Abstract
A series of novel influenza neuraminidase (NA) inhibitors based on thiazole core were synthesized and evaluated for their ability to inhibit NA of influenza A virus (H3N2). All compounds were synthesized in good yields starting from commercially available 2-amino-4-thiazole-acetic ester using a suitable synthetic strategy. These compounds showed moderate inhibitory activity against influenza A NA. The most potent compound of this series is compound 4d (IC50 = 3.43 μM), which is about 20-fold less potent than oseltamivir, and could be used to design novel influenza NA inhibitors that exhibit increased activity based on thiazole ring.
Keywords::
Introduction
Despite advances in the understanding of molecular and cellular aspects of influenza, the disease remains the major cause of mortality and morbidity among patients with respiratory diseases.(Citation1)
Influenza virus neuraminidase (NA, EC 3.2.1.18) is a major surface glycoprotein of the influenza A and B. Viral NA catalyses cleavage of the ketosidic linkage between terminal sialic acid (SA) and an adjacent sugar residue(Citation2,Citation3) via a oxocarbonium intermediate. Compounds that inhibit NA can protect the host from viral infection and retard its propagation and it is thought that rationally designed NA inhibitors are effective for the treatment of influenza. Also, because the structure of the active site of NA is highly conserved across all nine influenza A NA subtypes and influenza B subtypes, the NA inhibitors are well-tolerated against all influenza types.(Citation4)
The NA inhibitors have currently emerged as promising therapeutics for the treatment of influenza.(Citation5) Two such successful examples of structure-based drug design of NA inhibitors, Relenza (zanamivir, Glaxo Wellcome, Misslesex, UK/Biota, Notting Hill, Victoria, Australia) and Tamiflu (oseltamivir, Hoffman-La Roche, Basel, Switzerland/Gilead, Foster Ciy, CA), exhibit their antiviral effects through the inhibition of NA activities of the influenza A and B viruses and further underscored the importance of NA as a valid anti-influenza drug target.(Citation6,Citation7)
Oseltamivir is the predominant choice and is used worldwide for the treatment of influenza, but the generation and circulation of oseltamivir-resistant mutants of seasonal influenza, as well as H5N1 avian influenza, due to NA mutations of the viruses(Citation8–13) after Tamiflu treatment of humans infected with viruses containing different NA subtypes, have become major concerns. Moreover, some attractive structural features of H5N1 avian influenza virus NA(Citation14) have led us to search for new drug candidate and more effective antivirals.(Citation15–17)
Material and methods
Design of novel NA inhibitors based on thiazole scaffold
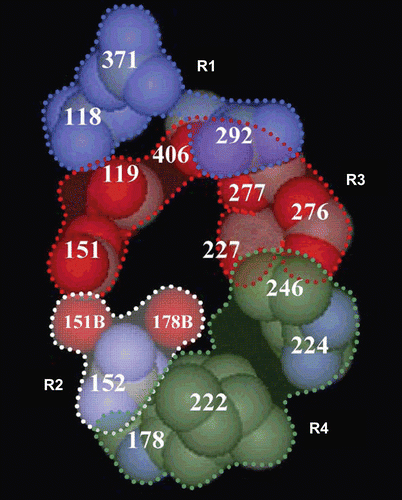
Earlier crystallographic and ensuing SAR studies have revealed that the active site of NA can be divided into four major binding sites. Abu Hammad et al.(Citation18) envisaged the binding compartment of NA as a shallow pouch comprised of four primary regions involved in binding with docked ligands (). Wang et al.(Citation19) have also derived an “airplane” model of the NA active site to summarize the basic structural requirements of a potent NA inhibitor. The first region (R1) is comprised of the positively charged guanidino groups of arginines 118, 292, and 371 and interacts with the carboxylate. Confronting R1 is a mainly hydrogen-bond-forming region (R2) consisting of the side chain of Arg 152 and the amidic carbonyl of Trp 178 and Asp 151. The negatively charged region R3 is comprised of Glu 119, Asp 151, and Glu 227, Glu 276, Glu 277, and Tyr 406 and interacts with the amino or guanidine. The hydrophobic region (R4) is situated adjacent to R2 and comprised of the side chains of Ala 246, Ile 222, Trp 178, and Arg 224 and binds to hydrophobic side chain. All binding regions were implicated in substrate recognition; however, Asp 151, Glu 277, and Tyr 406 are believed to play a critical role in the catalytic activity of NA.(Citation20,Citation21)
Figure 1. A presentation of binding pocket of neuraminidase (NA) showing the different binding regions. The crater of the binding pocket is the middle gap.(Citation17)
According to the studies on NA active site and SAR of published NA inhibitors, inhibition of the NA is mainly determined by the relative positions of the four substituents of the central ring.(Citation22) For example, in oseltamivir, the four substituents are carboxyl ethyl ester, amino, acetamino, and alkyl.
In our previous study,(Citation23,Citation24) we have reported several kinds of novel NA inhibitors based on pyrrolidine and benzyl scaffold. In our recent screening, we found that the commercially available 2-amino-4-thiazole-acetic ester 1 exhibited substantial NA inhibition (IC50 = 358 μM). Considering that there have not been thiazole derivatives reported as NA inhibitors, we chose compound 1 as the lead compound for further modification and optimization.
