Abstract
A novel series of 6-(2-chloroquinolin-3-yl)-4-substituted-phenyl-6H-1,3-oxazin-2-amines were synthesized and evaluated for in vitro antimalarial efficacy against chloroquine sensitive (MRC-02) as well as chloroquine resistant (RKL9) strains of Plasmodium falciparum. The activity tested was at nanomolar concentration. β-Hematin formation inhibition activity (BHIA50) of oxazines were determined and correlated with antimalarial activity. A reasonably good correlation (r = 0.49 and 0.51, respectively) was observed between antimalarial activity (IC50) and BHIA50. This suggests that antimalarial mode of action of these compounds seems to be similar to that of chloroquine and involves the inhibition of hemozoin formation. Some of the compounds were showing better antimalarial activity than chloroquine against resistant strain of P. falciparum and were also found to be active in the in vivo experiment.
Introduction
Frequency of malaria and other diseases is progressively increasing worldwide. New emerging strains of bacterium and resistance to currently available drugs make this field more conscientious and alarming. Malaria is the most common parasitic disease in tropical and subtropical regions and worldwide. Around 40% of the population is living in the malarial endemic area. According to the survey done by World Health Organization, it was found that a quantum of people living in these areas is at the risk of malarial transmission, which is literally alarming. Approximately 1.0–3.0 millions of population died due to the non-availability of proper chemotherapy.Citation1,Citation2 The situation regarding the control and treatment of malaria has progressively worsened with the spread of insecticide-resistant mosquito vectors and drug-resistant malarial parasites.Citation3 Therefore, the need for new agents that are active against multi-resistant Plasmodium strains, through the identification of new targets is very urgent.Citation4,Citation5 These points are critical for the disease process or essential for the survival of the parasite.
Plasmodium falciparum and Plasmodium vivax are the two major human malarial parasites. P. falciparum is responsible for the most deaths and it has developed resistance against almost all the available drugsCitation6 so far in the market. The search for novel antimalarial drugs against specific parasitic targets is thus an immense priorityCitation7,Citation8 for the medicinal chemists all over the world for the sake of humanity. The potential antimalarial activity of chalcones has generated a great interest.Citation9–12 Their antimalarial activity was first reported when licochalcone A, a natural product isolated from Chinese liquorice root, exhibited potent antimalarial activity.Citation9 Subsequently, a synthetic analogue 2,4-dimethoxy-4′-butoxy-chalcone was reported to have antimalarial activity.Citation10 Some chalcones inhibit falcipain cysteine proteases, but it is unclear whether the antimalarial activity of this class is primarily due to protease inhibitionCitation11 or others. Several oxygenated chalcones and bis-chalcones were reported to have antimalarial activity.Citation12 Chalcones have been used as intermediate for the preparations of compounds having multi-therapeutic value. The literature review reveals that chalcone derivatives exhibit diverse pharmacological activities, such as potential cytotoxic agents, antimalarial agents, antimicrobial agents, antiviral, anti-inflammatory, anesthetics, and mydriatics.Citation13–22
In this study, we have synthesized and characterized a series of novel quinoline-based chalcones and their oxazine derivatives. In this study, we have also reported the antimalarial activity of substituted oxazines against chloroquine (CQ) sensitive (MRC-02) as well as CQ resistant (RKL9) strains of P. falciparum. In this study, only the substitutions on “B” ring of the 6-(2-chloroquinolin-3-yl)-4 substituted phenyl-6H-1,3-oxazin-2-amine moiety was changed by keeping the basic skeleton intact. The compounds were also evaluated for β-hematin formation inhibition activity and correlated with their antiplasmodial activity against CQ sensitive as well as resistant strains of malaria parasite. From the correlations data, the mode of action of this class of compounds was proposed to follow haem detoxification inhibition pathway. Three compounds (4b, 4g, and 4i) were found most active in vitro against CQ-resistant malaria parasite were also tested in vivo against CQ-resistant N-67 strain of Plasmodium yoelli.
Chemistry
6-(2-Chloroquinolin-3-yl)-4 substituted phenyl-6-H-1,3-oxazin-2-amines 4(a–j) were synthesized according to the method as shown in . In the first step, synthesis of chalcones was carried out by the well-known Claisen–Schmidt reaction, and products were isolated in 60–70% yield after recrystallization. In the second step, chalcone and urea with catalytic amount of NaOH were refluxed in ethanol to obtain the desired products 4(a–j). Factors such as the structure and position of the substituents have profoundly influenced the rate of reaction. Halogenated chalcones viz. 3b, 3g, and 3f took 10 h for complete conversion into final oxazines, whereas unsubstituted 3a, methyl substituted (3e and 3d) and alkoxy substituted 3i chalcones took 12, 14, and 14.5 h, respectively. The general accepted interpretation regarding chemistry of this reaction involves the nucleophilic attack of oxygen on the carbon–carbon double bond at β position. Hence, the electropositive nature of β-carbon may control the overall rate of the reaction. The electropositive nature of β-carbon is controlled by the aromatic ring directly connected to it (ring C as shown in ). Halogens being electron withdrawing in nature significantly increase the positive character of β-carbon leading to faster reaction while electron donating alkyl and alkoxy groups contributed to execute slower reaction. However, this phenomenon was not observed in the case of nitro group substituents (compound 4h). The possible explanation might be the meta-directing nature of nitro group. In compound 4h, nitro group is placed at meta position of the phenyl ring and by virtue of its meta-directing nature, electron density at β-carbon increases and its electro-positivity decreases leading to longer time for the completion of reaction.
Scheme 1. Synthesis of substituted oxazines 4(a–j).
Figure 1. Structure of 6-(2-chloroquinolin-3-yl)-4 substituted phenyl-6H-1,3-oxazin-2-amine.
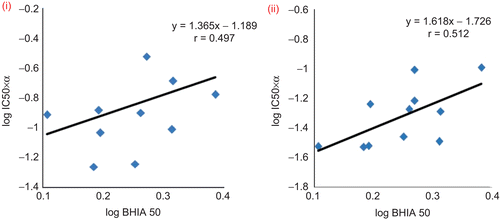
Figure 2. A Plot of the normalized antiplasmodial IC50 (IC50 X α) versus log of the activity against the inhibition of β-hematin formation. (i) Study involving chloroquine resistant (RKL9) strain with CQ diphosphate. (ii) Study involving chloroquine sensitive (MRC-02) strain with CQ diphosphate.
