Abstract
Eight selected sulfonamide drugs were investigated as inhibitors of heat shock protein 90 (Hsp90). The investigation included simulated docking experiments to fit the selected compounds within the binding pocket of Hsp90. The selected molecules were found to readily fit within the ATP-binding pocket of Hsp90 in low-energy poses. The sulfonamides torsemide, sulfathiazole, and sulfadiazine were found to inhibit the ATPase activity of Hsp90 with IC50 values of 1.0, 2.6, and 1.5 μM, respectively. Our results suggest that these well-established sulfonamides can be good leads for subsequent optimization into potent Hsp90 inhibitors.
Keywords::
Introduction
Heat shock protein 90 (Hsp90) belongs to a family of molecular chaperones that play a pivotal role in the conformational maturation, stability, and function of protein substrates within the cell. The ATPase activity of Hsp90 provides the necessary energy required for refolding of denatured cellular proteins.Citation1
Amongst the client proteins of Hsp90 are many oncogenes essential for the survival, proliferation, invasion, metastasis, and angiogenesis of tumors.Citation2 In fact, several oncogenic proteins have been shown to be dependent upon Hsp90 for conformational activation, including: telomerase, Her2 (erbB2), Raf-1, focal adhesion kinase, and the steroid hormone receptors.Citation3
The validity of Hsp90 as anticancer target for drug discoveryCitation4–16 was further established by emerging clinical and preclinical trials employing the potent Hsp90 inhibitor 17-allylamino-17-desmethoxygeldanamycinCitation17–27 and the natural Hsp90 inhibitors geldanamycin,Citation28–31 radicicol,Citation32 and other small molecules, for example, purinesCitation33 and pyrazoles.Citation34
However, despite the high cellular activity and clinical progression of 17-allylamino-17-desmethoxygeldanamycin, it has several limitations, for example, poor solubility, hepatotoxicity, and extensive metabolism.Citation17–27 These issues have led to significant efforts to identify novel small molecule inhibitors of Hsp90.Citation35–37
The recent discovery that certain hydroxynaphthalene-arylsulfonamide derivatives possess micromolar inhibitory actions against Hsp90Citation38,Citation39 combined with the moderate anticancer properties reported for antibacterial sulfonamides and their selective accumulation in cancerous cellsCitation40 prompted us to assess the inhibitory profiles of eight clinically established sulfonamides against the ATPase activity of Hsp90, namely, torsemide, sulfathiazole, sulfadiazine, sulfamethizole, sulfisoxazole, sulfadoxine, sulfaguanide, and sulfacetamide ().Citation41 The excellent safety profiles of the selected compounds should render them superior leads for subsequent optimization if found active. Docking studies into the ATP-binding site of Hsp90 were used to explain the inhibitory bioactivities of active compounds.
Figure 1. Chemical structures of tested sulfonamides.
The selected compounds were bioassayed against recombinant human Hsp90α (BioQuote, York, UK) employing malachite green-based detection of free phosphate released by the ATPase action of Hsp90Citation42–44 using geldanamycin as positive control to standardize our experimental setup. We believe malachite green assay is more appropriate to measure the inhibitory effects on ATPase activity of Hsp90 compared with other more biologically intensive methods, for example, western blotting, since these rely on living cell lines that might show problems related to compound permeability across cellular membranes and metabolism, which may not necessarily reflect real Hsp90 inhibition.Citation39
Materials and methods
All of the chemicals used in these experiments were of reagent grade and obtained from commercial suppliers. Torsemide, sulfadiazine, sulfathiazole, sulfamethizole, sulfisoxazole, sulfadoxine, sulfaguanide, and sulfacetamide were purchased from Sigma-Aldrich (Munich, Germany), ATP 100x solution, geldanamycin, and Hsp90α were purchased from BioQuote (UK); Quantichrom ATPase/GTPase assay kit was purchased from BioAssay Systems (Hayward, CA).