To contain the different substituents to interact with the four binding pockets of the NA active sites, we used the following chemical modifications: (1) compound 1 were coupled with a few kinds of amino acids with different hydrophobic side chains to introduce the amide and alkyl, aiming to form hydrogen bond with R2 and interact with the hydrophobic region R4, respectively; (2) carboxyl ethyl ester was kept or converted to carboxylic group, aiming to interact with R1; (3) BOC-protected amino group was kept or be converted to free amino group, aiming to interact with R3.
Then we will describe the synthesis, biological activity, and the docking study of the thiazole derivates as NA inhibitors.
Chemistry
All reactions were carried out by standard techniques for the exclusion of moisture. Solvents were distilled prior to use and flash chromatography was performed using silica gel (60 Å, 200 ± 300 mesh). All reactions were monitored by thin-layer chromatography on 0.25-mm silica gel plates (60GF-254) and visualized with UV light, or iodine vapour. Melting points were obtained on an electrothermal melting point apparatus and are uncorrected. Proton nuclear magnetic resonance (1H NMR) spectra were determined on a Brucker Avace 600 spectrometer using trimethylsilane (TMS) as an internal standard. Chemical shifts are reported in delta (δ) units, parts per million (ppm) downfield from TMS. High-resolution mass spectral (HRMS) data are reported as m/z (relative intensity).
2-(tert-Butoxycarbonylamino)acetic acid (N-BOC-l-glycine) (2)
The title compound was prepared as described by Nielsen et al.(Citation25)
General procedure for the synthesis of 3a–9a
Compound 2 (10 mmol) was dissolved in anhydrous CH2Cl2 and 1 equiv of Et3N was added to the solution. Then TBTU (11 mmol) was added slowly to the above solution under ice-bath. After stir for 10 min at 0°C, compound 1 (12 mmol) was added to the mixture and adjust to pH 7 with Et3N. After about 4 h stirring at room temperature, the mixture was filtrated and washed with 10% citric acid, brine, and saturated NaHCO3 solution, and then was dried with Na2SO4. The solvent was evaporated to give a residue that was chromatographed on a silica column to give the title compound 3a (yield: 85%) as a white solid, m.p. 154–156°C. HRMS: calcd for C14H21N3O5S [M+H]+ 344.1235. Found: 344.1286. 1H NMR (DMSO-d6): δ 1.18 (t, 3H, J = 7.2 Hz), 1.39 (s, 9H), 3.68 (s, 2H), 3.8 (d, 2H, J = 6.0 Hz), 4.08 (q, 2H, J = 7.2 Hz), 6.97 (s, 1H), 7.14 (t, 1H, J = 6.0 Hz), 12.2 (s, 1H).
Compounds 4a–7a were prepared following the general procedure as described above.
(S)-Ethyl 2-(2-(2-(tert-butoxycarbonylamino)propanamido)thiazol-4-yl)acetate (4a)
A white solid, yield 81%, m.p. 140–142°C. HRMS: calcd for C15H23N3O5S [M+H]+ 358.1392. Found: 358.1489. 1H NMR (DMSO-d6): δ 1.18 (t, 3H, J = 7.2 Hz), 1.24 (d, 3H, J = 7.2 Hz), 1.39 (s, 9H), 3.682 (s, 2H), 4.08 (q, 2H, J = 7.2 Hz), 4.20 (m, 1H), 6.98 (s, 1H), 7.22 (d, 1H, J = 6.6 Hz), 12.18 (s, 1H).
Ethyl 2-(2-((2S,3R)-2-(tert-butoxycarbonylamino)-3-methylpentanamido)thiazol-4-yl)acetate (5a)
A white solid, yield 87%, m.p. 64–66°C. HRMS: calcd for C18H29N3O5S [M+H]+ 400.1861. Found: 400.2092. 1H NMR (DMSO-d6): δ 0.83 (m, 6H), 1.16 (t, 3H), 1.24 (m, 1H), 1.37 (t, 9H), 1.45(m, 1H), 1.78 (m, 1H), 3.69 (s, 2H), 4.05(q, 1H), 4.07 (q, 2H), 6.99 (s, 1H), 7.10 (d, 1H), 12.20 (s, 1H).
(S)-Ethyl 2-(2-(2-(tert-butoxycarbonylamino)-3-methylbutanamido)thiazol-4-yl) acetate (6a)
A yellow liquid, yield 79%. HRMS: calcd for C22H38N4O7S [M+H]+ 386.1705. Found: 386.1777. 1H NMR (DMSO-d6): δ 0.85–0.91 (m, 6H, J = 7.2 Hz),1.17–1.38 (s, 9H), 1.96 (m, 1H), 3.62 (s, 2H), 4.01 (m, 1H), 4.08 (q, 2H), 6.99 (s, 1H), 7.07 (d, 1H, J = 7.2 Hz), 12.21 (s, 1H).
(S)-Ethyl 2-(2-(2-(tert-butoxycarbonylamino)-4-methylpentanamido)thiazol-4-yl) acetate (7a)
A white solid, yield 83%, m.p. 68–70°C. HRMS: calcd for C18H29N3O5S [M+H]+ 400.1861. Found: 400.1967. 1H NMR (DMSO-d6): δ 1.18 (t, 3H), 1.38 (s, 9H), 1.82–1.91 (m, 2H), 2.046 (m, 3H), 2.41–2.54 (m, 2H), 3.69 (s, 2H), 4.08 (q, 2H), 4.23 1(q, 1H), 6.99 (s, 1H), 7.27 (s, 1H),12.27 (s, 1H).