Structures of compounds 4(a–j) were confirmed by elemental analysis, IR, MS, and NMR techniques. All the substituted oxazine possesses similar basic skeletal structure. Proton NMR signals were assigned by comparing the spectra of the products 4(a–j) with their corresponding chalcones. The 1H-NMR spectrum of oxazine derivatives were recorded on Bruker Avance II 400 (400 MHz) NMR spectrometer in dimethyl sulphoxide (DMSO)-d6 using TMS as an internal standard: Peak at 2.31 ppm (singlet) was attributed to (2H) of –NH2. Doublets were observed at 5.21 and 6.31 ppm for 1H of (6-H) and (5-H), respectively, of oxazine ring in the spectrum. The NMR spectrum also showed multiplets around 7.13–8.30 ppm for aromatic protons. Mass spectral data provided useful information regarding the structural analysis. The molecular ion peak of 4a was found at 335 Da. There were some guide peaks in the IR spectrum of 4a, which were helping in the structural analysis regarding the different functionalities present in the moieties. Some of the guides peaks were: 3412 (–NH2), 1394 (Het–C–O), 1570 (C=C, ring skeleton Ar moiety), 1415 (C=C, ring skeleton oxazine moiety), 1246 (C–N, str for oxazine ring), and 853 (C–Cl, for quinoline skeleton).The other signals and peaks of 1H-NMR and IR are in complete agreement with the assigned structures.
Materials and methods
In vitro antimalarial assay
P. falciparum strains MRC-02 (CQ sensitive) and RKL9 (CQ resistant) were obtained from National Institute of Malaria Research, New Delhi, India, and were maintained in a continuous culture using the standard method described by Trager and Jensen.Citation23 Parasites were cultured in human B (+) erythrocytes in RPMI-1640 media supplemented with 25 mM HEPES buffer, 10% human AB (+) serum, and 0.2% sodium bicarbonate (Sigma). It was maintained at 5% CO2. Cultures containing predominantly early ring stages were synchronized by addition of 5% d-sorbitol (Sigma) lysis,Citation24 used for testing. Initial culture was maintained in small vials with 10% haematocrite (i.e. 10 μl erythrocytes containing 1.0% ring stage parasite in 100 μl complete media). The culture volume per vial for the assay was kept at 100 μl. Number of parasites for the assay was adjusted at 1–1.5% by diluting with fresh human B (+) RBC. Assay was done in 96-well microlitre flat-bottomed tissue culture plates, which were incubated at 37°C for 24 h in the presence of twofold serial dilutions of compounds and CQ diphosphate for their effect on schizont maturation. Compounds were dissolved in ethanol and further diluted with RPMI-1640 medium (the final ethanol concentration did not exceed 0.5%, which did not affect parasite growth). CQ diphosphate was dissolved in aqueous medium. Test was done in duplicate vials for each dose of the drugs. Solvent control culture containing the same concentrations of the solvent as present in the test vials was done with RPMI-1640 containing 10% AB (+) serum. Parasite growth was found unaffected at the solvent concentrations. Growth of the parasites from duplicate vials of each concentration was monitored in Giemsa-stained blood smears by counting number of schizont per 100 asexual parasites. Percent schizont maturation inhibition was calculated by the formula: (1 − Nt/Nc) × 100, where Nt and Nc represent the number of schizont in the test and control vial, respectively.
In vivo antimalarial assay
The in vivo efficacy was determined by Peter’s 4-day suppressive testCitation25 by using P. yoelli (N-67 strain) and male white Swiss albino mice (18–20 g of body weight). In brief, groups of five mice each were inoculated intraperitoneally with ∼30% of P. yoelli-infected erythrocytes from a donor mouse. Four hours later, selected test compounds were administered at a dose of 50 and 25 mg/kg/day intraperitoneally. CQ (8 mg/kg/day) was taken as standard for comparison. A total of four doses were given on days 0, 1, 2, and 3. Control groups received the same amount of solvent used to suspend the compounds. The tail blood smears were made on day 4 and day 7 stained with Giemsa and were examined microscopically. The percent parasitaemia and average percent suppression of parasitaemia in comparison with control groups were determined. Mean survival time (MST) was calculated for dead mice during the 28-day observation period.
In vitro cytotoxicity assay
Human cervical epithelial cells were routinely maintained at 37°C in Eagles Minimum Essential Medium (EMEM; Sigma) supplemented with foetal bovine serum (10%), l-glutamine (2 mM), and gentamycine (80 mg/l). Cytotoxicity was assessed using the neutral red (NR) dye uptake assayCitation26 using 96-well micro titer tissue (MT) culture plates (Greiner, Germany). Briefly, 2 × 103 cells were seeded into each well of a 96-well MT plate and incubated overnight at 37°C in a humidified atmosphere containing 5% CO2. All the compounds were dissolved in DMSO and subsequently serial twofold dilutions were prepared in EMEM to give a broad range of concentration. Before assessment of cytotoxicity, the preformed cell monolayer was washed with EMEM and replaced with drug solutions in duplicate keeping cell control including corresponding dilution of DMSO. The plates were incubated at 37°C and observed microscopically on daily routine basis for the appearance of any morphological changes. After 72-h incubation, the cells were fixed with 5% glutaraldehyde solution prepared in phosphate-buffer saline (PBS) kept at room temperature for 45 min. The fixed cells were washed with PBS, stained with 0.2% NR dye aqueous solution and incubated for 1 h at room temperature. After washing, the stained cells were subjected to acid alcohol lysis by adding 100 μl of 0.5% acetic acid (V/V) in 50% ethanol to each well followed by 10-min incubation. Absorbances of the resulting solutions were read at 550 nm and cell survival was calculated as the absorbance of the treated cells divided by the control (EMEM plus DMSO). Results were expressed in terms of the maximum non toxic dose values, that is, the maximum concentration of drug at which 100% cell survival was observed.
Determination of pKa values
Acid dissociation constants (pKa) were determined spectrophotometrically as previously describedCitation27 by using a Unicam UV-300 spectrometer of Thermo Spectronic. Absorbances of each compound (0.025 mg/ml) dissolved in buffers of different pH values ranging from 5.8 to 11.2 as well as in 0.1 M NaOH and 0.1 M HCl were determined. Buffers from pH 5.8 to 8.0 were prepared from 0.1 M solutions of monobasic potassium phosphate (KH2PO4) and dibasic potassium phosphate (K2HPO4) and from pH 8.4 to 11.2 were prepared from 0.1 M glycine and 0.1 M NaOH. Analytical wavelengths (223, 250, 260, and 367 nm) were chosen at which significant difference between the absorbance of molecules in 0.1 M HCl and 0.1 M NaOH was observed. To reduce error, pKa was determined in duplicate for each compound.