Docking
The binding site was generated from the cocrystallized ligand (GMD275) within Hsp90α protein (PDB code: 1YET). All eight ligands were docked employing the following docking configuration: (i) number of Monte Carlo search trials 25000, search step for torsions with polar hydrogens = 15°. (ii) The Root Mean Square Difference (RMS) threshold for ligand-to-binding site shape match was set to 2.5 employing a maximum of 1.0 binding site partitions and 1.0 site partition seed. (iii) The interaction energies were assessed employing Consistent Force Field (CFF) force field with a nonbonded cutoff distance of 10.0 Å and distance-dependent dielectric. An energy grid extending 5.0 Å from the binding site was implemented. (iv) Rigid body ligand minimization parameters were: 40 iterations of steepest descend (SD) minimization followed by 80 Broyden–Fletcher–Goldfarb–Shanno (BFGS) iterations applied to every successful orientation of the docked ligand. (v) A maximum of 10 diverse docked conformations/poses of optimal interaction energies were saved. The similarity threshold was set to a DockScore of 20 kcal/mol and an RMS value of 1.5 Å. (vi) The saved conformers/poses were further energy-minimized within the binding site for a maximum of 200 rigid-body iterations. The resulting docked poses were scored employing consensus scoring based on PLP1, PLP2, ligscore1, ligscore2, PMF, and JAIN.Citation45–51 The optimal docked poses of torsemide, sulfadiazine, sulfadoxine, and sulfaguanide achieved full consensus score from all six scoring functions, while the remaining compounds failed to do so. Therefore, sulfacetamide, sulfamethizole, sulfisoxazole, and sulfathiazole were superimposed against the docked poses of sulfadiazine and sulfadoxine. Subsequently, they were docked via rigid body docking employing the following settings: (ii) The RMS threshold for ligand-to-binding site shape match was set to 2.5 employing a maximum of 1.0 binding site partitions and 1.0 site partition seed. (iii) Interaction energies were assessed employing CFF force field with a nonbonded cutoff distance of 10.0 Å and distance-dependent dielectric. An energy grid extending 3.0 Å from the binding site was implemented. (iv) Rigid body ligand minimization parameters were: 10 iterations of SD minimization followed by 20 BFGS iterations applied to every successful orientation of the docked ligand. (vi) The saved poses were further energy-minimized within the binding site for a maximum of 100 rigid-body iterations.
Measurement of Hsp90 inhibition
The bioassay was performed as reported earlier.Citation52,Citation53 In brief, tested compounds were dissolved as dimethyl sulfoxide (DMSO) stock solutions (0.2 M). Bioassays were performed by mixing Hsp90α solution (6 µL, 25 µg/mL in assay buffer), 24 µL assay buffer, and 5 µL of the particular tested compounds to yield final inhibitor concentrations of 100, 10, 1, and 0.1 µM per well. The mixtures were incubated for 30 min at 37°C in ELISA plate shaker, and then ATP solutions (5 µL, 4 mM in assay buffer) were added to each mixture. Blank was prepared as earlier except 5 µL of 2% DMSO in distilled water (v/v%) was used instead of inhibitor solution. The mixtures were equilibrated to 37°C and incubated for 24 h. The enzymatic reaction was terminated by the addition of 80 µL malachite green ammonium molybdate–Tween-20 solution in 0.27 M H2SO4 and 10 µL of 34% sodium citrate. Color was allowed to develop at room temperature for 30 min, and sample absorbance were determined at λmax 620 nm using a plate reader (Bio-Tek Instruments ELx 800, Winooski, VT). Inhibition of recombinant Hsp90 was calculated as percent activity of the uninhibited control. DMSO concentrations were kept <1% in all experiments and controls. Samples and blanks were prepared in duplicates. Geldanamycin was tested as positive control, and negative controls were prepared by adding the substrate after reaction termination.
Results and discussion
shows the inhibitory profiles of tested compounds against ATPase activity of Hsp90. Interestingly, torsemide, sulfadiazine, and sulfathiazole illustrated low micromolar IC50 values, whereas sulfamethizole and sulfisoxazole exhibited mediocre IC50 values. On the other hand, sulfadoxine, sulfaguanide, and sulfacetamide exhibited poor Hsp90 inhibition profiles.
Table 1. Results of evaluated sulfonamides.
To probe this intriguing behavior, we decided to dock the different sulfonamides into the ATP-binding pocket of Hsp90. The docking experiment was conducted by employing LIGANDFITCitation45 and consensus scoring based on PLP1,Citation46 PLP2,Citation46 ligscore1,Citation47 ligscore2,Citation45 PMF,Citation48,Citation49 and JAINCitation50,Citation51 scoring functions. However, since individual scoring function(s) are generally incapable of correctly evaluating binding free energies due to the high complexity of the underlying molecular interactions,Citation54–56 we decided to select optimal docked conformers/poses based on consensus among the six scoring functions.Citation57,Citation58 The highest ranking docked conformer/pose was selected to represent the bound ligand. The consensus function assigned a value of 1 for any molecular pose ranked within the highest 40% by the particular scoring function; otherwise, it was assigned a zero value, that is, if it was within the lowest 60%. Subsequently, the consensus function summed up the scores for each molecular pose/conformer and ranked the molecular orientations accordingly. Consensus scoring should alleviate the inability of scoring functions to individually evaluate binding free energies correctly.Citation59–64
shows the score values, including consensus scores, of optimal docked conformers/poses of each tested sulfonamide.