(S)-Ethyl 2-(2-(2-(tert-butoxycarbonylamino)-4-(methylthio)butanamido)thiazol-4-yl) acetate (8a)
A yellow liquid, yield 85%. HRMS: calcd for C17H27N3O5S2 [M+H]+ 418.1426. Found: 418.1491. 1H NMR (DMSO-d6): δ 1.17 (t, 3H), 1.19 (s, 9H), 1.93 (m, 3H), 2.14 (m, 2H), 2.56 (m, 2H), 3.72 (s, 2H), 4.08 (q, 2H), 4.23 (q, 1H), 6.99 (s, 1H), 7.27 (d, 1H), 12.27 (s, 1H).
(S)-Ethyl 2-(2-(2-(tert-butoxycarbonylamino)-3-phenylpropanamido)thiazol-4-yl) acetate (9a)
A white solid, yield 78%, m.p. 116–118°C; HRMS: calcd for C21H27N3O5S [M+H]+ 434.1705. Found: 434.1857. 1H NMR (DMSO-d6): δ 1.18 (t, 3H), 1.31 (s, 9H), 2.80 (dd, 1H), 2.99 (dd, 1H), 3.69 (s, 2H), 4.09 (q, 2H), 4.40 (br, 1H), 7.00 (s, 1H), 7.20 (t, 1H), 7.28 (t, 2H),7.35 (d, 2H), 12.41 (s, 1H).
General procedure for the synthesis of 3b–9b
To a solution of compound 3a (10 mmol) in dry EtOAc at 0°C was added a solution of EtOAc (10 mL) saturated with dry HCl gas. The reaction solution was stirred at 0°C for 2 h, and then the temperature is raised to room temperature and the reaction proceeds for 5 h before being concentrated in vacuo. The product 3b was recrystallized with MeOH and EtOAc from the residue as a white solid. Yield 90%, m.p. 97–99°C. HRMS: calcd for C9H13N3O3S (M+H): 244.0711. found: 244.0753; 1H NMR (DMSO-d6): δ 1.18 (t, 3H, J = 7.2 Hz), 3.68 (s, 2H), 3.80 (d, 2H, J = 6.0 Hz), 4.08 (q, 2H, J = 7.2 Hz), 6.97 (s, 1H), 7.14 (t, 1H, J =6.0 Hz), 12.20 (s, 1H).
Compounds 4b–9b were prepared following the general procedure as described above.
(S)-Ethyl 2-(2-(2-aminopropanamido)thiazol-4-yl)acetate (4b)
A white solid, yield 93%, m.p. 145–147°C. HRMS: calcd for C10H15N3O3S [M+H]+ 258.0868. Found: 257.0817. 1H NMR (DMSO-d6): δ 12.18 (s, 1H), 8.52 (s, 3H), 7.10 (d, 1H), 4.20 (m, 1H), 4.08 (q, 2H), 3.68 (s, 2H), 1.24 (d, 3H), 1.18 (t, 3H).
Ethyl 2-(2-((2S,3R)-2-amino-3-methylpentanamido)thiazol-4-yl)acetate (5b)
A white solid, yield 89%, m.p. 178–180°C. HRMS: calcd for C13H21N3O3S [M+H]+ 300.1337. Found: 300.1439. 1H NMR (DMSO-d6): δ 0.85–0.93 (m, 6H), 1.13 (m, 1H), 1.19 (t, 3H), 1.57 (m, 1H), 1.98 (m, 1H), 3.73 (s, 2H), 3.94 (q, 1H), 4.09 (q, 2H), 7.09 (s, 1H), 8.67 (d, 3H), 12.20 (s, 1H).
(S)-Ethyl 2-(2-(2-amino-3-methylbutanamido)thiazol-4-yl)acetate (6b)
A white solid, yield 91%, m.p. 138–140°C. HRMS: calcd for C12H19N3O3S [M+H]+ 286.1181. Found: 286.1233. 1H NMR (DMSO-d6): δ 0.97 (m, 6H, J = 7.2 Hz), 1.19 (m, 3H), 2.11 (m, 1H), 3.62 (s, 2H), 3.87 (m, 1H), 4.08 (q, 2H), 4.37 (br, 1H), 6.99 (s, 1H), 8.56 (d, 3H, J = 4.2 Hz), 12.21 (s, 1H).
Ethyl 2-(2-((2S,3R)-2-amino-3-methylpentanamido)thiazol-4-yl)acetate (7b)
A white solid, yield 88%, m.p. 151–153°C. HRMS: calcd for C13H21N3O3S [M+H]+ 300.1337. Found: 300.1453. 1H NMR (DMSO-d6): δ 0.91 (t, 6H, J = 6.0 Hz), 1.66–1.68 (m, 3H), 3.72 (s, 2H), 3.89 (br, 1H), 4.03 (m, 1H), 4.09 (m, 2H), 7.10 (s, 1H), 8.59 (s, 3H), 12.70 (s, 1H).
