Abstract
Apixaban is a potent, direct, selective, and orally active inhibitor of coagulation factor Xa. Rate constants for apixaban binding to free and prothrombinase-bound factor Xa were measured using multiple techniques. The inhibition mechanism was determined in purified systems and in a plasma prothrombin clotting time assay. Apixaban inhibits factor Xa with a Ki of 0.25 nM at 37°C, an association rate constant of approximately 20 μM−1 s−1, and a dissociation half-life of 1–2 min. Under physiological conditions apixaban exhibits mixed-type inhibition and maintains high factor Xa affinity with a Ki of 0.62 nM and association rate constant of 12 μM−1 s−1 for prothrombinase, and a Ki of 1.7 nM and association rate constant of 4 μM−1 s−1 for the prothrombinase:prothrombin complex. Experiments in prothrombin depleted human plasma showed that the mechanism and kinetics of inhibition are maintained in plasma. The mechanistic detail derived from these experiments can be used to understand and interpret the pharmacodynamic action of apixaban.
Keywords::
Introduction
Thrombosis causes blood vessel occlusion disrupting the normal flow of oxygen and other nutrients to tissues. An occlusive thrombus may occur at sites of vascular injury or stasis due to a defect in a haemostatic regulatory mechanism or an overwhelming prothrombotic stimulusCitation1. The clinical manifestations of thrombosis include acute coronary syndrome, myocardial infarction, peripheral artery disease, stroke, and venous thromboembolism. These diseases represent a major cause of morbidity and mortality in developed countriesCitation2. Pharmacological agents for the prevention and treatment of thromboembolic diseases have been the subject of intense research over the past few decades. Inhibitors of blood coagulation represent one class of pharmacologic agents. ApixabanCitation3 targets activated coagulation factor X (FXa) and has demonstrated antithrombotic activity in preclinical animal modelsCitation4,Citation5,Citation6 and promising safety and efficacy results in clinical studiesCitation7,Citation8,Citation9,Citation10.
Factor X activation occurs via two pathways. The phospholipid membrane bound complexes tissue factor: factor VIIa (extrinsic Xase) and factor VIIIa: factor IXa (intrinsic Xase) each hydrolyze Arg52-Ile53 in factor X to form factor Xa. Factor Xa hydrolyzes Arg320-Ile321 and Arg271-Thr272 in prothrombin to form the active enzyme thrombin. Together factor Xa and factor Va (FVa) form the prothrombinase complex on a phospholipid membrane to achieve a 300 000-fold increase in catalytic efficiency over factor Xa alone for prothrombin activationCitation11,Citation12. Thrombin amplifies its own production through activation of factors V, VIII, and XI resulting in formation of more Xase and prothrombinase complexes. Thrombin converts soluble fibrinogen to insoluble fibrin. Thrombin also activates platelets causing platelet membranes to support Xase and prothrombinase complexes and stimulating platelet aggregationCitation1.
Inhibitors of factor Xa can be classified as indirect or direct. Indirect inhibitors including heparin, low molecular weight heparins, and synthetic low molecular weight heparins, accelerate the irreversible formation of antithrombin III complexes with factor Xa. Direct inhibitors bind and inhibit factor Xa independent of antithrombin IIICitation13,Citation14.
Information derived from detailed kinetic and mechanistic studies support interpretation of observed pharmacologic effectsCitation15,Citation16. Factor Xa exerts biological activity in multiple forms, i.e. free and in a phospholipid membrane bound complex with factor Va (prothrombinase). Therefore, microscopic rate constants and mode of inhibition of apixaban for factor Xa and prothrombinase were determined by multiple methods. Experiments were also conducted in prothrombin depleted human plasma to investigate the inhibition mechanism in a more physiological system.
Materials and methods
Apixaban was prepared by the Discovery Chemistry Department, Bristol-Myers Squibb Co. (Princeton, NJ). Human coagulation proteins factor X, factor Xa, factor Va, prothrombin, α-thrombin, and antithrombin III were purchased from Haematologic Technologies (Essex Junction, VT). Phosphatidylserine (PS) and phosphatidylcholine (PC) were purchased from Avanti Polar Lipids (Alabaster, AL). Normal and immunodepleted prothrombin deficient plasma were purchased from Affinity Biologicals (Ontario, Canada). Chromogenic substrates S2366 (pyroGlu-Pro-Arg-pNA) and S2765 (N-α-benzyloxycarbonyl-d-Arg-Gly-Arg-pNA) were purchased from Diapharma (West Chester, OH). Fluorogenic substrate Pefa-5534 [CH3SO2-(d)-cyclohexylAla-Gly-Arg-7-amino-4-methoxy coumarin (AMC)] was purchased from Centerchem, Inc. (Norwalk, CT). Heparin was purchased from Baxter (Deerfield, IL). Prothrombin time (PT) reagents thromboplastin C-Plus and Innovin were purchased from Dade-Behring (Deerfield, IL), Neoplastine CI Plus from Diagnostica Stago (Parsippany, NJ). Microtiter assays were performed using ultra low binding 96-well plates (Corning-Costar 3474, Corning, NY), Spectramax 384 Plus or Gemini EM plate readers, and SoftMax Pro (Molecular Devices, Sunnydale, CA) data acquisition software. Data analysis was performed using GraFit version 5 from Erithacus Software Ltd. (West Sussex, UK).
Preparation of phospholipid vesicles
Stock solutions of 14 mg/mL or 17.2 mM PS and 15.7 mg/mL or 20.7 mM PC were made in chloroform and stored at −20°C under nitrogen. PS (200 nmol) and PC (600 nmol) were combined in a 16 mm by 100 mm glass test tube. Chloroform was removed by drying under a mild stream of nitrogen for at least 10 min. Lipids were resuspended in 0.4 mL buffer, vortexed, then sonicated for 20–30 min in a sonication bath (Laboratory Supplies Company, Inc., Hicksville, NY).
Mechanism of inhibition
Inhibition of prothrombin cleavage by prothrombinase was assayed against six concentrations of prothrombin and six concentrations of apixaban. Assays were conducted in 50 mM HEPES, pH 7.4 at 37°C, 5 mM CaCl2, 0.15 M NaCl. Final reagent concentrations are shown in parentheses. Apixaban (0 and 0.375–6.0 nM in 2-fold increments), FVa (10 nM), CaCl2 (5.0 mM), and the thrombin substrate, S2366 (1.0 mM) were incubated with phospholipid vesicles (25 μM) for 10 min. Reactions were initiated by adding FXa (2 pM) and prothrombin (25–800 nM in 2-fold increments). The results were not significantly affected by initiation with FXa alone or with FXa plus the thrombin substrate. Activity was monitored continuously in this coupled assay by measuring the accelerating increase in absorbance at 405 nm due to linear thrombin release and subsequent hydrolysis of the largely thrombin specific S2366 substrate. Prothrombinase activity was derived from the parabolic change in absorbance over time according to the following equationCitation17,Citation18:
where: a is proportional to the rate of prothrombin activation; b is proportional to the hydrolysis of S2366 in the absence of factor Xa (nearly zero due to the lack of contaminating thrombin, poor activity with and low amount of FXa); c is the absorbance at t = 0. Data above an optical density of 1.4 were excluded from the fits due to S2366 depletion (established by where the first derivative versus time, approximated from successive five-point linear fits, began to deviate from linearity).
Inhibition of peptide substrate cleavage by FXa was assayed against six concentrations of S2765 and six concentrations of apixaban. Typical assays were conducted in 0.1 M phosphate buffer containing 0.2 M NaCl and 0.5% PEG 8000 adjusted to pH 7.4. Final reagent concentrations are shown in parentheses. Apixaban (0 and 0.5–8.0 nM in 2-fold increments) and S2765 (31.25–1000 μM by 2-fold increments) were incubated for 10 min. Reactions at 25°C were initiated by adding FXa (75 pM). For comparison with the prothrombinase reaction the assays were also conducted with S2765 at 25 and 37°C in a similar buffer: 20 mM HEPES, pH 7.5 containing 0.1% PEG 8000, 5 mM CaCl2, 0.15 M NaCl, and 25 μM PC:PS phospholipid vesicles with or without 2 nM FVa. Final reagent concentrations are shown in parentheses. Apixaban (0 and 0.25–4.0 nM in 2-fold increments) and S2765 (31.25–1000 μM by 2-fold increments) were incubated for 10 min. Reactions at 37°C were initiated by adding FXa (50 pM).