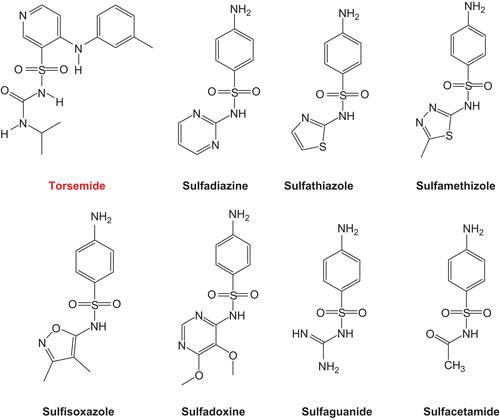
shows the docked poses of sulfonamides into the binding pocket of Hsp90. shows the binding interactions of torsemide and sulfadiazine as representatives of remaining sulfonamides.
Figure 2. Docked poses of tested sulfonamides in Hsp90 (PDB code: 1YET, resolution 1.9 Å). (A) and (B) showing the binding pocket with and without solvent accessible surface, respectively.
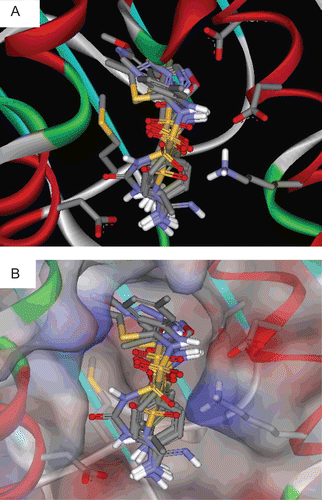
Figure 3. Docked poses of (A) torsemide and (B) sulfadiazine in Hsp90.
Apparently, all docked compounds, except sulfaguanide and sulfacetamide, share four critical features/interactions within the binding pocket: (i) an electron-deficient N-heterocycle stacking against the sulfide of MET98, (ii) a hydrogen-bonding interaction connecting the sulfonamidic NH (or aniline NH in torsemide) to the carboxylate of ASP54, (iii) a hydrogen-bonding interaction connecting the quaternary ammonium of LYS58 and sulfone oxygens in the ligands, and (iv) hydrogen-bonding interaction connecting aniline nitrogen (or urea NH in torsemide) to the carboxylate of ASP102.
The fact that potent and moderate inhibitors assume similar docking-based interactions suggested another factor to explain the apparent differences in activity. Upon evaluating the pKa values of tested sulfonamides,Citation65,Citation66 it became clear that potent inhibitors (torsemide, sulfadiazine, and sulfathiazole) are, on average, 1.2 pKa units higher than moderate inhibitors (sulfamethizole, sulfisoxazole, and sulfadoxine),Citation65,Citation66 suggesting certain role played by the ionizability of sulfonamidic NH in bioactivity.
Apparently, the greater acidity of moderate sulfonamides promotes ionization of sulfonamidic NH under pH 7.4 (bioassay pH) and therefore causes a loss of the critical hydrogen-bonding interaction with ASP54. Furthermore, ionization renders the sulfonamidic fragments negatively charged and hence electrostatically repulsive with ASP54 carboxylate.
Sulfadoxine seems to have another detrimental structural factor related to its inferior activity: its 4,5-dimethoxy-pyrimidine ring has higher electron density compared with corresponding heterocyclic rings in the other sulfonamides because of the electron-donating dimethoxy substituents. This property probably reduces the efficiency of π-stacking against the electron-rich sulfide of MET98 causing further reduction in bioactivity, that is, compared with sulfamethizole and sulfisoxazole.