Inhibition of β-hematin formation in vitro
The quantitative β-hematin inhibitory activity assay is based on the differential solubility of hematin and β-hematin in DMSO and NaOH solution, respectivelyCitation28,Citation29. The method determines a 50% inhibitory concentration for β-hematin formation inhibition (BHIA50) of the compound. In a micro-centrifuge tube, 100 μl of 6.4 mM solution of hematin freshly dissolved in 0.2 N NaOH solution, 50 μl of compound dissolved in ethanol (chloroquine diphosphate was dissolved in water), 200 μl of 3 M solution sodium acetate trihydrate (C2H3NaO2·3H2O), and 50 μl of glacial acetic acid were incubated at 37°C for 24 h. Concentration of compounds varied from 6 to 0.3 µM. Water and ethanol were taken as controls for water-soluble and ethanol-soluble compounds, respectively. After incubation, the tubes were centrifuged for 15 min. The supernatant was discarded and the pellet was reconstituted in DMSO and again centrifuged for 15 min at 3300g. The supernatant was discarded and β-hematin was obtained as a pellet. After centrifugation, to isolate DMSO insoluble β-hematin, the pellet was dissolved in 0.1 N NaOH and absorbance was recorded at 405 nM to calculate BHIA50 as previously described.Citation29
Experimental protocols
2-Chloroquinoline-3-carbaldehyde was synthesised starting from N-arylacetamides by Vilsmeier Haack cyclization.Citation30,Citation31 All the aromatic ketones were obtained from Sigma-Aldrich. Melting points were determined by open-tube capillaries method and are uncorrected. Mass spectra were recorded on a Micromass Q-To F high-resolution mass spectrometer equipped with electrospray ionization (ESI) on Masslynx 4.0 data acquisition system. ESI was used in positive ionization mode. Infrared spectra were recorded on a Schimadzu-IR Prestige 21 with a using (KBr) optics. 1H-NMR spectra were acquired on Bruker Avance II 400 (400 MHz) NMR spectrometer in DMSO-d6 as a solvent. The chemical shifts were expressed in the ppm (δ scale) downfield from TMS. Purity of chalcones and substituted oxazines was checked on thin layer chromatography (TLC; Merck Silica gel 60F254).
General method for synthesis of chalcones (3a–3j)
To the well-stirred solution of (8 mmol, 1.1 g) 2-chloroquinoline-3-carbaldehyde (1) in 5 ml of methanol in ice cold condition, freshly prepared 2 N methanolic NaOH solution (30 ml) was added and stirring was continued for further 10 min. To this, 8 mmol of appropriate ketones (2a–j) was added and the reaction mixture was stirred at room temperature for 24 h. The reaction mixture was cooled on an ice bath and neutralized with dilute hydrochloric acid. The precipitate thus formed was separated by filtration and washed three times with 50 ml distilled water to give the crude product. The product so obtained was recrystallized from methanol. The purity of the products was checked on TLC by using mixture of ethyl acetate and hexane as a mobile phase, which have shown single spot of the products in each case. The Rf values are given along with the spectral data below.
3-(2-Chloroquiolin-3-yl)-1-phenylprop-2-en-1-one (3a)
Prepared by the above method from 2 (8 mmol, 1.1 g) and acetophenone (8 mmol, 1.42 g); yield: 1.42 g, 61%; Rf = 0.44 in EtOAc/hexane, 3:7; yellow solid. mp:132–140°C; MS (M+), calcd.: 294.753, observed: 294.062; FTIR (cm−1):1732 (C=O), 1639 (CH=CH), 853 (C–Cl); 1H-NMR (400 MHz, DMSO-d6) δ/ppm 7.53 (1H, d, Hα), 8.52 (1H, d, Hβ), 7.40–8.31 (m, 10H, aromatic); Anal. Calcd: C18H12ClNO: C, 73.54; H, 4.17; N, 4.75. Found: C, 73.60; H, 4.24; N, 4.78.
3-(2-Chloroquinolin-3-yl)-1-(2,4-dichorophenyl)-)prop-2-en-1-one (3b)
Prepared by above method from 2 (8 mmol, 1.1 g) and 2,4-dichloroacetophenone (8 mmol, 0.89 g); yield: 1.21 g, 67%, Rf = 0.65 in EtOAc/hexane, 3:7. Pale yellow solid mp: 135–138°C; MS (M+):, calcd.: 360.682, observed: 360.981; FTIR (cm−1): 1732 (C=O), 1639 (CH=CH), 853 (C–Cl); 1H-NMR (400 MHz, DMSO-d6) δ/ppm: 7.65 (1H, d, Hα), 8.51 (1H, d, Hβ), 7.34–8.32 (m, 8H, aromatic).Anal. Calcd: C18H10Cl3NO: C, 59.60; H, 2.76; N, 3.81. Found: C, 59.65; H, 2.79; N, 3.86.
3-(2-Chloroquinolin-3-yl)-1-(3,4-dichlorophenyl)prop-2-en-1-one (3c)
Prepared by above method from 2 (8 mmol, 1.1 g) and 3,4-dichloroacetophenone (8 mmol, 1.12 g); yield: 1.20g, 63%, Rf = 0.61 in EtOAc/hexane, 3:7; Pale yellow solid. mp: 145–148°C; MS (M+): calcd.: 360.072, observed: 360.214; FTIR (cm−1): 1730 (C=O), 1643 (CH=CH), 853 (C–Cl); 1H-NMR (400 MHz, DMSO-d6) δ/ppm 7.70 (1H, d, Hα), 8.55 (1H, d, Hβ), 6.99–8.25 (m, 8H, aromatic)., Anal. Calcd: C18H10Cl3NO: C, 59.40; H, 2.76; N, 3.80. Found: C, 59.45; H, 2.78; N, 3.88.