(S)-Ethyl 2-(2-(2-amino-4-(methylthio)butanamido)thiazol-4-yl)acetate (8b)
A white solid, yield 85%, mp = 162–164°C. HRMS: calcd for C12H19N3O3S2 (M+H): 318.0901. Found: 318.0952. 1H NMR (DMSO-d6): δ 2.06 (m, 3H), 2.14 (m, 2H), 2.56 (m, 2H), 3.72 (s, 2H), 3.83 (br, 1H), 4.09 (q, 2H), 4.13 (q, 1H), 7.10 (s, 1H), 8.64 (s, 3H), 12.80 (s, 1H).
(S)-Ethyl 2-(2-(2-amino-3-phenylpropanamido)thiazol-4-yl)acetate (9b)
A white solid, yield 92%, m.p. = 238–239°C. HRMS: calcd for C16H19N3O3S [M+H]+ 334.1181. Found: 334.1252. 1H NMR (DMSO-d6): δ 1.19 (t, 3H), 3.11 (dd, 1H), 3.20 (t, 1H), 3.61 (q, 2H), 3.72 (s, 2H), 4.26 (m, 1H), 7.08(s, 1H), 7.25 (s, 1H), 7.27 (t, 1H), 7.33 (t, 2H), 7.55 (d, 2H ), 12.79 (s, 1H).
General procedure for the synthesis of 3c–9c
LiOH hydrate (12 mmol) was added in portions to a solution of 3b (10 mmol) in dioxane/H2O (3:2) (20 mL). The reaction mixture was stirred at room temperature until all the starting material had been consumed. The dioxane was evaporated and water was added. The aqueous layer was washed twice with CH2Cl2, neutralized with KHSO4 (12 mmol), then extracted with CH2Cl2, washed once with brine, and was dried with Na2SO4. The solvent was evaporated to give a residue that was chromatographed on a silica column to give the title compound 3c (yield: 72%) as a white solid. Yield 82%, m.p. 161–163°C. HRMS: calcd for C12H17N3O5S [M+H]+ 316.0966. Found: 316.0964. 1H NMR (DMSO-d6): δ 1.39 (s, 9H), 3.59 (s, 2H), 3.80 (d, 2H, J = 6.0 Hz), 7.14 (t, 1H, J = 6.0 Hz), 6.95 (s, 1H), 12.15 (s, 1H), 12.15 (s, 1H).
Compounds 4c–9c were prepared following the general procedure as described above.
(S)-2-(2-(2-(tert-Butoxycarbonylamino)propanamido)thiazol-4-yl)acetic acid (4c)
A white solid, yield 78%, m.p. 159–160°C. HRMS: calcd for C13H19N3O5S [M+H]+ 330.1179. Found: 330.1124. 1H NMR (DMSO-d6): δ 1.24 (d, 3H), 1.39 (s, 9H), 3.59 (s, 2H), 4.19 (d, 2H), 6.95 (s, 1H), 7.21 (t, 1H, J = 6.0 Hz), 12.15 (s, 1H), 12.16 (s, 1H).
2-(2-((2S,3R)-2-(tert-Butoxycarbonylamino)-3-methylpentanamido)thiazol-4-yl) acetic acid (5c)
A white solid, yield 82%, m.p. 122–124°C. HRMS: calcd for C16H25N3O5S [M+H]+ 372.1548. Found: 372.1594. 1H NMR (DMSO-d6): δ 0.83 (m, 6H), 1.27 (m, 1H), 1.37 (t, 9H), 1.46 (m, 1H), 1.75 (m, 1H), 3.60 (s, 2H), 4.07 (q, 1H),M 6.95 (s, 1H), 7.09 (d, 1H), 12.18 (s, 1H), 12.38 (s, 1H).
(S)-2-(2-(2-(tert-Butoxycarbonylamino)-3-methylbutanamido)thiazol-4-yl)acetic acid (6c)
A white solid, yield 85%, m.p. 173–175°C. HRMS: calcd for C20H34N4O7S [M+H]+ 358.1392. Found: 358.1453. 1H NMR (DMSO-d6): δ 1.25 (m, 6H, J = 7.2 Hz), 1.38 (s, 9H), 2.98 (m, 1H), 3.60 (s, 2H), 4.01 (m, 1H), 6.95 (s, 1H), 7.05 (d, 1H, J = 7.2 Hz),12.17 (s, 1H), 12.38 (s, 1H).
(S)-2-(2-(2-(tert-Butoxycarbonylamino)-4-methylpentanamido)thiazol-4-yl)acetic acid (7c)
A white solid, yield 78%, m.p. 138–140°C. HRMS: calcd for C16H25N3O5S [M+H]+ 372.1548. Found: 372.1595. 1H NMR (DMSO-d6): δ 0.88 (t, 6H, J = 7.2 Hz), 1.27 (m, 2H), 1.37 (s, 9H), 1.52 (m, 1H), 1.62 (t, 1H, J = 6.6 Hz), 3.33 (s, 2H), 4.21 (m, 1H), 6.95 (s, 1H), 7.15 (d, 1H, J = 7.2 Hz), 12.23 (s, 1H), 12.34 (s, 1H).
