the most active compound (1, ), the quaternary ammonium moiety could establish van der Waals contact with the indole ring of Trp84, and electrostatic interactions with Glu199. The spacer chain seems well accommodated along the hydrophobic cavity, and the aromatic system (the cinnamic conjugated system) forms a π–π contact with Tyr334. One of the methoxy groups is pointing toward the PAS, forming van der Waals interactions with the Trp279 and Tyr70 aromatic systems, although the other methoxy group is pointing to the opposite side of the cavity, forming weak interactions with the Ile287 and Phe331 side chains. Superimposition of the experimental conformations found for Dnp (on crystal structure 1EVE) and that calculated for compound 1 evidenced that the area of contact with the enzyme is larger for Dnp than for 1 (see Figures S1 and S3, Supplementary Material), thus explaining its concomitant higher inhibitory potency (IC50 values of 0.03 and 7.3 μM, respectively). In fact, although Dnp forms strong π–π and van der Waals contacts with the gorge wall residues (namely benzyl-Trp84 and piperidine-Phe330) and the peripheral site (dimethoxyphenyl-Trp279), in the 1-AChE complex these interactions are weaker. This can be due to differences on the spacer (the linker between the quaternary system and the phenolic aromatic ring), namely on the length and on the surface, which are smaller in compound 1 than in Dnp, thus accounting for weaker hydrophobic interactions with the gorge residues (Figure S3b).
Analyzing the docking of the cinnamic and caffeic derivatives (2 and 3) with AChE, in comparison with that found for compound 1, their binding conformations are very similar, namely with the spacers and the aromatic rings almost perfectly superimposed (Figure S4, Supplementary Material). Hence, most of the interactions observed are maintained. The only differences between these compounds are due to the substituent groups at the 3 and 4 positions of the benzene rings, and they are responsible for the differences in their inhibitory activities (which decreased 6.8- and 12.5-fold for compounds 2 and 3, with respect to that of compound 1). Concerning compound 2, the absence of the methoxy groups, with their favorable interactions with the enzyme (as found for compound 1), accounts for a lower stability of the respective ligand–enzyme complex, compared with that of compound 1. Regarding compound 3, the presence of two hydrophilic hydroxyl substituent groups hampered the hydrophobic interactions established with the methoxyl groups of compound 1. Instead, only a weak H bond may be established between the 3-hydroxy and the carbonyl oxygen of Tyr334, but, apparently, it is not enough to compensate the loss in hydrophobic interactions, thus resulting in a decrease in inhibitory activity.
Concerning the rosmarinic (4) and trolox (5) derivatives, comparison between the interactions of these compounds with the enzyme resulted a bit more difficult, because they have different main backbones and side chains. For compound 4 (Figure S5, Supplementary Material), the docking results indicated that one of its aromatic rings (the caffeic part) is able to establish a π–π contact with the aromatic residues on the peripheral site (with Trp279 and Tyr70), although the other aromatic ring forms weak van der Waals interactions with Phe331. The spacer between the choline and the aromatic moieties of compound 4 seems to favor a positional accommodation very similar to that of compound 1, although compound 4 seems to be able to establish more aromatic interactions with the enzyme than compound 1. Therefore, the decrease in its inhibitory activity with respect to compound 1 (41-fold decrease) may be explained mainly by the hydrophilic–hydrophobic repulsion caused by the hydroxyl substituent groups of the aromatic rings, which decreases the overall stability of the 4-AChE complex. Regarding compound 5, its aromatic rings seem to be well accommodated at the center of the gorge channel, apparently disabling the formation of any strong π–π stacking with the enzyme, but only van der Waals contacts (namely through the heterocyclic ring with Tyr334, and the methyl groups with the peripheral site aromatic residues). Furthermore, the hydroxyl group must contribute unfavorably for the binding interactions of this compound within the AChE active gorge, resulting in the lowest inhibitory activity among these new compounds.
Antioxidant activity
The choline-ester conjugates and the corresponding carboxylic analogues were screened for their antioxidant activities based on their interaction with the DPPH stable free radical, and the results are shown in .