3-(2-Chloroquinolin-3-yl)-1-p-tolyl prop-2-en-1-one (3d)
Prepared by above method from 2 (8 mmol, 1.1 g) and 4-methyl acetophenone (8 mmol, 1.15 g); yield: 1.14 g, 60%, Rf = 0.56 in EtOAc/hexane, 3:7; yellow crystalline solid. mp: 140–148°C; ESI-MS (M+): calcd.: 307.107, observed: 307.184; FTIR (cm−1): 1728 (C=O), 1643 (CH=CH), 853 (C–Cl); 1H-NMR (400 MHz, DMSO-d6) δ/ppm 2.38 (3H, s, CH3), 7.54 (1H, d, Hα), 8.59 (1H, d, Hβ), 7.29–8.33 (m, 9H, aromatic)., Anal. Calcd: C19H14ClNO: C, 74.12; H, 4.56; N, 4.49. Found: C, 74.15; H, 4.58; N, 4.18.
3-(2-Chloroqunolin-3-yl)-1-o-tolyl prop-2-en-1-one (3e)
Prepared by above method from 2 (8 mmol, 1.1 g) and 2-methyl acetophenone (8 mmol, 1.45 g); yield: 1.54 g, 65%, Rf = 0.51 in EtOAc /hexane, 3:7; yellow crystalline solid. mp: 132–140°C; ESI-MS (M+): calcd.: 307.128, observed: 307.161; FTIR (cm−1):1728 (C=O), 1643 (CH=CH), 853 (C–Cl); 1H-NMR (400 MHz, DMSO-d6) δ/ppm 2.38 (3H, s, CH3), 7.54 (1H, d, Hα), 8.51 (1H, d, Hβ), 7.29–8.27 (m, 9H, aromatic). Anal. Calcd: C19H14ClNO: C, 74.11; H, 4.56; N, 4.49. Found: C, 74.15; H, 4.58; N, 4.18.
3-(2-Chloroquinolin-3-yl)-1-(4-bromophenyl) prop-2-en-1-one (3f)
Prepared by above method from 2 (8 mmol, 1.13 g) and 4-bromoacetophenone (8 mmol, 1.1 g); yield: 1.55 g, 64%; Rf = 0.59 in EtOAc/hexane, 3:7; pale yellow crystalline solid. mp: 146–150°C; ESI-MS (M+Na): calcd.: 393.132, observed: 393.148; FTIR (cm−1): 1728 (C=O), 1643 (CH=CH), 853 (C–Cl), 588 (C–Br);1H-NMR (400 MHz, DMSO-d6) δ/ppm 7.54 (1H, d, Hα), 8.53 (1H, d, Hβ), 7.43–8.31 (m, 9H, aromatic).Anal. Calcd: C18H11ClBrNO: C, 58.01; H, 2.96; N, 3.74. Found: C, 58.04; H, 4.98; N, 3.78.
3-(2-Chloroquinolin-3-yl)-1-(2-bromophenyl) prop-2-en-1-one (3g)
Prepared by above method from 2 (8 mmol, 1 g) and 2-bromoacetophenone (8 mmol, 1.58 g); yield: 1.65 g, 66%, Rf = 0.58 in EtOAc/hexane, 3:7; yellow crystalline solid. mp: 166–170°C; ESI-MS (M+): calcd.: 370.123, observed: 370.190. FTIR (cm−1):1728 (C=O), 1643 (CH=CH), 853 (C–Cl), 588 (C–Br); 1H-NMR (400 MHz, DMSO-d6) δ/ppm 7.54 (1H, d, Hα), 7.90 (1H, d, Hβ), 7.43–8.32 (m, 9H, aromatic). Anal. Calcd: C18H11ClBrNO: C, 58.01; H, 2.96; N, 3.74. Found: C, 58.04; H, 4.98; N, 3.78.
3-(2-Chlroquinlin-3-yl)-1-(3-nitrophenyl) prop-2-en-1-one (3h)
Prepared by above method from 2 (8 mmol, 1.1 g) and 3-nitroacetophenone (8 mmol, 1.75 g); yield: 1.61 g, 60%, Rf = 0.54 in EtOAc/hexane, 4:6; pale yellow solid. mp: 145–150°C; ESI-MS (M+): calcd.: 338.012, observed:338.966; FTIR (cm−1): 1732 (C=O), 1645 (CH=CH), 853 (C–Cl), 1589(–NO2); 1H-NMR (400 MHz, DMSO-d6) δ/ppm 7.53 (1H, d, Hα), 8.51 (1H, d, Hβ), 7.23–8.10 (m, 9H, aromatic). Anal. Calcd: C18H11ClN2O3: C, 68.71; H, 3.26; N, 8.24. Found: C, 63.84; H, 3.28; N, 8.28.
3-(2-Chloroquinolin-3-yl)-1-(3,4,5-trimethoxyphenyl) prop-2-en-1-one (3i)
Prepared by above method from 2 (8 mmol, 1.1 g) and 3,4,5-trimethoxyacetophenone (8 mmol, 1.49 g); yield: 1.61 g, 67%, Rf = 0.48 in EtOAc/hexane, 3:7; yellow crystalline solid. mp: 174–175°C; ESI-MS (M+): calcd.: 383.132, observed: 383.401; FTIR (cm−1): 1732 (C=O), 1645 (CH=CH), 853 (C–Cl); 1H-NMR (400 MHz, DMSO-d6) δ/ppm 7.53 (1H, d, Hα), 8.57 (1H, d, Hβ), 3.80 (s, 6H, CH3), 3.64 (s, 3H, CH3), 7.43-8.28 (m, 7H, aromatic). Anal. Calcd: C21H18ClNO4: C, 65.71; H, 4.71; N, 3.64. Found: C, 65.84; H, 4.78; N, 3.68.
3-(2-Chloroquinolin-3-yl)-1-(5-bromo-2-methoxyphenyl) prop-2-en-1-one (3j)
Prepared by above method from 2 (8 mmol, 1.1 g) and 2-methoxy-5-bromoacetophenone (8 mmol, 1.2 g); yield: 1.31 g, 61%, Rf = 0.63 in EtOAc/hexane, 4:6; yellow solid. mp: 160–168°C; ESI-MS (M+):, calcd.: 400.850, observed: 400.991; FTIR (cm−1): 1736 (C=O), 1645 (CH=CH), 853 (C–Cl), 588 (C–Br); 1H-NMR (400 MHz, DMSO-d6) δ/ppm 7.53 (1H, d, Hα), 8.57 (1H, d, Hβ), 3.73 (s, 3H, CH3), 7.40–8.31 (m, 7H, aromatic). Anal. Calcd: C20H14ClBrNO2: C, 56.61; H, 3.21; N, 3.44. Found: C, 56.64; H, 3.25; N, 3.46.