Assays were conducted under conditions of excess substrate and inhibitor over enzyme. The steady state rates (linear region) of substrate hydrolysis after the initial curved time course region (slow binding, see below) were measured by continuous monitoring of the absorbance at 405 nm for 30 min.
For the calculation of thermodynamic binding parameters FXa inhibition by apixaban was determined in a similar manner in 0.1 M phosphate buffer containing 0.2 M NaCl and 0.5% PEG 8000 adjusted to pH 7.4 at 25°C, 31°C, 37°C, and 43°C. Reaction mixtures (200 μL) with six concentrations of S2765 (from 12.5 to 400 μM at 25°C and 31°C and from 31.25 to 800 μM at 37°C and 43°C) were prepared containing six fixed concentrations of apixaban (0 and from 0.25 to 4 nM at 25°C and 31°C, and from 0.5 to 8 nM at 37°C and 43°C). Hydrolysis was initiated by addition of FXa to final concentrations of 75 pM at 25°C and 31°C, 50 pM at 37°C, and 30 pM at 43°C. For comparison with the prothrombinase reaction the assays were also conducted with S2765 at 25 and 37°C in a similar buffer: 20 mM HEPES, pH 7.5 containing 0.1% PEG 8000, 5 mM CaCl2, 0.15 M NaCl, and 25 μM PC:PS phospholipid vesicles with or without 2 nM FVa.
Steady state velocity (vs) at varying substrate and inhibitor concentrations were fit to the equations for competitive, classical non-competitive, mixed-type, and uncompetitive inhibitionCitation19 using GraFit (Erithracus Software) with best fit established by the lowest standard deviations of the fitted parameters and lowest reduced χCitation2 value.
Equation 2A: competitive: vs=(Vmax*[S])/(Km*(1 + [I]/Ki) + [S])
Equation 2B: classical non-competitive: vs=(Vmax*[S])/((Km + [S])*(1 + [I]/Ki))
Equation 2C: mixed-type: vs=(Vmax*[S])/(Km*(1 + [I]/KiE) + [S]*(1 + [I]/KiES))
Equation 2D: uncompetitive: vs=(Vmax*[S])/(Km + [S]*(1 + [I]/Ki))
The following relationshipsCitation19 were used to calculate the thermodynamic binding parameters:
where: ΔG is the Gibbs free energy for binding; R is the gas constant; T is temperature; ΔH is enthalpy; ΔS is entropy.
The enthalpic contribution to binding was calculated from the slope of a van’t Hoff plot of ln Ki versus 1/T, (ΔH/R = enthalpy/gas constant) and the entropic contribution to binding was calculated from the intercept (−ΔS/R = −entropy/gas constant). The Km or kcat/T (Eyring plot) replaced the Ki in the above equation for calculations of the enthalpy and entropy of S2765 substrate binding and catalysis.
Kinetic components of binding—association rate constants (FXa and prothrombinase with peptide substrate)
The microscopic rate constants for binding of apixaban to FXa were determined by three methods. Observed association rate constants at each apixaban concentration were calculated from non-linear time courses of inhibition in the presence of varying concentrations of S2765 using the data from the mechanism of inhibition experiments at 25°C (see above), stopped-flow spectrophotometry in the presence of S2765 at 25 and 37°C, and by stopped-flow spectrofluorimetry in the absence of substrate at 25 and 37°C (direct measurement; see below). For the plate-based time course method the entire appearance of product absorbance progress curves (201 data points at 9-s intervals) were analyzed for slow binding inhibition by fitting to the slow binding inhibition equation described below (see equation 4). Data acquired over 30 min were corrected for the measured 25 s interval between the initiation of reactions and the onset of absorbance measurements.
For the stopped-flow method the increase in absorbance at 405 nm was also followed after rapidly mixing the factor Xa solution one-to-one with a premixed solution of 940 μM substrate S2765 and apixaban using the single-mixing mode of an Applied Photophysics (Leatherhead, UK) SX.18MV stopped-flow device equipped with a circulating water bath for temperature control. The data were acquired on a linear time base, creating a total of 1000 data points in each curve, using two to three repeated measurements at each inhibitor concentration. For a typical experiment in phosphate buffer (no FVa) syringe A contained 7 nM to 50 nM FXa, and syringe B contained 940 μM S2765 and apixaban concentrations from 4 to 20 000 nM. For a typical experiment in 20 mM HEPES buffer, pH 7.5, containing 25 μM phospholipids, 5 mM calcium chloride, and 0.1% PEG 8000 (prothrombinase conditions) syringe A contained 20 nM FXa and 40 nM FVa, while syringe B had 940 μM S2765 and apixaban from 12.5 to 200 nM. Due to the wide range of kobs, different time intervals were employed ranging from 0.5 s at 10 000 nM apixaban to 200 s at 4 nM apixaban. Time points near the dead time of the instrument (<2 ms) were excluded from the calculations. The SX.18MV instrument control software (Applied Photophysics) was used to analyze the large amounts of data in the time courses.
The Michaelis constants at 37°C used to correct for the effective inhibitor concentration were determined from independent initial velocity measurements of S2765 hydrolysis using FXa (Km = 100 μM) in phosphate buffer and FXa (Km = 136 μM) in HEPES buffer in the presence of phospholipids, FVa, and calcium chloride (prothrombinase) under the present experimental conditions. In all the stopped-flow experiments final concentrations of the reactants were half of those in the drive syringes and the experiments were conducted under pseudo-first-order conditions [(apixaban) ≥ 2.5-times (enzyme)].
Observed association rate constants in the absence of substrate were obtained by direct binding measurements of the quenching of the intrinsic FXa tryptophan fluorescence upon binding of inhibitor using stopped-flow spectrofluorimetry. The decrease in fluorescence after rapid mixing of equal volumes of FXa (414–516 nM; syringe A) and apixaban (1.25–20 μM by 2-fold increments; syringe B) was followed in a water bath equipped SX.18MV spectrometer (Applied Photophysics) using excitation at 285 nm and measuring broadband fluorescence (emission > 340 nm) with a long-pass filter in the emission beam. Experiments were performed in 0.1 M sodium phosphate buffer containing 0.2 M NaCl and 0.5% PEG 8000 at pH 7.4. The fluorescence signal was collected over at least five half-times at each inhibitor concentration (time intervals from 0.05 to 0.5 s). Monochromator entrance and exit slits were selected (1 mm) to minimize photobleaching during the collection period. Average rate constants were calculated from three replicate traces and duplicate determinations were obtained at each concentration of apixaban.
Kinetic components of binding—association rate constants (prothrombinase with prothrombin)
In 50 mM HEPES, pH 7.4 at 37°C containing 0.15 M NaCl, 5 mM CaCl2 and 0.1% PEG 8000, equal volumes of FXa (5 nM), FVa (28 nM) and phospholipids (26 μM) in syringe 1 were mixed with variable amounts (using the same buffer) of apixaban (20–160 nM), prothrombin (either 194 or 1940 nM), and phospholipids (26 μM) in syringe 2 using a KinTek (Austin, TX) Chemical Quench-Flow apparatus equipped with a circulating temperature controlled water bath. Duplicate reactions were quenched with 60 mM ethylenediaminetetraacetic acid in syringe C after incubation for 10-time points between 0.1 and 144 s using the push-pause-push mode with the appropriate reaction and exit loops. Aliquots were diluted into a standard 96-well assay for thrombin and the concentration of α-thrombin plus meizothrombin determined by comparing initial rates of S2366 (1.0 mM) hydrolysis to a standard curve of α-thrombin. The amount of meizothrombin formed was measured by determining amidolytic activity after incubating the reaction mixture for 2 min in the presence of heparin (10 μg/ mL) and antithrombin III (300 nM), which rapidly inactivates thrombin but not meizothrombinCitation20. Initial rates of thrombin formation were obtained by subtracting the rate of meizothrombin appearance from the initial rate of total active-site formation. The time courses were analyzed using the slow binding inhibition equation described below to obtain kobs values. Experiments were conducted with prothrombin at sub-saturating and at saturating prothrombin concentrations, 97 and 970 nM, respectively, to obtain on rate constants for apixaban into the prothrombinase and prothrombinase:prothrombin complexes. At sub-saturating prothrombin the on rate constant for apixaban was calculated from the slope of kobs versus apixaban times [1 + (prothrombin)/Km] for the competitive component; i.e. to extrapolate to zero substrate (prothrombinase on rate constant), while at saturating prothrombin the on rate constant was calculated from the slope times [1 + Km/(prothrombin)] for the uncompetitive component, i.e. to extrapolate to infinite substrate (prothrombinase:prothrombin complex on rate constant).