Among the carboxylic analogues, the rosmarinic acid was the best antioxidant, with EC50 value of 6.4 μM, which was very close to trolox activity (13.2 μM), whereas cinnamic acid was the weakest one (675 μM). As expected, the antioxidant activities of these compounds were not much affected by the introduction of a choline moiety, but actually some increases were observed, such as for the rosmarinylcholine, 4, which appeared as the strongest antioxidant (EC50 of 4.3 μM). This value is even lower than those observed for the standard antioxidants 3,5-di-tert-butyl-4-hydroxytoluene (BHT; an antioxidant food and cosmetic additive) and trolox (a vitamin E-like antioxidant, quite used in biochemical systems for oxidative stress protection). This may be due to the extra catechol group in the rosmarinic structure when compared with others. On the other hand, among the compounds in this study, the cinnamic derivatives (both for the carboxylic acid and the choline ester 2) displayed the lowest antioxidant activities. This is probably due to the absence of any phenol hydroxyl groups, which have an important role in the reactive oxygen species (ROS)-scavenging action of polyphenols (through high constant rates of the H-atom abstraction reactions).Citation43,Citation44 It is known that the presence of these phenolic hydroxyl groups increases the ROS-scavenging activity, but reduces the lipophilicity of the compounds. This fact may decrease their ability to penetrate the blood–brain barrier (BBB) and the cell membranes, and, as discussed before, also the anti-AChE activity. Hence, these two factors must be well balanced in the development of this type of drugs, to make them valuable as potential anti-neurodegenerative drugs.
In vitro neuroprotective activity
Preliminary assays were performed to evaluate the potential therapeutic action of a set of bifunctional phenolic-choline conjugates (compounds 1–3, which presented the best AChEI) on neuronal cells (SH-SY5Y) pretreated with the stressors MPP+ and Abeta1–42. SH-SY5Y cell line was selected because it has been shown to exhibit a neuronal phenotype and have several neurochemical markers.Citation45 On the other hand, Abeta1–42 and MPP+ treatments in SH-SY5Y cells constitute a reliable model for screening potential neuroprotective compounds, since it mimics some aspects of neurodegeneration underlying the AD and PD, respectively.Citation46,Citation47 Both stressors induce mitochondrial deficits, increase in ROS production, and apoptotic cell deathCitation48,Citation49; so all three compounds, due to their chemical properties, may act as strong antioxidants, and thus prevent ROS cellular damage.
The results depicted in evidenced a significant decrease in the cell viability after a 24-h treatment with 1 μM Abeta1–42 (ca. 30%) and 1 mM MPP+ (ca. 20%). The bifunctional phenolic-choline conjugates, by themselves, did not significantly affect the cell viability at the same end point (a maximum of 10% increase was achieved with compound 2). On the other hand, all the tested compounds provided some protection against the cell death induced by Abeta1–42 and MPP+ on cultured human SH-SY5Y cells. The Abeta1–42-treated cells showed the highest viability increments, approximately 19–22% increases relative to the absence of ligands. Compound 1 seemed to be slightly less effective than compounds 2 and 3 in preventing the Abeta1–42-mediated toxicity. However, the differences are not significant enough to establish good correlation with the previously cited biological properties, since compound 2 displayed the worst balance in terms of anti-AChE and antioxidant activities. Thus, other factors may contribute to protecting role of these compounds against cell Abeta1–42 injury, namely their ability for cell membrane crossing, which depend on several molecular parameters, such as the lipophilicity; also the caspase cascade stress-induced apoptosis, which was not evaluated herein.Citation50,Citation51
Figure 6. Effect of bifunctional phenolic-choline conjugates (1, 2, 3) on Abeta1–42 and MPP+ toxicity in SH-SY5Y cells. SH-SY5Y cells were treated with Abeta1–42 (1 μM) and MPP+ (1 mM) for 24 h, in the absence or the presence of 5 μM of compounds 1, 2, and 3. Results are expressed as the percentage of SH-SY5Y untreated cells, with the mean ± S.E.M. derived from six different experiments. P < 0.001, significantly different when compared with SH-SY5Y untreated cells; #P < 0.05; ##P < 0.01, significantly different when compared with Abeta1–42 treated cells; §P < 0.05; §§P < 0.01, significantly different when compared with MPP+ treated cells.
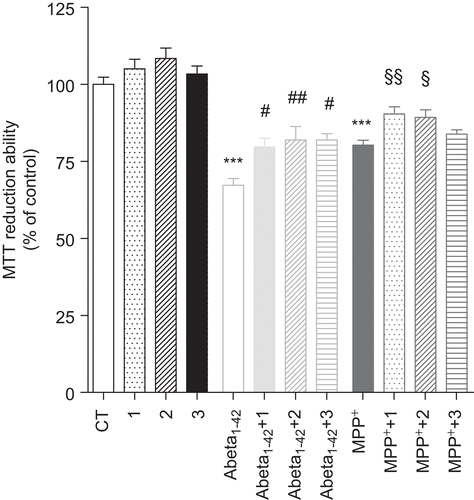
Concerning the effect of the compounds on MPP+-treated cells, the viability improvement was only of approximately 5–12%, compared with the absence of the compounds, and compound 3 presents the lowest protective activity. Differences between the cell permeability properties of this set of compounds may also, somehow, account for differences on their cell protective roles. Among this set of cell tested compounds, the calculated valuesCitation32 of log P and log BB (Table S1, Supplementary Material) was 5.82 and −0.057 for compounds 1, 5.18 and 0.47) for 2, and 3.84 and −1.07 for 3, suggesting the lowest lipophilicity and BBB permeability for compound 3. Therefore, this compound should present some extra difficulties to enter the cell, which may explain the lowest protective effects on MPP+-treated cells.