General method for synthesis of 6-(2-chloroquinolin-3-yl)-4 substituted phenyl-6H-1,3-oxazin-2-amine (4a–4j)
To the well-stirred solution of (4 mmol) appropriate chalcones (3a–3j) in 10 ml of ethanol, 4 mmol (0.55 g) of urea (5) and catalytic amount of NaOH was added and the reaction mixture was refluxed for 8–14 h. Conversion was monitored for every 60-min interval on pre-coated silica TLC plates by using mixture of acetone and petroleum ether (40:60 V/V) as mobile phase. The reaction mixture was cooled and poured into ice-cold water with excess stirring and kept in refrigerator for 1 h. The precipitate was filtered, washed five times with distilled water, and recrystallized with methanol. The purity of the products was checked on TLC by using mixture of ethyl acetate and hexane as mobile phase, which have shown single spot of the products in each case. The Rf values are given along with the spectral data below.
6-(2-chloroquinilin-3-yl)-4-phenyl-6H-1,3-oxazin-2-amine (4a)
Prepared by above method from 4a (4 mmol, 1.17 g) and 5 (4 mmol, 0.55 g) after 12 h reflux; yield: 1.61 g, 98%; brown amorphous solid. mp: 148–150°C; Rf = 0.48 in EtOAc/hexane, 3:7; ESI-MS (M+): calcd.:335.062, observed: 335.080; IR (KBr, cm−1): 3412 (–NH2), 1394 (Het–C–O), 1570 (C=C, ring skeleton phenyl and heterocyclic moiety), 1415 (C=C, ring skeleton oxazine moiety), 1246 (C–N, str, ring skeleton oxazine moiety), 853 (C–Cl, heterocyclic moiety); 1H-NMR(DMSO-d6, δ): 2.31 (2H, s, –NH2), 5.21 (1H, d, 6-H), 6.31 (1H, d, H), 7.21-8.08 (m, 10H, aromatic). Anal. Calcd: C19H14ClN3O: C, 67.94; H, 4.21; N, 12.44. Found: C, 67.98; H, 4.25; N, 12.46.
6-(2-Chloroquinolin-3-yl)-4-(2,4-dichlorophenyl)-6H-1,3-oxazin-2-amine (4b)
Prepared by above method from 4b (4 mmol, 0.89 g) and 5 (4 mmol, 0.55 g) after 10 h reflux; yield: 1.34 g, 98%; brown amorphous solid. mp: 145–150°C; Rf = 0.56 in EtOAc/hexane, 3:7; ESI-MS (M+): caltd.:403.139, observed: 403.312; IR (KBr, cm−1): 3400 (–NH2), 1396 (Ar–C–O), 1572 (C=C, ring skeleton phenyl and heterocyclic moiety), 1415 (C=C, ring skeleton oxazine moiety), 1246 (C–N, str, ring skeleton oxazine moiety),858 (C–Cl, heterocyclic moiety); 1H-NMR (DMSO-d6, δ): 2.32 (2H, s, –NH2), 5.19 (1H, d, 6-H), 6.21 (1H, d, 4H), 7.10–8.08 (m, 8H, aromatic). Anal. Calcd: C19H12Cl3N3O: C, 56.34; H, 2.92; N, 10.43. Found: C, 56.38; H, 2.99; N, 10.46.
6-(2-Chloroquinolin-3-yl)-4-(3,4-dichlorophenyl)-6H-1,3-oxazin-2-amine (4c)
Prepared by above method from 4c (4 mmol, 0.95 g) and 5 (4 mmol, 0.55 g) after 10-h reflux; yield: 1.38 g, 97%; brown amorphous solid.mp: 156–158 °C; Rf = 0.58 in EtOAc/hexane, 3:7; ESI-MS (M+):, calcd.: 403.155, observed: 403.075; IR (KBr, cm−1): 3398 (–NH2), 1396 (Ar–C–O), 1572 (C=C, ring skeleton Ar moiety), 1415 (C=C, ring skeleton oxazine moiety), 1246 (C–N, str),858 (C–Cl); 1H-NMR (DMSO-d6, δ): 2.32 (2H, s, –NH2), 5.19 (1H, d, 6-H), 6.21 (1H, d, 4-H), 7.12-8.08 (m, 8H, aromatic). Anal. Calcd.: C19H12Cl3N3O: C, 56.36; H, 2.92; N, 10.46.Found: C, 56.41; H, 2.99; N, 10.48.
6-(2-chloroquinolin-3-yl)-4-p-tolyl-6H-1,3-oxazin-2-amine(4d)
Prepared by above method from 4d (4 mmol, 0.95 g) and 5 (4 mmol, 0.55 g) after 14-h reflux; yield: 1.38 g, 97%; dark yellow crystalline solid.mp: 160–175°C; Rf = 0.52 in EtOAc/hexane, 3:7; ESI-MS (M+):, calcd.: 349.155, observed: 349.075; IR (KBr, cm−1): 3406 (–NH2), 1393 (Ar–C–O), 1569 (C=C, ring skeleton Ar moiety), 1418 (C=C, ring skeleton oxazine moiety), 1246 (C–N, str), 858 (C–Cl); 1H-NMR (DMSO-d6, δ): 2.34 (3H, s, CH3), 2.32 (2H, s, -NH2), 5.16 (1H, d, 6-H), 6.23 (1H, d, 4-H), 7.02-8.08 (m, 9H, aromatic).Anal.Calcd: C20H16ClN3O: C, 68.58; H, 4.60; N, 12.02. Found: C, 68.61; H, 4.58; N, 12.01.
6-(2-chloroquinolin-3-yl)-4-o-tolyl-6H-1,3-oxazin-2-amine (4e)
Prepared by above method from 4e (4 mmol, 1.19 g) and 5 (4 mmol, 0.55 g) after 14-h reflux; yield: 1.57 g, 95%;%; light brown crystalline solid. mp: 173–178°C; Rf = 0.58 in EtOAc/hexane, 3:7; ESI-MS (M+):, calcd.: 349.055, observed: 349.075; IR (KBr, cm−1): 3403 (–NH2), 1393 (Ar–C–O), 1569 (C=C, ring skeleton Ar moiety), 1418 (C=C, ring skeleton oxazine moiety), 1246 (C–N, str),858 (C–Cl); 1H NMR(DMSO-d6, δ): 2.34 (3H, s, CH3), 2.32 (2H, s, -NH2), 5.16 (1H, d, 6-H), 6.23 (1H, d, 4-H), 7.02–8.08 (m, 9H, aromatic). Anal. Calcd.: C20H16ClN3O: C, 68.58; H, 4.60; N, 12.02. Found: C, 68.61; H, 4.58; N, 12.01.