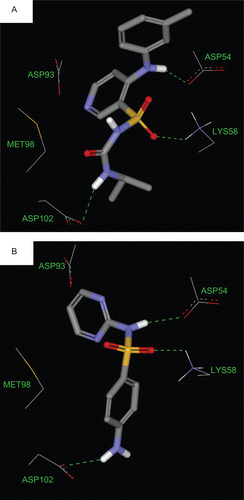
The poor inhibitory action of sulfacetamide is also explainable by the high acidity of its sulfonamidic NH due to its mixed amidic/sulfonamidic nature. Furthermore, sulfacetamide lacks electronically deficient heterocycle capable of stacking against MET98 sulfide, as shown in .
Figure 4. Docked poses of (A) sulfacetamide and (B) sulfaguanide in Hsp90.
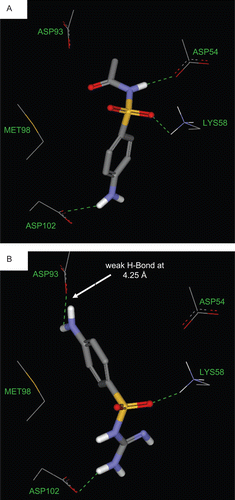
Finally, sulfaguanide seems to assume flipped pose (): its aniline NH2 is suboptimally hydrogen-bonded to ASP93 (at a distance of 4.25 Å), whereas the sulfonamidic oxygens are hydrogen-bonded to LYS58. Similarly, the terminal guanidino seems to interact with the carboxylate of ASP102. However, although the aromatic ring in sulfaguanide stacks against MET98 sulfur, the fact that it is electron-rich aniline suggests weaker attraction with MET98 sulfide compared with electron-deficient heterocycles in sulfadiazine, sulfathiazole, and torsemide. Furthermore, this flipped pose cost sulfaguanide a critical hydrogen-bonding interaction with ASP54. The critical significance of this interaction can be deduced from the consensus of other more potent sulfonamides (e.g. torsemide, sulfadiazine, and sulfathiazole).
Moreover, the alkaline nature of the guanidine moiety of sulfaguanide (pKa = 9.7)Citation65,Citation66 suggests that it is positively charged under bioassay conditions (pH = 7.4) and hence should be heavily hydrated, particularly as it resides at the outer rim of the binding site. This factor probably further contributes to the poor Hsp90 inhibition of sulfaguanide.
These structure–activity trends suggest that other sulfonamide-related linkers can be useful to generate and optimize new potent Hsp90 inhibitors, for example, sulfonylurea. Furthermore, it is anticipated that introduction of small amidic substituents at the para-position of the aromatic heterocycle (e.g. pyrimidine in sulfadiazine) should allow hydrogen-bonding interactions with ASP93 carboxylate without increasing the electron density of the heterocycle, which might jeopardize aromatic stacking against MET98 sulfide.
Conclusion
The current research shows promising Hsp90 inhibition for some clinically established sulfonamides. Furthermore, docking experiments illustrated the significance of four critical amino acids in binding to Hsp90, namely MET98, ASP54, ASP102, and LYS58. Our findings and explanations pave the way for subsequent optimization of new potent sulfonamidic Hsp90 inhibitors.
Declaration of interest
The authors thank the Deanship of Scientific Research and Hamdi-Mango Center for Scientific Research at the University of Jordan for their generous funds. The authors also thank Al-Zaytoonah Private University of Jordan for support.
Related Research Data
References
- Mahalingam D, Swords R, Carew JS, Nawrocki ST, Bhalla K, Giles FJ. Targeting HSP90 for cancer therapy. Br J Cancer 2009, 100, 1523–1529.
- Chiosis G, Rodina A, Moulick K. Emerging Hsp90 inhibitors: from discovery to clinic. Anticancer Agents Med Chem 2006, 6, 1–8.
- Dymock BW, Drysdale MJ, McDonald E, Workman P. Inhibitors of Hsp90 and other chaperones for the treatment of cancer. Expert Opin Ther Pat 2004, 14, 837–847.
- Isaacs JS, Xu W, Neckers L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell 2003, 3, 213–217.
- Workman P. Altered states: selectively drugging the Hsp90 cancer chaperone. Trends Mol Med 2004, 10, 47–51.
- Chiosis G, Vilenchik M, Kim J, Solit D. Hsp90: the vulnerable chaperone. Drug Discov Today 2004, 9, 881–888.
- Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer 2005, 5, 761–772.
- Janin YL. Heat shock protein 90 inhibitors. A text book example of medicinal chemistry? J Med Chem 2005, 48, 7503–7512.
- Chiosis G. Targeting chaperones in transformed systems–a focus on Hsp90 and cancer. Expert Opin Ther Targets 2006, 10, 37–50.