In summary, our preliminary results on ex vivo cells indicate that the new phenolic-choline hybrids 1–3 exert some protective effects on the death of SH-SY5Y cells induced by Abeta1–42 or MPP+. However, the mechanism of their cell-protecting roles still needs to be clarified, namely in relation with their biological activity as AChE inhibitors and antioxidative agents. The neuroprotective properties are also likely to be improved with the lipophilicity of the design compounds, which can be predicted by computational tools. Thus, from this work, a new type of bifunctional derivatives of naturally occurring compounds appeared with proved neuroprotective roles, and they represent a challenge to pursuit with further structural optimizations to improve both target activities and their membrane-cross abilities.
Conclusions
A small set of new bifunctional compounds has been developed and studied on their potential antineurodegenerative roles. The design strategy was based on conjugating known naturally occurring antioxidant phenolic acids (three caffeic acid derivatives, rosmarinic acid, and trolox) with choline through ester formation, to enable the AChEI.
These conjugates demonstrated hybrid properties, namely AChE inhibitory and antioxidant activities in micromolar range, two drug properties of relevance for combating the symptoms and the progression of multifactorial neurodegenerative diseases, namely AD. Concerning the AChEI activity, 3,4-dimethoxycinnamoylcholine was identified as the most potent inhibitor, from our set, with IC50 of 7.3 μM. Interestingly, it contains a catechol group protected with methyl groups, and in that moiety it is similar to the FDA-approved drug Dnp. Among these compounds, the rosmarinic and trolox derivatives, presented the lowest AChEI activities, although they are among the strongest antioxidants (EC50 values of 4.3 and 38.4 μM). Their different activities are rationalized on the basis of their structures and docking simulations. The most active AChE inhibitors (the caffeic acid derivatives) were also assessed for protective effects on the death of neuroblastoma SH-SY5Y cells treated with Abeta1–42 and MPP+. These compounds proved to reduce the cell viability loss induced by each of the stressors, thus evidencing neuroprotective roles. Further studies are envisaged to optimize the structure of the bifunctional analogous compounds as potential anti-neurodegeneratives.
Acknowledgements
The authors thank Portuguese NMR Network (IST-UTL Center) for providing access to the NMR facility. The mass spectra were obtained at the IST Node, which is part of the National Mass Spectrometry Network (RNEM) created by the Portuguese Foundation for Science and Technology (FCT). The authors thank Leonel Gil Nogueira for helping with Ionex HPLC.
Declaration of interest
The authors are grateful to the Portuguese Foundation for Science and Technology (FCT) (Postdoc grant SFRH/BPD/42511/2007, PhD grant SFRH/BD/37547/2007, PhD grant SFRH/BD/32470/2006, and PhD grant SFRH/BD/38743/2007) for financial support. The authors are also thankful to Centro de Neurociências e Biologia Celular da Universidade de Coimbra (Portugal) for financial support. All molecular modeling figures were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH Grant P41 RR-01081).
Related Research Data
References
- Casetta I, Govoni V, Granieri E. Oxidative stress, antioxidants and neurodegenerative diseases. Curr Pharm Des 2005;11:2033–2052.
- Lau I-F Brodney, MA. Therapeutic approaches for the treatment of Alzheimer disease: an Overview. In: Lau I-F Brodney, MA (eds), Alzheimer’s Disease. Berlin: Springer-Verlag, 2008, pp. 1–25.
- Jakob-Roetne R, Jacobsen H. Alzheimer’s disease: from pathology to therapeutic approaches. Angew Chem Int Ed Engl 2009;48:3030–3059.
- Mimica N, Presecki P. Current treatment options for people with Alzheimer’s disease in Croatia. Chem Biol Interact 2010;187:409–410.
- Tumiatti V, Bolognesi ML, Minarini A, Rosini M, Milelli A, Matera R, Melchiorre C. Progress in acetylcholinesterase inhibitors for Alzheimer’s disease: an update. Expert Opin Ther Patents 2008;18:387–401.