(S)-2-(2-(2-(tert-Butoxycarbonylamino)-4-(methylthio)butanamido)thiazol-4-yl)acetic acid (8c)
A white solid, yield 86%, m.p. 153–155°C. HRMS: calcd for C15H23N3O5S2 [M+H]+ 390.1113. Found: 390.1161. 1H NMR (DMSO-d6): δ 1.26 (t, 3H), 1.83 (s, 9H), 1.90 (m, 2H), 2.03 (m, 3H), 3.59 (m, 2H), 3.69 (s, 2H), 4.08 (q, 2H), 4.23 (q, 1H), 6.95 (s, 1H), 7.25 (d, 1H), 12.24 (s, 1H), 12.57 (s, 1H).
(S)-2-(2-(2-(tert-Butoxycarbonylamino)-3-phenylpropanamido)thiazol-4-yl)acetic acid (9c)
A white solid, yield 83%, m.p. = 180–182°C. HRMS: calcd for C19H23N3O5S [M+H]+ 406.1392. Found: 406.1448. 1H NMR (DMSO-d6): δ 1.19 (t, 3H), 1.30 (s, 9H), 2.51 (t, 1H), 2.51 (dd, 1H), 3.60 (s, 2H), 4.38 - 4.42 (m, 1H), 6.96 (s, 1H),7.20 (s, 1H), 7.25 (t, 1H), 7.28 (t, 2H), 7.34 (d, 2H), 12.39 (s, 2H).
General procedure for the synthesis of 3d–9d
To a solution of compound 3c (10 mmol) in dry EtOAc at 0°C was added a solution of EtOAc (10 mL) saturated with dry HCl gas. The reaction solution was stirred at 0°C for 2 h, and then the temperature is raised to room temperature and the reaction proceeds for 5 h before being concentrated in vacuo. The product 3d was recrystallized with MeOH and EtOAc from the residue as a white solid. Yield 88%, m.p. 185–187°C. HRMS: calcd for C7H9N3O3S [M+H]+ 216.0398. Found: 216.0344. 1H NMR (DMSO-d6): δ 3.62 (s, 2H), 3.86 (q, 2H), 7.04 (s, 1H), 8.53 (t, 3H).
Compounds 4d–9d were prepared following the general procedure as described above.
(S)-2-(2-(2-Aminopropanamido)thiazol-4-yl)acetic acid (4d)
A white solid, yield 90%, m.p. 101–103°C. HRMS: calcd for C8H11N3O3S [M+H]+ 230.0655. Found: 230.0607. 1H NMR (DMSO-d6): δ 1.47 (d, 3H, J = 7.2 Hz), 4.08 (m, 1H), 3.63 (s, 2H), 7.06 (s, 1H), 8.51 (d, 3H, J = 4.2 Hz), 12.70 (s, 1H).
2-(2-((2S,3R)-2-Amino-3-methylpentanamido)thiazol-4-yl)acetic acid (5d)
A white solid, yield 88%, m.p. 172–174°C. HRMS: calcd for C11H17N3O3S [M+H]+ 272.1024. Found: 272.1080. 1H NMR (DMSO-d6): δ 0.83 (m, 6H), 1.27 (m, 1H), 1.46 (m, 1H), 1.75 (m, 1H), 3.60 (s, 2H), 4.07 (q, 1H), 6.95 (s, 1H), 7.09 (d, 1H), 8.59 (s, 3H), 12.38 (s, 1H).
(S)-2-(2-(2-Amino-3-methylbutanamido)thiazol-4-yl)acetic acid (6d)
A white solid, yield 92%, m.p. 194–196°C. HRMS: calcd for C10H15N3O3S [M+H]+ 258.0868. Found: 258.0909. 1H NMR (DMSO-d6): δ 1.25 (m, 6H, J = 7.2 Hz), 2.98 (m, 1H), 3.60 (s, 2H), 4.01 (m, 1H), 6.95 (s, 1H), 7.05 (d, 1H), 8.55 (s, 3H), 12.38 (s, 1H).
((S)-2-(2-(2-Amino-4-methylpentanamido)thiazol-4-yl)acetic acid (7d)
A white solid, yield 88%, m.p. 205–206°C. HRMS: calcd for C11H17N3O3S [M+H]+ 272.1024. Found: 272.1070. 1H NMR (DMSO-d6): δ 0.91 (t, 6H), 1.66–1.68 (m, 3H), 3.63 (s, 2H), 3.74 (s, 1H), 4.09 (m, 1H), 7.10 (s, 1H), 8.59 (s, 3H), 12.70 (s, 1H).
(S)-2-(2-(2-Amino-4-(methylthio)butanamido)thiazol-4-yl)acetic acid (8d)
A white solid, yield 90%, m.p. 190–192°C. HRMS: calcd for C10H15N3O3S2 [M+H]+ 290.0588. Found: 290.0624. 1H NMR (DMSO-d6): δ 2.06 (m, 3H), 2.15 (m, 2H), 2.56 (m, 2H), 3.72 (s, 2H), 4.09 (q, 1H), 4.15 (m, 1H), 7.10 (d, 1H), 8.74 (s, 3H), 12.34 (s, 1H).
(S)-2-(2-(2-Amino-3-phenylpropanamido)thiazol-4-yl)acetic acid (9d)
A white solid, yield 88%, m.p. 225–227°C. HRMS: calcd for C14H15N3O3S [M+H]+ 306.0868. Found: 306.0914. 1H NMR (DMSO-d6): δ 3.11–3.20 (m, 2H), 3.61 (dd, 2H), 3.72 (s, 1H), 4.26 (t, 1H), 4.26 (t, 1H), 7.08 (s, 1H), 7.25 (s, 1H), 7.27 (t, 2H), 7.33 (d, 2H), 8.55 (s, 3H), 12.79 (s, 1H).