Kinetic components of binding—dissociation rate constants
The FXa:apixaban complex was formed by incubation of 1 nM FXa with 2 nM apixaban in 50 mM HEPES, pH 7.4 at 37°C, 5 mM CaCl2, 0.15 M NaCl, and 0.5% PEG 8000 for 30 min at 37°C. Based on the steady state assay the enzyme was fully inhibited under these conditions. The time-dependent increase in FXa activity was measured by fluorescence after the pre-formed FXa:apixaban complex was diluted 200-fold into reaction mixtures containing 1 mM CH3SO2-(d)-cyclohexylAla-Gly-Arg-AMC in the same buffer such that the final concentration of FXa was 5 pM and of apixaban was 10 pM (>10 times below the Ki). The relative fluorescence changes were monitored at 9-s intervals at 460 nm with excitation at 355 nm. Data acquired over 30 min were corrected for the measured 20-s interval between the initiation of reactions and the onset of fluorescence measurements. Under these conditions the observed rate constant from a fit to the slow binding equation below is a close approximation of the off-rate constant. Similar conditions were used to measure the off-rate constant at 25°C.
Kinetic components of binding—data analysis
Non-linear time course data were fit to the slow binding inhibition equationCitation21,Citation22:
where: product is absorbance of pNA at 405 nm or fluorescence of AMC at 460 nm, or prothrombin activation products; offset is the initial absorbance or fluorescence; vinitial and vfinal are initial and final velocities, respectively; kobs is the observed rate constant.
The stopped-flow association rate experiments with apixaban showed that the initial velocity was equal to the uninhibited rate up to 10 μM apixaban and could be locked at that value, which was especially useful for analysis of curved plate reader data at different concentrations of peptide substrate. The kobs versus apixaban data at all levels of substrate was fit to a modified competitive inhibition equationCitation21,Citation22:
Equation 5: kobs=koff + (kon*[I])/(1 + [S]/Km)
where: koff (dissociation rate constant) is the y-intercept intercept and kon (association rate constant) is the slope times ![]() .
.
In the preincubation-dilution dissociation rate experiments the initial rate is locked at zero, the final rate reaches >90% of the uninhibited rate, and kobs ≈ off-rate constant due to the dilution of the inhibitor below its Ki and competition with saturating levels of substrate in the assay.
In the direct binding experiments the observed rate constants at each inhibitor concentration were obtained from a fit of the fluorescence quenching data to a single exponential:
where: amplitude = Signalinitial − Signalfinal.
On rates constants were calculated from the slopes of a linear secondary plot of kobs versus inhibitor (and multiplied by ![]() to adjust for competition with the substrate when S2765 substrate was present). Off-rate constants were determined by the dilution experiment described above, or were calculated from the simple relationship:
to adjust for competition with the substrate when S2765 substrate was present). Off-rate constants were determined by the dilution experiment described above, or were calculated from the simple relationship:
Equation 7:
koff = final Ki × kon, or from the intercept of kobs versus inhibitor concentration plots when inhibitor concentrations were near the Ki (less than 100 × Ki) or from the global fit to the modified competition equation shown above.
PT assays
Prothrombin clotting time assays were performed in duplicate according to the reagent manufacturer’s directions using an automated coagulation analyzer (Sysmex CA-1500, Dade-Behring, Newark, DE). For PT, 50 μL plasma was incubated at 37°C. After 2 min 100 μL PT reagent was added and clotting time was recorded by the automated analyzer. Plasma prothrombin concentrations were varied by combining pooled normal plasma and prothrombin immunodepleted plasma. Alternatively, plasma purified prothrombin was added to prothrombin depleted plasma to obtain final prothrombin concentrations of 70, 140, 280, 700, and 1400 nM, or 5, 10, 20, 50, and 100 percent of the normal human prothrombin concentrationCitation23, respectively. Apixaban was spiked into plasma beginning with a 10 mM dimethyl sulfoxide stock solution followed by serial dilutions with plasma.
In enzyme kinetic terms the clotting time represents an inverse of the overall reaction rate (kcat−1 or v−1), i.e. the time required to form a critical amount of fibrinCitation24. PT results from experiments where prothrombin and apixaban concentrations were varied independently were analyzed by three methods. The first method was a linear regression of PT versus apixaban concentration at each prothrombin concentration. The second method was a non-linear fit of reciprocal clotting time (v) versus plasma prothrombin concentration (S) to the Michaelis-Menten equations (Equation 2A–D). A Dixon plot consists of inverse reaction rate versus inhibitor concentration. Therefore, a plot of clotting time versus apixaban concentration is analogous to the Dixon plot. The third method was a simultaneous fit of PT (v−1) at each prothrombin (S) and apixaban concentration (I) to the following equationCitation19.
The PT reagent and plasma constitute a matrix where the free, effective apixaban concentration (unbound to plasma protein and other reagent components) is unknown. Although enzyme kinetic terms can be calculated, they represent apparent kinetic constants subject to the influence of the matrix with an undetermined effect on the mechanism of inhibition.
Results
Mechanism of apixaban inhibition of factor Xa and prothrombinase
Michaelis–Menten kinetics were observed for prothrombinase and prothrombin with a Km of approximately 150 nM (n = 6). Apixaban inhibited prothrombin activation by prothrombinase with a best fit of the data to the equation for mixed-type inhibition (Equation 2C) with KiE for prothrombinase of 0.62 ± 0.06 nM and KiES for the prothrombinase:prothrombin complex of 1.68 ± 0.15 nM (). Since the apixaban Ki for prothrombinase determined at 37°C was higher than previously found for purified factor Xa and the peptide substrate S2765 at 25°C (0.075 nMCitation3), we repeated the assays with purified FXa and S2765 at four temperatures. The apixaban Ki values from global fits of the data to the equation for competitive inhibition (Equation 2A) were 0.075 ± 0.0031 nM at 25°C, 0.14 ± 0.0056 nM at 31°C, 0.25 ± 0.011 nM at 37°C () and 0.40 ± 0.013 nM at 43°C (). At 25, 31, 37, and 43°C the measured Km’s of S2765 for FXa were 31 ± 1.4, 48 ± 2.0, 62 ± 2.9, and 92 ± 3.0 μM, respectively.
Table 1. Effect of temperature on Ki and rate constants for apixaban.
Figure 1. Mechanism of apixaban inhibition of factor Xa and prothrombinase. (A) Lineweaver–Burk plot of steady state hydrolysis rate versus different concentrations of S2765 (31.25–1000 μM by 2-fold increments) at fixed concentrations of apixaban [0 (open circle), 0.5 nM (closed circle), 1.0 nM (open square), 2.0 nM (closed square), 4.0 nM (open triangle), and 8.0 nM (closed triangle)] in 0.1 M phosphate buffer, pH 7.4 at 37°C, containing 0.2 M NaCl and 0.5% PEG 8000. The lines drawn through the experimental data are to global fits of the data to the non-linear Equation 2A for competitive inhibition versus S2765 with fitted parameters of Vmax = 6.6 ± 0.070 (1000 × OD at 405 nm/min), Km = 62 ± 2.9 μM and Ki = 0.25 ± 0.011 nM. (B) Lineweaver–Burk plot of prothrombin activation rate versus different concentrations of prothrombin (25–800 nM by 2-fold increments) at fixed concentrations of apixaban [0, (open circle), 0.375 nM (closed circle), 0.75 nM (open square), 1.5 nM (closed square), 3 nM (open triangle), and 6 nM (closed triangle)]. Apixaban inhibition of the prothrombinase complex was measured by addition of factor Xa (2 pM) and different concentrations of prothrombin to assay mixtures containing 50 mM HEPES, pH 7.4 at 37°C, 5 mM CaCl2, 0.15 M NaCl, 10 nM factor Va, 25 μM phosphatidylcholine (PC)/phosphatidylserine (PS) (75:25) vesicles, 1 mM S2366 and fixed concentrations of apixaban. Thrombin generation was monitored continuously via hydrolysis of S2366 and the resultant parabolic increase in absorbance was analyzed as described in ‘Materials and Methods’. The lines drawn through the experimental data are to global fits of the data to the non-linear Equation 2C for mixed-type inhibition versus prothrombin with fitted parameters of Vmax = 19200 ± 380 (arbitrary units of absorbance/s2), Km = 140 ± 8.1 nM, KiE = 0.62 ± 0.064 nM and KiES = 1.7 ± 0.15 nM.