- Kamal A, Boehm MF, Burrows FJ. Therapeutic and diagnostic implications of Hsp90 activation. Trends Mol Med 2004, 10, 283–290.
- Neckers L, Neckers K. Heat-shock protein 90 inhibitors as novel cancer chemotherapeutics—an update. Expert Opin Emerg Drugs 2005, 10, 137–149.
- Drysdale MJ, Brough PA, Massey A, Jensen MR, Schoepfer J. Targeting Hsp90 for the treatment of cancer. Curr Opin Drug Discov Devel 2006, 9, 483–495.
- Powers MV, Workman P. Targeting of multiple signalling pathways by heat shock protein 90 molecular chaperone inhibitors. Endocr Relat Cancer 2006, 13(Suppl. 1), S125–S135.
- Xu W, Neckers L. Targeting the molecular chaperone heat shock protein 90 provides a multifaceted effect on diverse cell signaling pathways of cancer cells. Clin Cancer Res 2007, 13, 1625–1629.
- Workman P, Burrows F, Neckers L, Rosen N. Drugging the cancer chaperone HSP90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann N Y Acad Sci 2007, 1113, 202–216.
- Pearl LH, Prodromou C, Workman P. The Hsp90 molecular chaperone: an open and shut case for treatment. Biochem J 2008, 410, 439–453.
- Maloney A, Workman P. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. Expert Opin Biol Ther 2002, 2, 3–24.
- Schulte TW, Neckers LM. The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemother Pharmacol 1998, 42, 273–279.
- Banerji U, O’Donnell A, Scurr M, Pacey S, Stapleton S, Asad Y, Simmons L, Maloney A, Raynaud F, Campbell M, Walton M, Lakhani S, Kaye S, Workman P, Judson I. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. J Clin Oncol 2005, 23, 4152–4161.
- Goetz MP, Toft D, Reid J, Ames M, Stensgard B, Safgren S, Adjei AA, Sloan J, Atherton P, Vasile V, Salazaar S, Adjei A, Croghan G, Erlichman C. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. J Clin Oncol 2005, 23, 1078–1087.
- Weigel BJ, Blaney SM, Reid JM, Safgren SL, Bagatell R, Kersey J, Neglia JP, Ivy SP, Ingle AM, Whitesell L, Gilbertson RJ, Krailo M, Ames M, Adamson PC. A phase I study of 17-allylaminogeldanamycin in relapsed/refractory pediatric patients with solid tumors: a Children’s Oncology Group study. Clin Cancer Res 2007, 13, 1789–1793.
- Modi S, Stopeck AT, Gordon MS, Mendelson D, Solit DB, Bagatell R, Ma W, Wheler J, Rosen N, Norton L, Cropp GF, Johnson RG, Hannah AL, Hudis CA. Combination of trastuzumab and tanespimycin (17-AAG, KOS-953) is safe and active in trastuzumab-refractory HER-2 overexpressing breast cancer: a phase I dose-escalation study. J Clin Oncol 2007, 25, 5410–5417.
- Egorin MJ, Rosen DM, Wolff JH, Callery PS, Musser SM, Eiseman JL. Metabolism of 17-(allylamino)-17-demethoxygeldanamycin (NSC 330507) by murine and human hepatic preparations. Cancer Res 1998, 58, 2385–2396.
- Kelland LR, Sharp SY, Rogers PM, Myers TG, Workman P. DT-Diaphorase expression and tumor cell sensitivity to 17-allylamino, 17-demethoxygeldanamycin, an inhibitor of heat shock protein 90. J Natl Cancer Inst 1999, 91, 1940–1949.
- Kaur G, Belotti D, Burger AM, Fisher-Nielson K, Borsotti P, Riccardi E, Thillainathan J, Hollingshead M, Sausville EA, Giavazzi R. Antiangiogenic properties of 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin: an orally bioavailable heat shock protein 90 modulator. Clin Cancer Res 2004, 10, 4813–4821.
- Ge J, Normant E, Porter JR, Ali JA, Dembski MS, Gao Y, Georges AT, Grenier L, Pak RH, Patterson J, Sydor JR, Tibbitts TT, Tong JK, Adams J, Palombella VJ. Design, synthesis, and biological evaluation of hydroquinone derivatives of 17-amino-17-demethoxygeldanamycin as potent, water-soluble inhibitors of Hsp90. J Med Chem 2006, 49, 4606–4615.