- Kao J, Grossberg G. Cholinesterase inhibitors. In: Lau I-F Brodney, MA (eds), Alzheimer’s Disease. Berlin: Springer-Verlag, 2008, pp. 26–51.
- Cornelli U. Treatment of Alzheimer’s disease with a cholinesterase inhibitor combined with antioxidants. Neurodegener Dis 2010;7:193–202.
- Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature 2004;430:631–639.
- Sayre LM, Perry G, Smith MA. Oxidative stress and neurotoxicity. Chem Res Toxicol 2008;21:172–188.
- Gaeta A, Hider RC. The crucial role of metal ions in neurodegeneration: the basis for a promising therapeutic strategy. Br J Pharmacol 2005;146:1041–1059.
- Biran Y, Masters CL, Barnham KJ, Bush AI, Adlard PA. Pharmacotherapeutic targets in Alzheimer’s disease. J Cell Mol Med 2009;13:61–86.
- Rosini M, Andrisano V, Bartolini M, Bolognesi ML, Hrelia P, Minarini A et al. Rational approach to discover multipotent anti-Alzheimer drugs. J Med Chem 2005;48:360–363.
- Bolognesi ML, Cavalli A, Valgimigli L, Bartolini M, Rosini M, Andrisano V et al. Multi-target-directed drug design strategy: from a dual binding site acetylcholinesterase inhibitor to a trifunctional compound against Alzheimer’s disease. J Med Chem 2007;50:6446–6449.
- Bolognesi ML, Matera R, Minarini A, Rosini M, Melchiorre C. Alzheimer’s disease: new approaches to drug discovery. Curr Opin Chem Biol 2009;13:303–308.
- Zheng H, Youdim MB, Fridkin M. Site-activated multifunctional chelator with acetylcholinesterase and neuroprotective-neurorestorative moieties for Alzheimer’s therapy. J Med Chem 2009;52:4095–4098.
- Arduino D, Silva D, Cardoso SM, Chaves S, Oliveira CR, Santos MA. New hydroxypyridinone iron-chelators as potential anti-neurodegenerative drugs. Front Biosci 2008;13:6763–6774.
- Koufaki M, Theodorou E, Galaris D, Nousis L, Katsanou ES, Alexis MN. Chroman/catechol hybrids: synthesis and evaluation of their activity against oxidative stress induced cellular damage. J Med Chem 2006;49:300–306.
- Sanbongi C, Takano H, Osakabe N, Sasa N, Natsume M, Yanagisawa R et al. Rosmarinic acid inhibits lung injury induced by diesel exhaust particles. Free Radic Biol Med 2003;34:1060–1069.
- Yang JQ, Zhou QX, Liu BZ, He BC. Protection of mouse brain from aluminum-induced damage by caffeic acid. CNS Neurosci Ther 2008;14:10–16.
- Snape MF, Misra A, Murray TK, De Souza RJ, Williams JL, Cross AJ et al. A comparative study in rats of the in vitro and in vivo pharmacology of the acetylcholinesterase inhibitors tacrine, donepezil and NXX-066. Neuropharmacology 1999;38:181–193.
- Alemany A, Baluja G, Corral C, Leon JLF. Inhibidores de colinesterasa. 1. Influencia de la estructura del grupo acilo en la inhibicion de colinesterasa por esteres de colina. Anales Real Soc Espan Fis Quim 1962;B58:531–538.
- Grigoryan HA, Hambardzumyan AA, Mkrtchyan MV, Topuzyan VO, Halebyan GP, Asatryan RS. alpha,beta-Dehydrophenylalanine choline esters, a new class of reversible inhibitors of human acetylcholinesterase and butyrylcholinesterase. Chem Biol Interact 2008;171:108–116.
- Armarego WLF, Perrin DD. Purification of Laboratory Chemicals, 4th edn. Oxford: Butterworth-Heinemann, 1996.
- Ingkaninan K, Temkitthawon P, Chuenchom K, Yuyaem T, Thongnoi W. Screening for acetylcholinesterase inhibitory activity in plants used in Thai traditional rejuvenating and neurotonic remedies. J Ethnopharmacol 2003;89:261–264.
- Jung YH, Kim JD. Mechanism studies on the CSI reaction with allyl ethers by varying p-substituent. Arch Pharm Res 2003;26:667–678.
- Bisogno F, Mascoti L, Sanchez C, Garibotto F, Giannini F, Kurina-Sanz M et al. Structure-antifungal activity relationship of cinnamic acid derivatives. J Agric Food Chem 2007;55:10635–10640.