6-(2-chloroquinolin-3-yl)-4-(4-bromophenyl)-6H-1,3-oxazin-2-amine (4f)
Prepared by above method from 4f (4 mmol, 1.20 g) and 5 (4 mmol, 0.55 g) after 10-h reflux; yield: 1.66 g, 98%; brown solid. mp: 158–160°C. Rf = 0.61 in EtOAc/hexane, 4:6; ESI-MS (M+):, calcd.: 412.945, observed: 412.992; IR (KBr, cm−1): 3410 (–NH2), 1393 (Ar–C–O), 1569 (C=C, ring skeleton Ar moiety), 1418 (C=C, ring skeleton oxazine moiety), 1246 (C–N, str), 858 (C–Cl), 588 (C–Br); 1H-NMR(DMSO-d6, δ): 2.02 (2H, s, –NH2), 5.19 (1H, d, 6-H), 6.32 (1H, d, 4-H), 7.19-8.08 (m, 9H, aromatic). Anal. Calcd.: C19H13ClBrN3O: C, 55.08; H, 3.16; N, 10.12. Found: C, 55.16; H, 3.18; N, 10.18.
6-(2-Chloroquinolin-3-yl)-4-(2-bromophenyl)-6H-1,3-oxazin-2-amine (4g)
Prepared by above method from 4g (4 mmol, 1.25 g) and 5 (4 mmol, 0.54 g) after 10-h reflux; yield: 1.68 g, 97%; dark brown solid. mp: 184–186°C; Rf = 0.68 in EtOAc/hexane, 3:7; ESI-MS (M+):, calcd.: 412.935, observed: 412.982; IR (KBr, cm−1): 3418 (–NH2), 1393 (Ar–C–O), 1569 (C=C, ring skeleton Ar moiety), 1418 (C=C, ring skeleton oxazine moiety), 1246 (C–N, str), 858 (C–Cl), 588 (C-Br); 1H-NMR (DMSO-d6, δ): 2.02 (2H, s, –NH2), 5.19 (1H, d, 6-H), 6.32 (1H, d, 4-H), 7.19–8.08 (m, 9H, aromatic). Anal. Calcd: C19H13ClBrN3O: C, 55.03; H, 3.16; N, 10.13. Found: C, 55.16; H, 3.18; N, 10.18.
6-(2-chloroquinolin-3-yl)-4-(3-nitrophenyl)-6H-1,3-oxazin-2-amine (4h)
Prepared by above method from 4h (4 mmol, 1.32 g) and 5 (4 mmol, 0.54 g) after 16-h reflux. Yield: 1.73 g, 96%; reddish yellow solid. mp: 168–172°C; Rf = 0.58 in EtOAc/hexane, 4:6; ESI-MS (M+):, calcd.: 380.075, observed: 380.098; IR (KBr, cm−1): 3407 (–NH2), 1398 (Ar–C–O), 1565 (C=C, ring skeleton Ar moiety), 1416 (C=C, ring skeleton oxazine moiety), 1256 (C–N, str), 858 (C–Cl), 1589 (–NO2); 1H-NMR (DMSO-d6, δ): 2.22 (2H, s, –NH2), 5.11 (1H, d, 6-H), 6.52 (1H, d, 4-H), 7.47–8.28 (m, 9H, aromatic). Anal. Calcd: C19H13ClBrN4O3: C, 59.90; H, 3.41; N, 14.73. Found: C, 59.96; H, 3.48; N, 14.78.
6-(2-Chloroquinolin-3-yl)-4-(3,4,5-trimethoxyphenyl)-6H-1,3-oxazin-2-amine (4i)
Prepared by above method from 4i (4 mmol, 1.21 g) and 5 (4 mmol, 0.55 g) after 8-h reflux; yield: 1.65 g, 98%; brown amorphous solid. mp: 184–186°C; Rf = 0.48 in EtOAc/hexane, 3:7; ESI-MS (M+):, calcd.: 425.175, observed: 425.198; IR (KBr, cm−1): 3397 (–NH2), 1398 (Ar–C–O), 1565 (C=C, ring skeleton Ar moiety), 1416 (C=C, ring skeleton oxazine moiety), 1256 (C–N, str), 858 (C–Cl); 1H-NMR (DMSO-d6, δ): 2.22 (2H, s, –NH2), 5.11 (1H, d, 6-H), 6.52 (1H, d, 4-H), 3.73 (s, 6H, CH3), 3.64 (s, 3H, CH3), 6.47–8.28 (m, 7H, aromatic). Anal. Calcd: C22H20ClN3O4: C, 62.03; H, 4.71; N, 9.73.Found: C, 62.09; H, 4.78; N, 9.87.
6-(2-Chloroquinolin-3-yl)-4-(5-bromo-2-methoxyphenyl)-6H-1,3-oxazin-2-amine (4j)
Prepared by above method from 4j (4 mmol, 1.21 g) and 5 (4 mmol, 0.55 g) after 8-h reflux; yield: 1.65g, 98%; brown crystalline solid. mp: 174–180°C; Rf = 0.57 in EtOAc/hexane, 3:7; ESI-MS (M+):, calcd.: 443.717, observed: 443.798; IR (KBr, cm−1): 3412 (–NH2), 1398 (Ar–C–O), 1565 (C=C, ring skeleton Ar moiety), 1416 (C=C, ring skeleton oxazine moiety), 1256 (C-N, str), 858 (C–Cl), 588 (C–Br); 1H-NMR(DMSO-d6, δ): 2.12 (2H, s, –NH2), 5.19 (1H, d, 6-H), 6.12 (1H, d, 4-H), 3.73 (s, 3H, CH3), 6.61-8.08 (m, 8H, aromatic). Anal. Calcd: C20H15ClBrN3O2: C, 54.03; H, 3.39; N, 9.43. Found: C, 54.09; H, 3.42; N, 9.84.