![Figure 1. Mechanism of apixaban inhibition of factor Xa and prothrombinase. (A) Lineweaver–Burk plot of steady state hydrolysis rate versus different concentrations of S2765 (31.25–1000 μM by 2-fold increments) at fixed concentrations of apixaban [0 (open circle), 0.5 nM (closed circle), 1.0 nM (open square), 2.0 nM (closed square), 4.0 nM (open triangle), and 8.0 nM (closed triangle)] in 0.1 M phosphate buffer, pH 7.4 at 37°C, containing 0.2 M NaCl and 0.5% PEG 8000. The lines drawn through the experimental data are to global fits of the data to the non-linear Equation 2A for competitive inhibition versus S2765 with fitted parameters of Vmax = 6.6 ± 0.070 (1000 × OD at 405 nm/min), Km = 62 ± 2.9 μM and Ki = 0.25 ± 0.011 nM. (B) Lineweaver–Burk plot of prothrombin activation rate versus different concentrations of prothrombin (25–800 nM by 2-fold increments) at fixed concentrations of apixaban [0, (open circle), 0.375 nM (closed circle), 0.75 nM (open square), 1.5 nM (closed square), 3 nM (open triangle), and 6 nM (closed triangle)]. Apixaban inhibition of the prothrombinase complex was measured by addition of factor Xa (2 pM) and different concentrations of prothrombin to assay mixtures containing 50 mM HEPES, pH 7.4 at 37°C, 5 mM CaCl2, 0.15 M NaCl, 10 nM factor Va, 25 μM phosphatidylcholine (PC)/phosphatidylserine (PS) (75:25) vesicles, 1 mM S2366 and fixed concentrations of apixaban. Thrombin generation was monitored continuously via hydrolysis of S2366 and the resultant parabolic increase in absorbance was analyzed as described in ‘Materials and Methods’. The lines drawn through the experimental data are to global fits of the data to the non-linear Equation 2C for mixed-type inhibition versus prothrombin with fitted parameters of Vmax = 19200 ± 380 (arbitrary units of absorbance/s2), Km = 140 ± 8.1 nM, KiE = 0.62 ± 0.064 nM and KiES = 1.7 ± 0.15 nM.](/cms/asset/6b9305c0-4dcb-4c3c-aa2c-040f1ae91ced/ienz_a_535793_f0001_b.gif)
Competitive inhibition of FXa versus S2765 in the HEPES buffer used for prothrombin activation studies was also observed in the presence of FVa and phospholipids with a Ki of 0.41 ± 0.018 nM at 25°C and a Ki of 0.72 ± 0.028 nM at 37°C (). Under these conditions the measured Km’s of S2765 for FXa at 25 and 37°C were 128 ± 5.8 and 142 ± 5.4 μM, respectively. The difference in Ki values between 25 and 37°C is largely mitigated by the presence of factor Va since the Ki ratio of 37–25°C is reduced from 3.3 (phosphate buffer) or 3.5 (HEPES buffer, data not shown) to 1.8 in the presence of FVa and phospholipids. Thus, the difference between the prothrombin KiE and the previously reported FXa Ki can be attributed to temperature and FVa.
Thermodynamic properties of apixaban inhibition of factor Xa
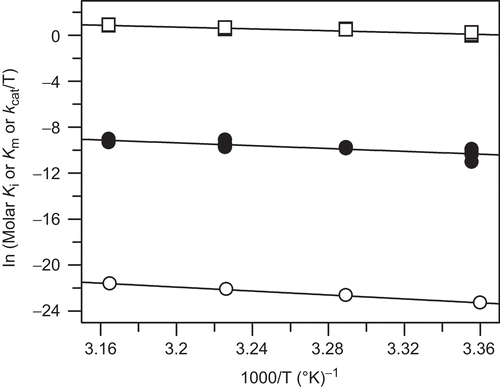
The thermodynamic parameters derived from four temperatures are shown in . At equilibrium the binding of apixaban to FXa in the absence of phospholipids, calcium, and FVa is driven entirely by enthalpy. The van’t Hoff plots of the ln (Ki) and ln (Km) versus 1/T were linear as was the Eyring plot of ln (kcat/T) versus 1/T. The linearity of these relationships (see legend for regression coefficients) suggests there is only a very small temperature dependent change in heat capacity in the temperature range studied. To estimate the enthalpy and the ΔCp from the apixaban Ki values using the integrated van’t Hoff relationshipCitation25 is not reliable under our experimental conditions due to the restricted number of data points, the narrow temperature interval, and the exceptionally good linear relationship (r2 > 0.99). Instead, the enthalpic contribution to binding was calculated from the slope of the apixaban van’t Hoff plot (ΔH/R = enthalpy/gas constant) and ΔH = −17,040 ± 410 cal/mole. From Equation 3 the calculated entropy is −11 ± 1.3 cal/mole, −T × ΔS = +3280 cal/mole at 25°C or unfavourable, and the free energy of binding is −13,760 ± 412 cal/mole.
Figure 2. Thermodynamic properties of apixaban: effect of temperature on the kinetic parameters of factor Xa catalyzed cleavage of S2765 and apixaban inhibtion of factor Xa. Van’t Hoff plots for the apixaban Ki (open circle) and substrate Km (closed circle). Eyring plot for the substrate kcat (open square). Lines shown are from linear fits of the data with slope = −8.581 ± 0.207, intercept = 5.54 ± 0.677 for ln (Ki) versus 1000/T, slope = −6.17 ± 0.813, intercept = 10.39 ± 2.66 for ln (Km) versus 1000/T, and slope = −4.00 ± 0.281, intercept = 13.59 ± 0.919 for ln (kcat/T) versus 1000/T. The correlation coefficients were 0.999, 0.879, and 0.961, respectively. The Ki data used in this analysis is from for 0.1 M phosphate buffer, pH 7.4 at 37°C, containing 0.2 M NaCl and 0.5% PEG 8000, however, the Km and kcat data are from multiple determinations in the same phosphate buffer.
Kinetic components of binding
Non-linear time courses for apixaban inhibition were observed in the plate reader assays at several concentrations of the peptide substrate. Curvature in the plate reader data progress curves in the presence of apixaban indicated that steady state hydrolysis was not achieved within a few minutes after initiating substrate hydrolysis with FXa at 25°C. Since equilibrium is established within the time courses and the steady state rates are non-zero at concentrations of inhibitor near the Ki, apixaban is a readily reversible inhibitor at any of the temperatures. In stopped-flow experiments a wider apixaban concentration range could be studied and a greater number of absorbance measurements recorded in the curved portion.
Stopped-flow assay data in the presence of S2765 (see ) were fit to the slow binding inhibition equation (Equation 4) with vinitial unlocked to obtain kobs values. The data from secondary plots of kobs versus apixaban concentrations at a fixed concentration of S2765 were linear with correlation coefficients >0.99 and were fit to a modified equation for competition (Equation 5) to obtain on rate constant values at 37°C of 20 ± 0.83 μM−1s−1 for FXa in phosphate buffer and 13 ± 0.62 μM−1s−1 for FXa in HEPES buffer with FVa, phospholipids and calcium. The on and off-rate constant data for apixaban and FXa determined in the presence of the peptide substrate at 25°C and 37°C are summarized in . From these results it is clear that the on rate constants do not change significantly with temperature and the ∼3-fold increase in Ki at 37°C compared to 25°C is primarily due to faster off-rate constants.
Determination of the rate constants of inhibition of FXa from progress curves of substrate hydrolysis can be limited by potential artefacts introduced by the kinetics of the enzyme – substrate interaction. Therefore, the kinetics of apixaban binding to FXa were also monitored by the change in intrinsic fluorescence of the protease upon rapid mixing using stopped-flow techniques. To detect transient fluorescence changes in the protein, time resolved emission was collected above 340 nM using a cut-off filter to improve sensitivity. Upon binding of apixaban to FXa, the fluorescence intensity decreased by ∼23% at 37°C (and ∼29% at 25°C) in a single exponential manner (). Similar experiments were conducted with zymogen factor X, but no change in fluorescence was detected at any concentration of apixaban up to 10 μM at 25°C suggesting that apixaban does not bind to factor X (data not shown).