- Tepe B, Daferera D, Sokmen A, Sokmen M, Polissiou M. Antimicrobial and antioxidant activities of the essential oil and various extracts of Salvia tomentosa Miller (Lamiaceae). Food Chem 2005;90:333–340.
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H et al. The Protein Data Bank. Nucleic Acids Res 2000;28:235–242.
- Maestro, version 7.5; Schrödinger Inc.: Portland, OR, 2005.
- Macromodel, version 8.5; Schrödinger Inc.: Portland, OR, 1999.
- Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 1997;267:727–748.
- QikProp, version 2.5, Schrödinger, LLC, New York, NY, 2005.
- Di L, Kerns EH, Carter GT. Strategies to assess blood–brain barrier penetration. Expert Opin Drug Discov 2008;3:677–687.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63.
- López S, Bastida J, Viladomat F, Codina C. Acetylcholinesterase inhibitory activity of some Amaryllidaceae alkaloids and Narcissus extracts. Life Sci 2002;71:2521–2529.
- Giacobini E. Drugs that target cholinesterase. In: Buccafusco J (ed), Cognitive Enhancing Drugs. Switzerland: Birkhauser Verlag, 2004, pp. 11–37.
- Bolognesi ML, Bartolini M, Cavalli A, Andrisano V, Rosini M, Minarini A et al. Design, synthesis, and biological evaluation of conformationally restricted rivastigmine analogues. J Med Chem 2004;47:5945–5952.
- Wlodek ST, Antosiewicz J, Briggs JM. On the Mechanism of acetylcholinesterase action: the electrostatically induced acceleration of the catalytic acylation step. J Am Chem Soc 1997;119:8159–8165.
- Tõugu V. Acetylcholinesterase: mechanism of catalysis and inhibition. Curr Med Chem CNS Agents 2001;1:155–170.
- Colletier J-P Fournier, D, Greenblatt HM, Stojan J, Sussman JL, Zaccai G, Silman I, Weik M. Structural insights into substrate traffic and inhibition in acetylcholinesterase. EMBO J 2006;25:2746–2756.
- Kryger G, Silman I, Sussman JL. Structure of acetylcholinesterase complexed with E2020 (Aricept): implications for the design of new anti-Alzheimer drugs. Structure 1999;7:297–307.
- Harel M, Schalk I, Ehret-Sabatier L, Bouet F, Goeldner M, Hirth C et al. Quaternary ligand binding to aromatic residues in the active-site gorge of acetylcholinesterase. Proc Natl Acad Sci USA 1993;90:9031–9035.
- Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact 2006;160:1–40.
- Denisov ET, Afanas’ev IB. Oxidation and Antioxidants in Organic Chemistry and Biology. Boca Raton, FL: CRC Press, 2005:835–894.
- Youdim MB, Amit T, Bar-Am O, Weinstock M, Yogev-Falach M. Amyloid processing and signal transduction properties of antiparkinson-antialzheimer neuroprotective drugs rasagiline and TV3326. Ann N Y Acad Sci 2003;993:378–86; discussion 387.
- Cardoso SM, Moreira PI, Agostinho P, Pereira C, Oliveira CR. Neurodegenerative pathways in Parkinson’s disease: therapeutic strategies. Curr Drug Targets CNS Neurol Disord 2005;4:405–419.
- Pereira C, Agostinho P, Moreira PI, Cardoso SM, Oliveira CR. Alzheimer’s disease-associated neurotoxic mechanisms and neuroprotective strategies. Curr Drug Targets CNS Neurol Disord 2005;4:383–403.
- Esteves AR, Domingues AF, Ferreira IL, Januário C, Swerdlow RH, Oliveira CR et al. Mitochondrial function in Parkinson’s disease cybrids containing an nt2 neuron-like nuclear background. Mitochondrion 2008;8:219–228.
- Cardoso SM, Santos S, Swerdlow RH, Oliveira CR. Functional mitochondria are required for amyloid beta-mediated neurotoxicity. Faseb J 2001;15:1439–1441.
- Troy CM, Shelanski ML. Caspase-2 redux. Cell Death Differ 2003;10:101–107.
- Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J Cell Biol 2004;165:347–356.
- Otoguro K, Kuno F, Omura S. Arisugacins, selective acetylcholinesterase inhibitors of microbial origin. Pharmacol Ther 1997;76:45–54.
- Mata T, Proença C, Ferreira AR, Serralheiro MLM, Nogueira JMF, Araújo MEM. Antioxidant and antiacetylcholinesterase activities of five plants used as Portuguese food spices. Food Chem 2007;103:778–786.