Results and discussion
In vitro antimalarial activity
The In vitro antimalarial activity () of 4a–c and 4f, 4g, 4i were found comparable with CQ diphosphate (IC50 = 0.021 μM) against CQ sensitive strain (MRC-02) of P. falciparum. Compounds 4b, 4c, 4e, 4d, 4g, and 4i of this series have shown better activity than CQ diphospahte (IC50 = 0.177 μM) against CQ-resistant strain (RKL9) of parasite. Compound 4i was found to be most potent against resistant (IC50 = 0.0435 μM) as well as sensitive (IC50 = 0.0265 μM) strains of P. falciparum. For compound 4i, R2, R3, and R4 are methoxy groups. Interestingly, compound 4a, for which R1 to R5 are all hydrogen, also showed antiplasmodial efficacy close to 4i against CQ-sensitive strain (IC50 = 0.0302μM). However, against resistant strain, compound 4a (IC50= 0.1305 μM) was found to be less active than 4i (IC50 = 0.0435μM). 4b, 4c, 4g, and 4f are the four halogen containing compounds showed very good activity against both CQ-sensitive and -resistant strains. Methyl group in place of R1 (4e) and R3 (4d) also delivered good activities. 4j is the compound containing two electron withdrawing groups each but showed moderate to poor in vitro antiplasmodial activity.
Table 1. Cytoxicity, in vitro antimalarial activity of 6-(2-chloroquinolin-3-yl)-4 substitutedphenyl-6H-1,3-oxazin-2-amine.
In vitro cytotoxicity
The maximum concentration of DMSO in any cell was 0.1% and did not affect cell growth. To compare all compounds, we have used the 50% inhibitory concentration IC50 and selectivity index (SI), which is defined as the ratio of cytotoxicity over antimalarial activity, each one expressed with IC50. summarizes IC50 and SI of all the compounds. For all the compounds, the SI values were >1, indicating better selectivity against P. falciparum even though a long cell drug exposure time (72 h) was given for cytotoxicity test. SI of two most potent antimalarial compounds 4g and 4i were 59.56 and 19.48, respectively, for resistant strain of P. falciparum. In case of sensitive strain, those values were 140.14 and 31.24, respectively. The most potent substituted oxazine 4i was found to be most toxic and second best active 4g was found to be least toxic among the 10 compounds under study. SI of 4g was found best for both sensitive and resistant strains of malaria parasite.
In vivo antimalarial activity
The three most active compounds (4b, 4g, and 4i) found in the in vitro antiplasmodial assays were tested in vivo activity in Swiss mice against P. yoelli (N-67 strain) intraperitoneally (). Compound 4b showed 69.93% average suppression at 50 mg/kg/day in comparison with 100% suppression by CQ at 8 mg/kg/day. Similarly, for compounds 4g and 4i in vivo antimalarial activity followed the same trend and showed a correlation. The MST was found in accordance with inhibition data.
Table 2. In vivo antimalarial activity against Chloroquine resistant N-67 strain of P. yoelli in Swiss mice.
Correlation of antiplasmodial activities with β-hematin formation inhibition
To ascertain the mode of action, vacuolar accumulation ratio (VAR) of all the compounds were theoretically calculated from experimentally determined pKa values () using equation 1.Citation32
Table 3. Relationship between pKa, normalized antiplasmodial IC50 values, and inhibition of β-hematin formation for substituted oxazine and CQ diphosphate.
The two pKa values of CQ diphosphate were determined experimentally and were found close to the previously reported values.Citation32 In equation 1, VAR represents the ratio of drug inside and outside of the parasite food vacuole. pHi = pH inside food vacuole (assumed to be pH 5.3) and pHe = pH externally (assumed to be pH 7.2). The calculated VAR and the VAR relative to 4a (α) are shown in . Compound 4a was taken as reference molecule to calculate α because this molecule was not substituted at B ring. VAR values for the compounds of this series are calculated in the range of 603 (4h) to 2457 (4g). For CQ diphosphate, the value was 5890. A significant difference between VAR values were observed between 4g (VAR = 2457) and 4f (VAR = 1545) in which bromine is present at ortho and meta positions, respectively. In case of 4j (VAR = 1551) bromine is present at Meta position and its VAR value was found closer to 4f. The relative VAR (α) values for all the compounds were calculated in the range 0.48–1.95 and that for CQ diphospahte was 4.69. Due to higher pKa values, CQ diphosphate achieved 2.5–9 times better accumulation in the parasite food vacuole in comparison with the substituted oxazine 4(a–j). Normalized IC50 values (IC50 × α) were obtained by multiplying experimentally obtained antimalarial IC50 with α. Normalized IC50 values are the theoretically expected IC50values, if the compounds accumulate in the vacuole to the same extent as 4a. Having adjusted for the differential accumulation of the compounds, the normalized antiplasmodial IC50 values and the inhibition of β-hematin formation (BHIA50) should show a clear correlation. Hawley et al.Citation33 and Kaschula et al.Citation32 reported direct proportionality between normalized antiplasmodial activity and inhibition of β-hematin formation of quinoline antimalarials using chloroquine-sensitive strains of P. falciparum. In this study, we have taken both CQ-sensitive (MRC-02) and CQ-resistant (RKL9) strains of P. falciparum. The quantities of the compounds required to inhibit the formation of β-hematin by 50% (BHIA50) were found in the range 1.53 (4b) to 2.43 µM (4e). Unsubstituted 4a showed comparable β-hematin polymerization inhibition activity (1.56 µM). Exceptionally, compound 4e containing a methyl group showed better BHIA50 than 4i (1.72 µM), which contains three methoxy groups. In case of 4g and 4f, unlike VAR, the position of bromine did not make any significant difference between their BHIA50 values. CQ diphosphate showed a comparable value of 1.87 µM. Although chloroquine showed lower β-hematin inhibition activity, due to better VAR value it was found to be more potent than 4i against chloroquine sensitive P. falciparum. The normalized IC50 (IC50 × α) values for all the compounds including CQ diphosphate were calculated for both sensitive and resistant strains. A plot of log (IC50 × α) against log BHIA50 () showed some correlation present between the normalized IC50 and the ability of the compounds to inhibit β-hematin formation. The r values were found decreased to 0.51 and 0.49 for sensitive and resistant strains respectively when chloroquine was included. This indicates that chloroquine and substituted oxazines 4(a–j) have similar antimalarial mode of action. These findings further strengthened the hypothesis that haem polymerization inhibition may be a possible mode of action for this series of compounds 4(a–j).