NA inhibition assay
NA inhibitory activity was determined by the commercial NA inhibitory screening kit (Beyotime Institute of Biotechnology, Jiangsu, China). Although the NA for enzyme assay is not originated from avian influenza A/H5N1 strain, their sequences are highly conserved. Therefore, this kit is suitable for the high-throughput screening of NA inhibitors in vitro.
The compound 2′-(4-methylumbelliferyl)-a-d-acetyl neuraminic acid (MUNANA) is the substrate of NA. And cleavage of this substrate by NA produces a fluorescent product, which can emit an emission wavelength of 460 nm with an excitation wavelength of 355 nm. The intensity of fluorescence can reflect the activity of NA sensitively.
The reaction mixture containing the buffer, NA enzyme, test compounds, or oseltamivir carboxylate (which was prepared according to literature method(Citation26)) and the substrate were incubated at 37°C. Terminate the reaction by adding 150 μL stop solution to all wells including the blank row. Read the plate within 20 min of adding stop solution detecting fluorescence using an excitation wavelength of 355 nm and an emission wavelength of 460 nm. The IC50 was calculated by plotting percent inhibition versus the inhibitor concentration and determination of each point was performed in duplicate. The data are expressed as the mean of three independent experiments.(Citation27)
Result and discussion
Chemistry
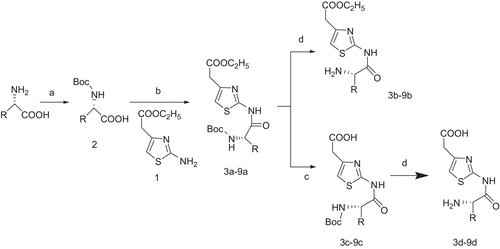
The synthetic route of target compounds was shown in . Different amino acids with hydrophobic side chains are first protected by BOC2O, and then the BOC-protected amino acids 2 were coupled with the lead compound 1 using TBTU as the coupling agent. Compounds 3a–9a were obtained and the BOC group was easily cleaved by using 6 N HCl in EtOAc to provide compounds 3b–9b. The esters of 3a–9a were hydrolyzed to carboxylic acid by LiOH/H2O in dioxane to obtain compounds 3c–9c. Then through deprotecting the BOC– group using 6 N HCl in EtOAc, compounds 3d–9d were obtained.
Scheme 1. Reagents and conditions: a. (BOC)2O/THF; b. TBTU, Et3N/anhydrous CH2Cl2, 0°C; c. 1 M LiOH, dioxane/H2O; and d. HCl/anhydrous EtOAc.
NA inhibition assay
All the target compounds (28 compounds) were tested for their ability to inhibit NA. Preliminary results showed that most of the compounds (25 compounds) displayed enhanced inhibitory activities compared with the lead compound (). It seems that the compounds with proper hydrophobic side chain show better inhibitory activity as three such compounds (4d, 5d, and 8d) exhibit good activity (3.43, 4.27, and 7.78 μM, respectively) and the methyl group is the most suitable. As expected, compounds with –COOH group showed better activities than those with –COOEt group. Compounds with –NH2 group showed increased activities than those with BOC–NH– group. On the other hand, the incorporation of natural amino acids would generate compounds with similar or better inhibitory activities compared with the parent compound.
Table 1. The structure and in vitro inhibitory activities of target compounds against neuraminidase (NA).
In order to determine the interaction between thiazole derivatives and the NA active site, compound 4d was docked into the active sites of NA (PDB entry: 2HU4) using SYBYL 7.0 and optimized using Powell’s method with the Tripos force field with convergence criterion set at 0.05 kcal/(Å·mol), and assigned with Gasteiger-Hückel method. The docking study performed using Sybyl/FlexX module, the residues in a radius of 7.0 Å around oseltamivir in the co-crystal structure (PDB ID: 2HU4) were selected as the active site. The binding of compound 4d in the active site of NA is shown in , and we found that the –COOH group of the target compounds interacts with the positively charged region R1 of NA active site by making charge–charge interactions with Arg 292 and Arg 371 of this subsite. This correlates with the fact that compounds with –COOH group showed better activities than those with –COOEt group. The reason for which the –COOH group is important for development of novel inhibitors is that it is well-established that two or three Arg residues in the immediate vicinity of the carboxylic group of NA inhibitors play a key role in orienting and stabilizing various inhibitors.(Citation28,Citation29)
Figure 2. Flex X-docked result of compound 4d in the active site of neuraminidase (PDB ID: 2HU4). The yellow lines and numbers show the potential hydrogen bonds and bond length.