Figure 3. Determination of apixaban on rate constants to prothrombinase at 37°C from stopped-flow absorbance reaction time courses in the presence of S2765 and from stopped-flow fluorescence reaction time courses for direct binding of apixaban to factor Xa. (A) Stopped-flow absorbance time courses for S2765 hydrolysis by prothrombinase at 37°C were obtained with final concentrations of factor Xa (10 nM), factor Va (20 nM) and phospholipid vesicles (25 μM) in HEPES buffer at 37°C in the presence of 12.5 nM (open circle), 25 nM (open square), 50 nM (open triangle), 100 nM (open inverse triangle) and 200 nM (◊) apixaban. Time courses were obtained after rapidly mixing the prothrombinase solution one-to-one with a premixed solution of 400 μM substrate S2765, 25 μM phospholipids, and apixaban using three repeated measurements at each inhibitor concentration. Due to the wide range of kobs, different time intervals were employed ranging from 10 s at 200 nM apixaban to 100 s at 12.5 nM apixaban (intermediate data points removed for clarity). The mean absorbance data versus time from triplicate determinations were fitted to Equation 4 and the fitted lines in the figure had the following parameters at 12.5 nM apixaban: vinitial = 0.00844 OD/s, vfinal = 0.00152 OD/s, kobs = 0.0687 s−1; at 25 nM apixaban: vinitial = 0.00884 OD/s, vfinal = 0.000867 OD/s, kobs = 0.143 s−1; at 50 nM apixaban: vinitial = 0.00827 OD/s, vfinal = 0.000464 OD/s, kobs = 0.269 s−1; at 100 nM apixaban: vinitial = 0.0116 OD/s, vfinal = 0.000384 OD/s, kobs = 0.646 s−1; at 200 nM apixaban: vinitial = 0.00737 OD/s, vfinal = 0.000150 OD/s, kobs = 1.001 s−1; (B) time dependent quenching of the relative intrinsic fluorescence of 0.26 μM factor Xa at 37°C is shown for 0.625–5 μM apixaban in 0.1 M phosphate, pH 7.4 containing 0.2 M NaCl and 0.5% PEG 8000. Solid lines are fits to a single exponential decay to a non-zero value (Equation 5) The fitted parameters are: kobs = 9.35 ± 0.37 s−1, Signalinitial = 0.124 ± 0.003, and Signalfinal = 0.0023 ± 0.0006 for 0.625 μM apixaban (upper left); kobs = 17.43 ± 0.76 s−1, Signalinitial = 0.114 ± 0.003 and Signalfinal = 0.0099 ± 0.0007 for 1.25 μM apixaban (upper right); kobs = 36.15 ± 2.12 s−1, Signalinitial = 0.110 ± 0.0036 and Signalfinal = −0.0141 ± 0.0010 for 2.5 μM apixaban (lower left); kobs = 76.65 ± 5.64 s−1, Signalinitial = 0.1022 ± 0.0043 and Signalfinal = −0.0135 ± 0.0011 for 5 μM apixaban (lower right).
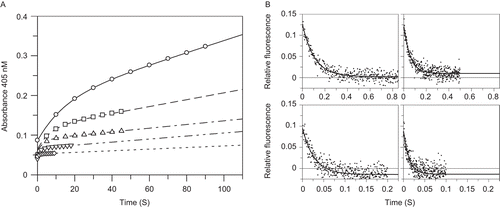
In these experiments, pseudo-first-order conditions (I ≥ 2.5E) were maintained. The dependence of the observed rate constant on the inhibitor concentration was examined. The experiments were conducted at a constant offset of ∼ 4 V by adjusting the photomultiplier (PM) voltage from 463 V at 5 μM apixaban to 548 V at 0.625 μM apixaban. Under the experimental conditions apixaban contributes a significant amount of fluorescent background especially at the higher apixaban concentrations. However, experiments conducted at constant PM voltage, where the Signalinitial increased with increasing apixaban, gave nearly identical on rate constants. At constant PM voltage the decrease in fluorescence intensity was monophasic and the amplitudes (>Equation 6: Signalinitial − Signalfinal) were also constant with apixaban concentrations up to 5 μM, suggesting that the apixaban fluorescence did not interfere with the quenching of FXa fluorescence. The secondary plot of kobs versus apixaban concentrations was linear with a correlation coefficient of 0.999 and is consistent with the results and mechanisms discussed above in the presence of S2765. The slope of the line yields an independent measurement for the bimolecular rate constant for the formation of the FXa:apixaban complex of 16 ± 0.38 μM−1s−1 at 37°C in excellent agreement with the determinations made in the presence of S2765 (). The observed rate constants were independent of FXa concentration which verifies the pseudo-first-order conditions. The y-intercepts were poorly defined (−1.4 ± 1.1 s−1 at 37°C) and inhibitor concentrations were too far removed from the Ki to use as a measure of the dissociation rate constant unlike the results obtained in the presence of S2765.
Since the secondary plots were linear and the initial velocities were approximately equal to the uninhibited velocities up to 10 μM inhibitor at 25°C (data not shown), apixaban binds according to a one step model or a two step model where the initial complex formation is extremely weak (initial Ki >> 10 μM). Therefore, the off-rate constants could be calculated from the simple relationship: koff = (final Ki) × (kon).
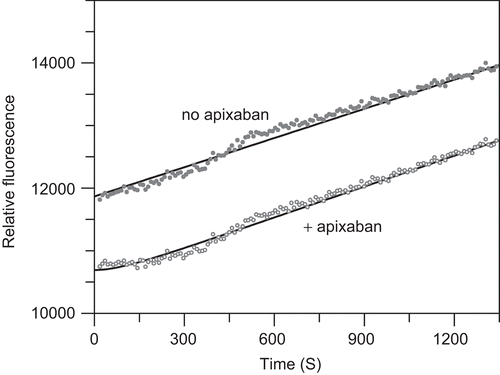
Reversibility of factor Xa inhibition is also demonstrated by the recovery of factor Xa activity upon 200-fold dilution of a pre-formed FXa:apixaban complex into substrate as shown in . Analysis of the non-linear time course of this reaction revealed an off-rate constant of 0.0113 ± 0.0014 s−1 and a dissociation half-life of 61 s similar to the values determined from the kobs versus I y-intercept and those calculated from the on rate constant and final Ki.
Figure 4. Off-rate constant from time courses following dilution of pre-formed factor Xa-apixaban complex into a fluorescent substrate plate reader assay. The factor Xa:apixaban complex was formed by incubation of 1 nM factor Xa with 2 nM apixaban in 50 mM HEPES, pH 7.4 at 37°C, 5 mM CaCl2, 0.15 M NaCl, and 0.5% PEG 8000 for 30 min at 37°C. Enzyme activity was monitored after 200-fold dilution of this complex (1 μL into 200 μL) into reaction mixtures containing 1 mM CH3SO2-(d)-cyclohexylAla-Gly-Arg-7- amino-4-methoxy coumarin (AMC) in the same buffer. The relative fluorescence was monitored at 460 nM with excitation at 355 nm for 30 min at 9-s interval. Replicate measurements (n = 6) were averaged and compared to factor Xa activity in the absence of apixaban. Fluorescence data in the absence of apixaban was fit to a linear equation with slope = 1.56 ± 0.01 units/s and y-intercept = 11870 ± 10 units. Fluorescence data for the recovery of factor Xa activity from the pre-formed factor Xa:apixaban complex was the fit to Equation 4 with kobs = 0.0113 ± 0.0014 s−1, offset or initial relative fluorescence = 10690 ± 18, vss = 1.64 ± 0.01 units/s, and vi fixed at 0 units/s. Under these conditions the vss was 105% of the uninhibited buffer control rate and kobs ∼ off-rate constant.
Since apixaban is a mixed-type inhibitor versus prothrombin with slightly different affinities for prothrombinase (E) or the prothrombinase:prothrombin complex (ES), we wanted to determine whether the onset of inhibition differed in either case, therefore, quenched-flow experiments were conducted at two levels of prothrombin. Quenched-flow time courses of apixaban inhibition of FXa in the prothrombinase complex in the presence of sub-saturating prothrombin (97 nM) are depicted in , and at saturating prothrombin (970 nM) in .