Conclusions
We have reported the synthesis and evaluation of substituted oxazines as potential antimalarial agents. The molecules were found active against in vitro culture of both CQ-sensitive and CQ-resistant strains of P. falciparum. Compound 4i was found to be most active as well as most cytotoxic. The second best active compound 4g was found to be least cytotoxic among the 10 compounds. Three compounds (4b, 4g, and 4i) were tested against P. yoelli (N-67 strain) in mouse model found active. A good correlation was observed between normalized antimalarial activity (IC50 × α) and β-hematin formation inhibition (BHIA50) against both sensitive and resistant strains. These observations indicated that the substituted oxazines are antimalarial and their mode of action may be through inhibition of haem detoxification process.
Acknowledgement
The authors thank Head of Department of Chemistry, Rashtrasant Tukadoji Maharaj Nagpur University, Nagpur (India), for providing necessary laboratory facilities and Director, SAIF Lucknow (India) for providing necessary spectral and biological data of compounds.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- Fidock DA, Rosenthal PJ, Croft SL, Brun R, Nwaka S. Antimalarial drug discovery: efficacy models for compound screening. Nat Rev Drug Discov 2004;3:509–520.
- Wiesner J, Ortmann R, Jomaa H, Schlitzer M. New antimalarial drugs. Angew Chem Int Ed Engl 2003;42:5274–5293.
- Winstaley PA, Parasitol. Today. 2000;16:136–153.
- Bremen J, Trop A J. Med. Hyg. 2001;64:1–11.
- Domínguez JN. Chemotherapeutic agents against malaria: what next after chloroquine? Curr Top Med Chem 2002;2:1173–1185.
- White NJ. Drug resistance in malaria. Br Med Bull 1998;54:703–715.
- Rosenthal PJ, Miller LH, In Antimalarial Chemotherapy: Mechanisms of Action, Resistance, and New Directions in Drug Discovery; P.J., Rosenthal, Ed, Humana Press Inc.: Totowa, NJ, 2001: pp 3–13.
- Domínguez JN, Lo´pez S, Charris J, Iarruso L, Lobo G, Semenov A, Olson JE, Rosenthal PJ. J. Med. Chem. 1997;40:2726–2732.
- Chen M, Theander TG, Christensen SB, Hviid L, Zhai L, Kharazmi A. Licochalcone A, a new antimalarial agent, inhibits in vitro growth of the human malaria parasite Plasmodium falciparum and protects mice from P. yoelii infection. Antimicrob Agents Chemother 1994;38:1470–1475.
- Chen M, Brøgger Christensen S, Zhai L, Rasmussen MH, Theander TG, Frøkjaer S et al. The novel oxygenated chalcone, 2,4-dimethoxy-4’-butoxychalcone, exhibits potent activity against human malaria parasite Plasmodium falciparum in vitro and rodent parasites Plasmodium berghei and Plasmodium yoelii in vivo. J Infect Dis 1997;176:1327–1333.
- Li R, Kenyon GL, Cohen FE, Chen X, Gong B, Dominguez JN et al. In vitro antimalarial activity of chalcones and their derivatives. J Med Chem 1995;38:5031–5037.
- Liu M, Wilairat P, Croft SL, Tan AL, Go ML. Structure-activity relationships of antileishmanial and antimalarial chalcones. Bioorg Med Chem 2003;11:2729–2738.
- Domı´nguez JN, Charris JE, Lobo G, Gamboa-Domínguez N, Moreno MM, Riggione F, Sa´nchez E, Olson J, Rosenthal PJ. Eur. J. Med. Chem. 2001; 36: 555–560.
- Vibhute YB and Basser MA. Ind.J. of Chem. 2003;42B:202–205.
- Bhat BA, Dhar KL, Puri SC, Saxena AK, Shanmugavel M, Qazi GN. Synthesis and biological evaluation of chalcones and their derived pyrazoles as potential cytotoxic agents. Bioorg Med Chem Lett 2005;15:3177–3180.
- Edwards ML, Stemerick DM, Sunkara PS. Chalcones: a new class of antimitotic agents. J Med Chem 1990;33:1948–1954.
- Kalirajan R, Palanivelu M, Rajamanickam V, Vinothapooshan G, Anandarajagopal K. Int. J. of Chem. Sci. 2007;5:73–80.
- Udupi RH, Bhat R, Kumar K. Indian J. Het.Chem. 1998;8:143–146.
- Pande A and Saxena VK. Ind.J.of Chem. 1987;26B:390–392.
- Gupta U, Sareen V, Khatri V, Chugh S. Indian J. Het.Chem. 2005;14:265–266.
- Pandey VK, Gupta VD, Tiwari DN. Indian J. of Het.Chem. 2004;13:399–400.
- Mishra RM and Wahab A. Indian J. Het.Chem. 2003;13:29–32.
- Trager W, Jensen JB. Human malaria parasites in continuous culture. Science 1976;193:673–675.
- Lambros C, Vanderberg JP. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol 1979;65:418–420.
- Peters W, Portus JH, Robinson BL, Ann. Trop. Med. Parasitol. 1975;69:155–171.
- Borenfreund E, Puerner JA. Toxicity determined in vitro by morphological alterations and neutral red absorption. Toxicol Lett 1985;24:119–124.
- Albert A, Serjeant EP, The Determination of Ionization Constants. Chapman and Hall Ltd: London 1971: pp 94.
- Basilico N, Pagani E, Monti D, Olliaro P, Taramelli D. A microtitre-based method for measuring the haem polymerization inhibitory activity (HPIA) of antimalarial drugs. J Antimicrob Chemother 1998;42:55–60.
- Baelmans R, Deharo E, Bourdy G, Muñoz V, Quenevo C, Sauvain M et al. A search for natural bioactive compounds in Bolivia through a multidisciplinary approach. Part IV. Is a new haem polymerisation inhibition test pertinent for the detection of antimalarial natural products? J Ethnopharmacol 2000;73:271–275.
- Meth-Cohn O, Narine B, Tarnowski B. J chem. Soc., Perkin Trans. 1981;1:1520.
- Ali MM, Tasneem Rajanna, KC, Sai Prakash PK. Synlett. 2001;2:251.
- Kaschula CH, Egan TJ, Hunter R, Basilico N, Parapini S, Taramelli D et al. Structure-activity relationships in 4-aminoquinoline antiplasmodials. The role of the group at the 7-position. J Med Chem 2002;45:3531–3539.
- Hawley SR, Bray PG, Mungthin M, Atkinson JD, O’Neill PM, Ward SA. Relationship between antimalarial drug activity, accumulation, and inhibition of heme polymerization in Plasmodium falciparum in vitro. Antimicrob Agents Chemother 1998;42:682–686.