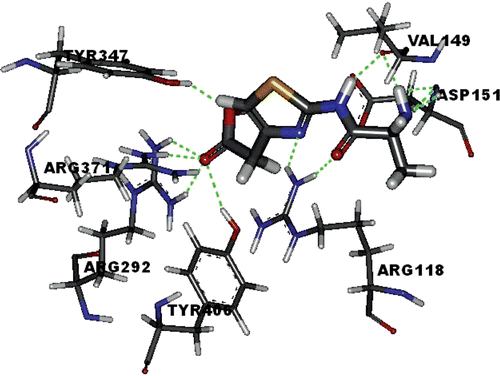
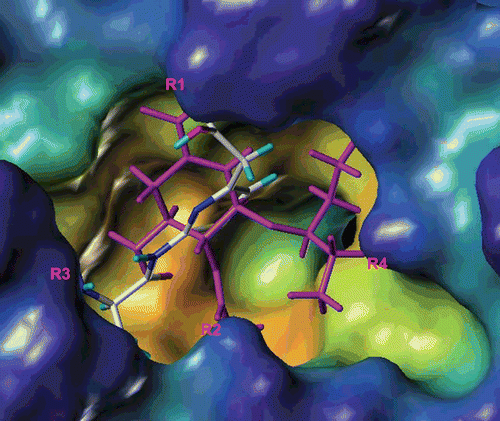
The –NH2 group binds to the negatively charged region R3 by interaction with Asp 151 and Glu 119. The –CO–NH– group forms hydrogen bond with Asp 151 of the region R2. In addition, the –COOH group of compound 4d could also interact with Tyr 347 and Tyr 406 residues, the –NH2 group could interact with Arg 156 residue by hydrogen bond for stabilizing the interaction. Also, as shown in , the methyl group may accommodate a small hydrophobic pocket next to region R3 instead of the major hydrophobic region R4. And considering the actual activities, we can conclude that the methyl group is just the most suitable for this small hydrophobic pocket. Although R2 and R4 could not be occupied well, the result still indicated that compound 4d could be a lead compound to develop new thiazole-based derivatives as novel NA inhibitors.
Figure 3. Docking result of compound 4d shown by SYBYL 7.0 (zanamivir shown in magenta line).
Conclusion
A series of novel influenza NA inhibitors based on core were synthesized and evaluated for their ability to inhibit NA of influenza A virus.
In conclusion, we have described the synthesis and properties of a series of thiazole derivatives as influenza NA inhibitors. Several compounds were shown to possess moderate influenza NA inhibitory activity, although in all cases, measured activity was lower than that of oseltamivir. The most potent compound of the series is compound 4d (IC50 = 3.43 μM), about 20-fold less potent than oseltamivir.
The binding of compound 4d in the active site of NA is shown in . Although the four regions of the active site of NA were not occupied as well as oseltamivir to establish a consistent binding orientation and the inhibitory activity is not so potent, our study still indicated that thiazole derivatives can show potent NA inhibitory activity and this finding could be used to design novel influenza NA inhibitors that exhibit increased activity based on thiazole ring. Both NA enzyme inhibition and X-ray crystallography data suggest that the strategy of designing an inhibitor of NA that binds to the highly conserved active site of the NA achieves the desired goal of activity against all influenza NA subtypes, N1–N9, and influenza B viruses.(Citation30,Citation31)
Declaration of interest
We thank the financial support from the National Natural Science Foundation of China (Grant No. 30672541).
Related Research Data
References
- Moscona, A. Neuraminidase inhibitors for influenza. N. Engl. J. Med. 2005, 353, 1363–1373.
- Gong, J., Xu, W., Zhang, J. Structure and functions of influenza virus neuraminidase. Curr. Med. Chem. 2007, 14, 113–122.
- Chong, A.K., Pegg, M.S., Taylor, N.R., von Itzstein, M. Evidence for a sialosyl cation transition-state complex in the reaction of sialidase from influenza virus. Eur. J. Biochem. 1992, 207, 335–343.
- Liu, Y., Zhang, J., Xu, W. Recent progress in rational drug design of neuraminidase inhibitors. Curr. Med. Chem. 2007, 14, 2872–2891.
- Oxford, J.S., Novelli, P., Sefton, A., Lambkin, R. New millennium antivirals against pandemic and epidemic influenza: the neuraminidase inhibitors. Antivir. Chem. Chemother. 2002, 13, 205–217.
- Wilson, J.C., von Itzstein, M. Recent strategies in the search for new anti-influenza therapies. Curr. Drug Targets 2003, 4, 389–408.
- Dreitlein, W.B., Maratos, J., Brocavich, J. Zanamivir and oseltamivir: two new options for the treatment and prevention of influenza. Clin. Ther. 2001, 23, 327–355.
- Collins, P.J., Haire, L.F., Lin, Y.P., Liu, J., Russell, R.J., Walker, P.A., Skehel, J.J., Martin, S.R., Hay, A.J., Gamblin, S.J. Crystal structures of oseltamivir-resistant influenza virus neuraminidase mutants. Nature 2008, 453, 1258–1261.
- Moscona, A. Global transmission of oseltamivir-resistant influenza. N. Engl. J. Med. 2009, 360, 953–956.
- Mihajlovic, M.L., Mitrasinovic, P.M. Another look at the molecular mechanism of the resistance of H5N1 influenza A virus neuraminidase (NA) to oseltamivir (OTV). Biophys. Chem. 2008, 136, 152–158.
- Mihajlovic, M.L., Mitrasinovic, P.M. Some novel insights into the binding of oseltamivir and zanamivir to H5N1 and N9 influenza virus neuraminidases: a homology modeling and flexible docking study. J. Serb. Chem. Soc. 2009, 74, 1–13.