Figure 5. Quenched-flow time courses for inhibition of prothrombinase by apixaban at sub-saturating prothrombin. Quenched-flow time courses for the activation of sub-saturating prothrombin (97 nM; 0.64 × Km) to α-thrombin were obtained with final concentrations of factor Xa (2.5 nM), factor Va (14 nM) and phospholipid vesicles (26 μM) in HEPES buffer at 37°C in the presence of 0 (open circle, top curve, solid line), 10 nM (open square), 20 nM (open triangle), 40 nM (open inverted triangle), and 80 nM (◊) apixaban as described in the ‘Materials and Methods’. The data were fitted to the Equation 4 and the fitted lines in the figure had the following parameters at 0 nM (solid line): slope of 0.43 nM/s, intercept = −0.031 nM; at 10 nM (dashed line): offset = −0.066 nM, vi = 0.46 nM/s, vf = 0.0022 nM/s, kobs = 0.21 s−1; at 20 nM (dotted line): offset = 0.00024 nM, vi = 0.42 nM/s, vf = 0.0041 nM/s, kobs = 0.28 s−1; at 40 nM (dashed and dotted line): offset = −0.046 nM, vi = 0.39 nM/s, vf = 0.0034 nM/s, kobs = 0.43 s−1; at 80 nM (bottom curve, small dotted line): offset = −0.031 nM, vi = 0.27 nM/s, vf = 0.0027 nM/s, kobs = 0.71 s−1. Time courses for and calculations of the observed rate constants for the formation of total thrombin and meizothrombin were also determined as described in the ‘Materials and Methods’ (data not shown).
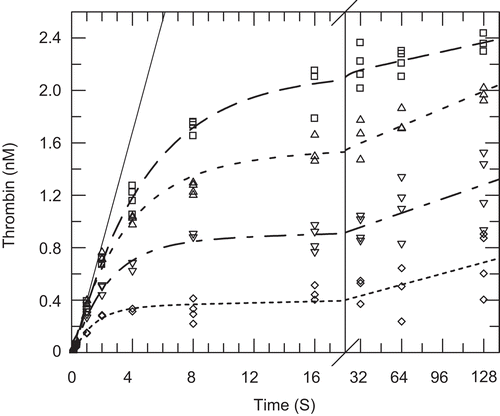
Figure 6. Quenched-flow time courses for inhibition of prothrombinase by apixaban at saturating prothrombin. (A) Quenched-flow time courses for the activation of saturating prothrombin (970 nM; 6.4 × Km) to α-thrombin were obtained with final concentrations of factor Xa (2.5 nM), factor Va (14 nM) and phospholipid vesicles (26 μM) in HEPES buffer at 37°C in the presence of 0 (open triangle), 10 nM (open square), 20 nM (open triangle), 40 nM (open inverse triangle), and 80 nM (◊) apixaban as described in the ‘Materials and Methods’. The data were fitted to Equation 4 and the fitted lines in the figure had the following parameters at 0 nM (solid line): slope of 1.32 nM/s, intercept = −0.093 nM; at 10 nM (dashed line): offset = −0.15 nM, vi = 1.23 nM/s, vf = 0.023 nM/s, kobs = 0.10 s−1; at 20 nM (dotted line): offset = −0.13 nM, vi = 1.31 nM/s, vf = 0.026 nM/s, kobs = 0.15 s−1; at 40 nM (dashed and dotted line): offset = −0.082 nM, vi = 1.03 nM/s, vf = 0.016 nM/s, kobs = 0.20 s−1; at 80 nM (bottom curve, small dotted line): offset = −0.21 nM, vi = 1.08 nM/s, vf = 0.012 nM/s, kobs = 0.35 s−1. Time courses for and calculations of the observed rate constants for the formation of total thrombin and meizothrombin were also determined as described in the ‘Materials and Methods’. (B) Quenched-flow time courses for the activation of saturating prothrombin (970 nM; 6.4 × Km) to meizothrombin were obtained with final concentrations of factor Xa (2.5 nM), factor Va (14 nM) and phospholipid vesicles (26 μM) in HEPES buffer at 37°C in the presence of 0 (open circle), 10 nM (open square), 20 nM (open triangle), 40 nM (open inverse triangle), and 80 nM (◊) apixaban as described in the ‘Materials and Methods’. The data were fitted to the Equation 4 and the fitted lines in the figure had the following parameters at 0 nM (solid line): slope of 1.53 nM/s, intercept = 0.28 nM; at 10 nM (dashed line): offset = 0.49 nM, vi = 1.68 nM/s, vf = 0.01 nM/s, kobs = 0.22 s−1; at 20 nM (dotted line): offset = 0.44 nM, vi = 1.70 nM/s, vf = 0.0088 nM/s, kobs = 0.27 s−1; at 40 nM (dashed and dotted line): offset = 0.32 nM, vi = 1.53 nM/s, vf = 0.012 nM/s, kobs = 0.35 s−1; at 80 nM (bottom curve, small dotted line): offset = 0.28 nM, vi = 1.58 nM/s, vf = 0.0078 nM/s, kobs = 0.59 s−1.
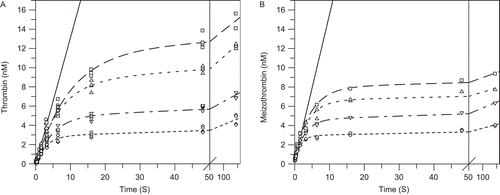
Two proteolytic cleavages are required to form α-thrombin from prothrombinCitation26. Because the hydrolysis at R320 to form meizothrombin is kinetically preferred over initial cleavage at R271 to form prethrombin-2, and meizothrombin has similar catalytic properties to α-thrombin with peptide substrates, but different properties towards macromolecular substrates, we wanted to demonstrate that the onset of inhibition was similar regardless of the product formed. As described in the Methods, the meizothrombin component can be separated from the total thrombin and the kobs values for the net α-thrombin and meizothrombin formed at both sub-saturating and saturating prothrombin were plotted versus apixaban. All four of these graphs were clearly linear and the on rate constants could be calculated from the slopes after linear regression analysis and extrapolation to zero prothrombin for the sub-saturating case and to infinite prothrombin in the saturating case as described in the Methods. The linear fits of the data shown in from 10 nM to 80 nM apixaban gave a slope of 0.0071 ± 0.000065 nM−1 s-1, y-intercept = 0.14 ± 0.003 s−1 and correlation coefficient of 0.9999 for α-thrombin and from 20 nM to 80 nM apixaban gave a slope of 0.0072 ± 0.0004 nM−1 s-1, y-intercept = 0.33 ± 0.021 s−1 and correlation coefficient of 0.998 for meizothrombin. Linear fits of the data shown in from 10 nM to 80 nM apixaban gave a slope of 0.0034 ± 0.00012 nM−1 s−1, y-intercept = 0.072 ± 0.0054 s−1 and correlation coefficient of 0.999 for α-thrombin and gave a slope of 0.0052 ± 0.00031 nM−1 s-1, y-intercept = 0.16 ± 0.014 s−1 and correlation coefficient of 0.996 for meizothrombin. The rate constants derived from the slopes and intercepts as described in the ‘Materials and Methods’ are summarized in .
Table 2. Microscopic rate constants for apixaban inhibition of prothrombinase.
PT assays
Experiments were performed with three PT reagents with graphical results for one reagent, thromboplastin C-Plus depicted in . Similar results were found for the other PT reagents tested. Apixaban produced a concentration dependent increase in the PT (). Linear regression of PT versus apixaban concentration at each prothrombin concentration showed the apixaban plasma concentration required to produce a two-fold increase in the clotting time decreased slightly from 3.7 μM at normal prothrombin concentration to 3.2 μM at 5% of normal prothrombin concentration. The reciprocal clotting time (s−1) versus prothrombin concentration (substrate) expressed as a percent of normal plasma prothrombin concentration was best fit to the equation for mixed-type inhibition with prothrombin Km = 2.98 ± 0.16% normal, KiE = 2.52 ± 0.24 μM, and KiES = 3.48 ± 0.10 μM (). Dixon plot analysis of clotting time versus apixaban concentration using Equation 8 yielded nearly identical kinetic parameters with prothrombin Km = 2.98 ± 0.15%, KiE = 2.49 ± 0.22 μM, and KiES = 3.52 ± 0.10 μM (). The similarity of the concentration required to produce a two-fold increase in PT from the linear analysis (3.7 μM) and the KiES value from the Dixon plot analysis (3.5 μM) supports the validity of the kinetic analytical method. Likewise analysis of data from the two other PT reagents yielded comparable results with prothrombin Km = 6.15 ± 0.23%, KiE = 5.39 ± 0.74 μM, and KiES = 8.25 ± 0.44 μM for Innovin and prothrombin Km = 4.84 ± 0.36%, KiE = 0.84 ± 0.12 μM, and KiES = 3.50 ± 0.29 μM for Neoplastin CI Plus.
Figure 7. Prothrombin time (PT) assays. Plasma prothrombin concentrations were varied by combining pooled normal plasma and prothrombin immunodepleted plasma. Apixaban was spiked into plasma beginning with a 10 mM dimethyl sulfoxide stock solution followed by serial dilutions with plasma. Symbols represent the average of duplicate clotting time determinations. Panels A and B are two representations of the PT data collected at each prothrombin and apixaban concentration pair. The results shown are from thromboplastin C-plus PT reagent (Dade-Behring). (A) Plasma prothrombin concentration as a percent of normal plasma are 100 (open circle), 50 (closed circle), 20 (open square), 10 (closed square), 5 (open triangle). Dashed lines represent linear fit to the PT versus apixaban concentration at individual prothrombin concentrations. Solid lines are drawn according to the mixed-type inhibition parameters calculated using Equation 8 as described in ‘Methods’ with apparent Vmax = 0.086 ± 0.001 s−1, Km = 2.98 ± 0.15% of normal prothrombin, apparent apixaban KiE = 2.49 ± 0.22 μM, and apparent KiES = 3.52 ± 0.10 μM. (B) Apixaban concentrations (μM) are 0 (open circle), 0.156 (closed circle), 0.313 (open square), 0.625 (closed square), 1.25 (open triangle), 2.5 (closed triangle), 5 (open inverse triangle), 10 (closed inverse triangle). Solid lines are drawn according to the mixed-type inhibition parameters calculated using Equation 2C as described in ‘Methods’ with prothrombin apparent Km = 2.98 ± 0.16% of normal prothrombin, apparent apixaban KiE = 2.52 ± 0.24 μM, and apparent KiES = 3.48 ± 0.10 μM.
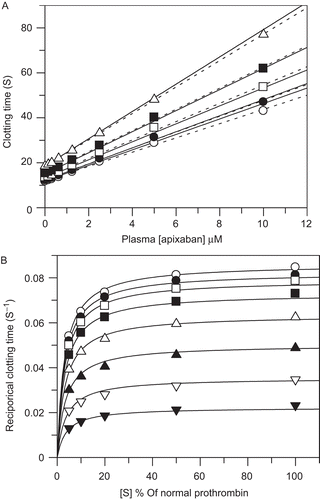
Plasma purified prothrombin was added in varying amounts to prothrombin depleted plasma and PT was determined with thromboplastin C-Plus only. Nearly identical kinetic parameters were derived as compared to the experiments conducted by mixing with normal plasma with prothrombin Km = 44 ± 1.6 nM or approximately 3% of normal, KiE = 2.57 ± 0.30 μM, and KiES = 3.40 ± 0.10 μM.
Discussion
Prothrombinase, a macromolecular complex of FXa and FVa formed on phospholipid surfaces in the presence of calcium, catalyzes the activation of prothrombin with 300 000-fold higher efficiency than FXa aloneCitation11,Citation12. The increase in catalytic efficiency is achieved through an increase in local prothrombin concentration at the membrane surface and increased substrate (prothrombin) recognition due to macromolecular structural changes induced by FVa and phospholipidsCitation27. Antithrombin-dependent, indirect FXa inhibitors exhibit markedly reduced ability to block prothrombin activation relative to their ability to block free FXaCitation28. Apixaban binds in the active-site of factor Xa (see reference 3 for crystal structure) and inhibits free FXa (Ki 0.25 nM at 37°C) and the prothrombinase complex (Ki 0.72 nM at 37°C) competitively versus a peptide substrate with similar affinity indicating that structural changes within the active site are either minor or have little consequence relative to apixaban binding. As reported for other direct, active-site FXa inhibitorsCitation17,Citation28, apixaban inhibits prothrombinase by a mixed-type mechanism versus prothrombin with similar affinity for prothrombinase (Ki 0.62 nM at 37°C) and the prothrombinase:prothrombin enzyme:substrate complex (Ki 1.7 nM at 37°C). The advantage of this inhibitory mechanism is that apixaban can bind to prothrombinase with high affinity and not be displaced by the high local prothrombin concentrations achieved at the membrane surface. Similarly, prothrombin retains affinity for apixaban-bound prothrombinase due to protein-protein interactions at regions distant from the active site (exosites)17. Prothrombin activation, however, is precluded by apixaban occupancy of the enzyme active site. Theoretically, a pure competitive inhibitor of prothrombinase activation of prothrombin would need to overcome the higher local prothrombin concentrations achieved at the membrane surface by a factor of {1 + [(prothrombin)/Km]}, where Km is the Michaelis-Menten constant for prothrombin binding to prothrombinase. Using a mean plasma concentration of 1.4 μM prothrombinCitation23 and a Km of 0.15 μM, a pure competitive inhibitor would require an inhibitor concentration of ∼10-times Ki [1 +(1.4/0.15)] to achieve 50% inhibition of prothrombinase, whereas a pure non-competitive inhibitor would require an inhibitor concentration equal to Ki to achieve 50% inhibition of prothrombinase.
Apixaban Ki’s obtained in both phosphate and HEPES buffers in the absence of macromolecular substrate and cofactor show increases of ∼3-fold at the higher temperature between 25 and 37°C. Under these conditions the increase in Ki with temperature was due almost entirely to slower dissociation of the FXa:apixaban complex at 25°C. At equilibrium the binding of apixaban to FXa in the absence of phospholipids, calcium, and FVa is driven entirely by enthalpy. However, in HEPES buffer containing phospholipids, calcium and FVa, the Ki increases less than 2-fold between 25 and 37°C, and in the presence of prothrombin the temperature shift in either Ki or KiES is negligible suggesting either much smaller enthalpic contributions to binding or non-linear van’t Hoff plots (heat capacity changes) in the presence of macromolecules. Thus, the difference in Ki values between 25 and 37°C is largely mitigated by the presence of FVa and prothrombin. Despite these subtle differences in temperature effects due to the presence of various components, apixaban is clearly a potent inhibitor of FXa with Ki values ranging from 75 to 720 pM under a variety of conditions.
While the equilibrium dissociation constant (Ki) is a determinant of inhibitor pharmacodynamic effects the microscopic rate constants underlying the equilibrium may also be important determinantsCitation29. Coagulation is a dynamic process with multiple reactions leading to thrombin production and ultimately fibrin formation. The PT measurement concludes with clotting in normal plasma occurring within 20 s of reaction initiation. Since no FXa is present in plasma prior to the initiation of coagulation all the FXa required for prothrombin activation forms within this period. To demonstrate efficacy in prolonging the prothrombin clotting time a direct FXa inhibitor must bind to and inhibit FXa before sufficient thrombin is generated. A relatively low concentration of thrombin (<10 nM) is required to cleave fibrinogen to fibrin and cause plasma to clotCitation30. Clotting is detected by optical or mechanical methods and is defined by exceeding a predefined threshold, e.g. turbidity for optical methods. In a one-stage clotting assay, such as PT, the plasma clotting time is inversely proportional to the velocity of the clotting reactionCitation24. On this basis PT experiments were analyzed using standard enzyme kinetic equations. These experiments showed the apparent Km for prothrombin is approximately 3% of the normal prothrombin concentration and that apixaban retains near equal affinity for factor Xa and prothrombin bound prothrombinase in a plasma-based system. The Ki values obtained from the PT experiments differ by 1000-fold as compared to the Ki values obtained from steady state reactions in purified enzyme systems. This difference may be attributed to effects of plasma protein binding of apixaban, non-specific binding of apixaban to PT reagent components, and the rapid reaction kinetics which lead to in vitro coagulation.
Physiologically, the rate of factor X activation is determined by the concentrations of extrinsic Xase (tissue factor: factor VIIa complex) and intrinsic Xase (factor VIIIa: factor IXa complex) produced at the site of vascular injury. Newly formed FXa rapidly associates with FVa on a negatively charged phospholipid membrane surface to form prothrombinase followed by rapid association with prothrombinCitation31. Since the prothrombinase:prothrombin complex forms rapidly, it is reasonable to conclude that a prothrombinase inhibitor must bind rapidly to inhibit prothrombin activation before a critical amount of thrombin forms.