- Mihajlovic, M.L., Mitrasinovic, P.M. Applications of the ArgusLab4/AScore protocol in the structure-based binding affinity prediction of various inhibitors of group-1 and group-2 influenza virus neuraminidases (NAs). Mol Simulat 2009, 35, 311–324.
- Wang, M.Z., Tai, C.Y., Mendel, D.B. Mechanism by which mutations at his274 alter sensitivity of influenza a virus n1 neuraminidase to oseltamivir carboxylate and zanamivir. Antimicrob. Agents Chemother. 2002, 46, 3809–3816.
- Russell, R.J., Haire, L.F., Stevens, D.J., Collins, P.J., Lin, Y.P., Blackburn, G.M., Hay, A.J., Gamblin, S.J., Skehel, J.J. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature 2006, 443, 45–49.
- Du, Q.S., Wang, S.Q., Chou, K.C. Analogue inhibitors by modifying oseltamivir based on the crystal neuraminidase structure for treating drug-resistant H5N1 virus. Biochem. Biophys. Res. Commun. 2007, 362, 525–531.
- Mitrasinovic, P.M. On the structure-based design of novel inhibitors of H5N1 influenza A virus neuraminidase (NA). Biophys. Chem. 2009, 140, 35–38.
- Mitrasinovic, P.M. Advances in the structure-based design of the influenza A neuraminidase inhibitors. Curr. Drug Targets 2010, 11, 315–326.
- Abu Hammad, A.M., Afifi, F.U., Taha, M.O. Combining docking, scoring and molecular field analyses to probe influenza neuraminidase–ligand interactions. J. Mol. Graph. Model. 2007, 26, 443–456.
- Wang, G.T., Chen, Y., Wang, S., Gentles, R., Sowin, T., Kati, W., Muchmore, S., Giranda, V., Stewart, K., Sham, H., Kempf, D., Laver, W.G. Design, synthesis, and structural analysis of influenza neuraminidase inhibitors containing pyrrolidine cores. J. Med. Chem. 2001, 44, 1192–1201.
- Zhang, J., Xu, W. Recent advances in anti-influenza agents with neuraminidase as target. Mini Rev. Med. Chem. 2006, 6, 429–448.
- Bossart-Whitaker, P., Carson, M., Babu, Y.S., Smith, C.D., Laver, W.G., Air, G.M. Three-dimensional structure of influenza A N9 neuraminidase and its complex with the inhibitor 2-deoxy 2,3-dehydro-N-acetyl neuraminic acid. J. Mol. Biol. 1993, 232, 1069–1083.
- Babu, Y.S., Chand, P., Bantia, S., Kotian, P., Dehghani, A., El-Kattan, Y., Lin, T.H., Hutchison, T.L., Elliott, A.J., Parker, C.D., Ananth, S.L., Horn, L.L., Laver, G.W., Montgomery, J.A. BCX-1812 (RWJ-270201): discovery of a novel, highly potent, orally active, and selective influenza neuraminidase inhibitor through structure-based drug design. J. Med. Chem. 2000, 43, 3482–3486.
- Zhang, J., Wang, Q., Fang, H., Xu, W., Liu, A., Du, G. Design, synthesis, inhibitory activity, and SAR studies of pyrrolidine derivatives as neuraminidase inhibitors. Bioorg. Med. Chem. 2007, 15, 2749–2758.
- Zhang, J., Wang, Q., Fang, H., Xu, W., Liu, A., Du, G. Design, synthesis, inhibitory activity, and SAR studies of hydrophobic p-aminosalicylic acid derivatives as neuraminidase inhibitors. Bioorg. Med. Chem. 2008, 16, 3839–3847.
- Nielsen, P.E., Haaina, G., Lohse, A., Buchardt, O. Peptide nucleic acids (PNAs) containing thymine monomers derived from chiral amino acids: hybridization and solubility properties of d-lysine PNA. Angew Chem. Int. Ed. Engl. Comm. 1995, 35, 1939–1942.
- Lindegårdh, N., Hien, T.T., Farrar, J., Singhasivanon, P., White, N.J., Day, N.P. A simple and rapid liquid chromatographic assay for evaluation of potentially counterfeit Tamiflu. J. Pharm. Biomed. Anal. 2006, 42, 430–433.
- http://www.nisn.org/documents/Zambon—VIRGIL IC50 SOP.pdf
- Wang, T., Wade, R.C. Comparative binding energy (COMBINE) analysis of influenza neuraminidase–inhibitor complexes. J. Med. Chem. 2001, 44, 961–971.
- Ortiz, A.R., Pisabarro, M.T., Gago, F., Wade, R.C. Prediction of drug binding affinities by comparative binding energy analysis. J. Med. Chem. 1995, 38, 2681–2691.
- Govorkova, E.A., Leneva, I.A., Goloubeva, O.G., Bush, K., Webster, R.G. Comparison of efficacies of RWJ-270201, zanamivir, and oseltamivir against H5N1, H9N2, and other avian influenza viruses. Antimicrob. Agents Chemother. 2001, 45, 2723–2732.
- Roberts, N.A., Govorkova, E.A. The activity of neuraminidase inhibitor oseltamivir against all subtypes of influenza viruses. In: Mitrasinovic, P.M., Ed., Global View of the Fight against Influenza. New York: Nova Science Publishers Inc., 2009, pp. 93–118.