Apixaban exhibits rapid association with free FXa (13–20 μM−1 s−1) and prothrombinase (4–12 μM−1 s−1) even in the presence of saturating prothrombin. These association rate constants are comparable to the association rate constants published for the physiological substrates of FXa and prothrombinaseCitation32,Citation33. This indicates that apixaban is expected to rapidly neutralize free FXa and prothrombinase in a physiological setting.
Conclusions
These experiments characterize apixaban binding to free FXa and the prothrombinase complex with and without prothrombin bound. Apixaban associates rapidly and exhibits high affinity binding to all forms of FXa independent of the presence of prothrombin. Apixaban exhibits mixed-type inhibition of prothrombin activation with approximately equal affinity for prothrombinase alone and prothrombinase in complex with prothrombin. PT experiments with normal and prothrombin deficient plasma demonstrate pharmacodynamic effects consistent with both the mechanism of inhibition and the microscopic rate constants derived from experiments with purified proteins. These properties support the investigation of apixaban as an antithrombotic agent with a predictable pharmacologic mechanism of action.
Acknowledgements
Charles Kettner, Ross Stein, and Lawrence Mersinger conducted initial enzyme kinetic studies with apixaban, phospholipid membrane preparations, and interpretation of the mechanistic observations. Mark Hixon provided a detailed derivation of Equation 1 validating its application to the prothrombin activation experiments. Jeff Bozarth provided technical assistance.
Declaration of interest
The authors are current or former employees of Bristol-Myers Squibb. This study was sponsored by Bristol-Myers Squibb and Pfizer Inc.
Related Research Data
References
- Colman RW, Clowes AW, Goldhaber SZ, Marder VJ, George JN. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. 5th edn. Philadelphia, PA: Lippincott Williams & Wilkins, 2006:1–16.
- Lloyd-Jones D, Adams R, Carnethon M, Simone GD, Ferguson TB, Flegal K et al. Heart Disease and Stroke Statistics – 2009 Update. A Report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2009;119:e21–e181.
- Pinto DJ, Orwat MJ, Koch S, Rossi KA, Alexander RS, Smallwood A et al. Discovery of 1-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine-3-carboxamide (apixaban, BMS-562247), a highly potent, selective, efficacious, and orally bioavailable inhibitor of blood coagulation factor Xa. J Med Chem 2007;50:5339–5356.
- Wong PC, Crain EJ, Xin B, Wexler RR, Lam PY, Pinto DJ et al. Apixaban, an oral, direct and highly selective factor Xa inhibitor: in vitro, antithrombotic and antihemostatic studies. J Thromb Haemost 2008;6:820–829.
- Wong PC, Watson CA, Crain EJ. Arterial antithrombotic and bleeding time effects of apixaban, a direct factor Xa inhibitor, in combination with antiplatelet therapy in rabbits. J Thromb Haemost 2008;6:1736–1741.
- Wong PC, Crain EJ, Watson CA, Xin B. Favorable therapeutic index of the direct factor Xa inhibitors, apixaban and rivaroxaban, compared with the thrombin inhibitor dabigatran in rabbits. J Thromb Haemost 2009;7:1313–1320.
- Lassen MR, Raskob GE, Gallus A, Pineo G, Chen D, Hornick P; ADVANCE-2 investigators. Apixaban versus enoxaparin for thromboprophylaxis after knee replacement (ADVANCE-2): a randomised double-blind trial. Lancet 2010;375:807–815.
- Lassen MR, Raskob GE, Gallus A, Pineo G, Chen D, Portman RJ. Apixaban or enoxaparin for thromboprophylaxis after knee replacement. N Engl J Med 2009;361:594–604.
- Alexander JH, Becker RC, Bhatt DL, Cools F, Crea F, Dellborg M, et al; APPRAISE Steering Committee and Investigators. Apixaban, an oral, direct, selective factor Xa inhibitor, in combination with antiplatelet therapy after acute coronary syndrome: results of the Apixaban for Prevention of Acute Ischemic and Safety Events (APPRAISE) trial. Circulation 2009;119:2877–2885.
- Buller H, Deitchman D, Prins M, Segers A; Botticelli Investigators, Writing Committe. Efficacy and safety of the oral direct factor Xa inhibitor apixaban for symptomatic deep vein thrombosis. The Botticelli DVT dose-ranging study. J Thromb Haemost 2008;6:1313–1318.
- Rosing J, Tans G, Govers-Riemslag JW, Zwaal RF, Hemker HC. The role of phospholipids and factor Va in the prothrombinase complex. J Biol Chem 1980;255:274–283.
- Nesheim ME, Kettner C, Shaw E, Mann KG. Cofactor dependence of factor Xa incorporation into the prothrombinase complex. J Biol Chem 1981;256:6537–6540.
- Harenberg J, Wehling M. Current and future prospects for anticoagulant therapy: inhibitors of factor Xa and factor IIa. Semin Thromb Hemost 2008;34:39–57.
- Zikria JC, Ansell J. Oral anticoagulation with factor Xa and thrombin inhibitors: on the threshold of change. Curr Opin Hematol 2009;16:347–356.
- Nilsson T, Sjöling-Ericksson A, Deinum J. The mechanism of binding of low-molecular weight active site inhibitors to α-thrombin. J Enzyme Inhibition 1988;13:11–29.
- Mann KG, Brummel K, Butenas S. What is all that thrombin for? J Thromb Haemost 2003;1:1504–1514.
- Krishnaswamy S, Betz A. Exosites determine macromolecular substrate recognition by prothrombinase. Biochemistry 1997;36:12080–12086.
- Christensen U, Müllertz S. Kinetic studies of the urokinase catalysed conversion of NH2-terminal lysine plasminogen to plasmin. Biochim Biophys Acta 1977;480:275–281.
- Segel I. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. New York: John Wiley & Sons, Inc. 1993:100–178 and 931–940.
- Schoen P, Lindhout T. The in situ inhibition of prothrombinase-formed human alpha-thrombin and meizothrombin(des F1) by antithrombin III and heparin. J Biol Chem 1987;262:11268–11274.
- Cha S, Agarwal RP, Parks RE Jr. Tight-binding inhibitors-II. Non-steady state nature of inhibition of milk xanthine oxidase by allopurinol and alloxanthine and of human erythrocytic adenosine deaminase by coformycin. Biochem Pharmacol 1975;24:2187–2197.
- Morrison JF, Stone SR. Approaches to the study and analysis of the inhibition of enzymes by slow and tight-binding inhibitors. Comments Mol Cell Biophys 1985;2:347–368.
- Mann KG. Prothrombin. Methods Enzymol 1976;45:123–156.
- Hemker HC, Vermeer C, Govers-Riemslag J. Kinetic aspects of the interaction of blood clotting enzymes. VII. The relation between clotting time and prothrombin concentration. Thromb Haemost 1977;37:81–85.
- Weber G. Van’t Hoff revisited: enthalpy of association of protein subunits. J Phys Chem 1995;99:1052–1059.
- Rosing J, Zwaal RF, Tans G. Formation of meizothrombin as intermediate in factor Xa-catalyzed prothrombin activation. J Biol Chem 1986;261:4224–4228.
- Qureshi SH, Yang L, Manithody C, Rezaie AR. Membrane-dependent interaction of factor Xa and prothrombin with factor Va in the prothrombinase complex. Biochemistry 2009;48:5034–5041.
- Rezaie AR. Prothrombin protects factor Xa in the prothrombinase complex from inhibition by the heparin-antithrombin complex. Blood 2001;97:2308–2313.
- Tummino PJ, Copeland RA. Residence time of receptor-ligand complexes and its effect on biological function. Biochemistry 2008;47:5481–5492.
- Brummel KE, Paradis SG, Butenas S, Mann KG. Thrombin functions during tissue factor-induced blood coagulation. Blood 2002;100:148–152.
- Krishnaswamy S, Jones KC, Mann KG. Prothrombinase complex assembly. Kinetic mechanism of enzyme assembly on phospholipid vesicles. J Biol Chem 1988;263:3823–3834.
- Krishnaswamy S, Mann KG, Nesheim ME. The prothrombinase-catalyzed activation of prothrombin proceeds through the intermediate meizothrombin in an ordered, sequential reaction. J Biol Chem 1986;261:8977–8984.
- Giesen PL, Willems GM, Hermens WT. Production of thrombin by the prothrombinase complex is regulated by membrane-mediated transport of prothrombin. J Biol Chem 1991;266:1379–1382.