- Sydor JR, Normant E, Pien CS, Porter JR, Ge J, Grenier L, Pak RH, Ali JA, Dembski MS, Hudak J, Patterson J, Penders C, Pink M, Read MA, Sang J, Woodward C, Zhang Y, Grayzel DS, Wright J, Barrett JA, Palombella VJ, Adams J, Tong JK. Development of 17-allylamino-17-demethoxygeldanamycin hydroquinone hydrochloride (IPI-504), an anti-cancer agent directed against Hsp90. Proc Natl Acad Sci USA 2006, 103, 17408–17413.
- Chiosis G, Caldas Lopes E, Solit D. Heat shock protein-90 inhibitors: a chronicle from geldanamycin to today’s agents. Curr Opin Investig Drugs 2006, 7, 534–541.
- DeBoer C, Meulman PA, Wnuk RJ, Peterson DH. Geldanamycin, a new antibiotic. J Antibiot 1970, 23, 442–447.
- Supko JG, Hickman RL, Grever MR, Malspeis L. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother Pharmacol 1995, 36, 305–315.
- Neckers L, Schulte TW, Mimnaugh E. Geldanamycin as a potential anti-cancer agent: its molecular target and biochemical activity. Invest New Drugs 1999, 17, 361–373.
- Soga S, Shiotsu Y, Akinaga S, Sharma SV. Development of radicicol analogues. Curr Cancer Drug Targets 2003, 3, 359–369.
- Chiosis G, Kang Y, Sun W. Discovery and development of purine-scaffold Hsp90 inhibitors. Expert Opinion on Drug Discov 2008, 3, 99–114.
- McDonald E, Jones K, Brough PA, Drysdale MJ, Workman P. Discovery and development of pyrazole-scaffold Hsp90 inhibitors. Curr Top Med Chem 2006, 6, 1193–1203.
- Taldone T, Gozman A, Maharaj R, Chiosis G. Targeting Hsp90: small-molecule inhibitors and their clinical development. Curr Opin Pharmacol 2008, 8, 370–374.
- Messaoudi S, Peyrat JF, Brion JD, Alami M. Recent advances in Hsp90 inhibitors as antitumor agents. Anticancer Agents Med Chem 2008, 8, 761–782.
- Biamonte MA, Van de Water R, Arndt JW, Scannevin RH, Perret D, Lee WC. Heat shock protein 90: inhibitors in clinical trials. J Med Chem 2010, 53, 3–17.
- Ganesh T, Thepchatri P, Li L, Du Y, Fu H, Snyder JP, Sun A. Synthesis and SAR study of N-(4-hydroxy-3-(2-hydroxynaphthalene-1-yl)phenyl)-arylsulfonamides: heat shock protein 90 (Hsp90) inhibitors with submicromolar activity in an in vitro assay. Bioorg Med Chem Lett 2008, 18, 4982–4987.
- Barril X, Brough P, Drysdale M, Hubbard RE, Massey A, Surgenor A, Wright L. Structure-based discovery of a new class of Hsp90 inhibitors. Bioorg Med Chem Lett 2005, 15, 5187–5191.
- Abel G, Connors TA, Goddard P, Hoellinger H, Nguyen-Hoang-Nam, Pichat L, Ross WC, Wilman DE. Cytotoxic sulphonamides designed for selective deposition in malignant tissue. Eur J Cancer 1975, 11, 787–793.
- Casini A, Scozzafava A, Mastrolorenzo A, Supuran LT. Sulfonamides and sulfonylated derivatives as anticancer agents. Curr Cancer Drug Targets 2002, 2, 55–75.
- Avila C, Hadden MK, Ma Z, Kornilayev BA, Ye QZ, Blagg BS. High-throughput screening for Hsp90 ATPase inhibitors. Bioorg Med Chem Lett 2006, 16, 3005–3008.
- Lanzetta PA, Alvarez LJ, Reinach PS, Candia OA. An improved assay for nanomole amounts of inorganic phosphate. Anal Biochem 1979, 100, 95–97.
- Christopher A, Boris A K, Brian S J. Development and optimization of a useful assay for determining Hsp90s inherent ATPase activity. Bioorg Med Chem 2006, 14, 1134–1142.
- Venkatachalam CM, Jiang X, Oldfield T, Waldman M. LigandFit: a novel method for the shape-directed rapid docking of ligands to protein active sites. J Mol Graph Model 2003, 21, 289–307.
- Böhm HJ. The development of a simple empirical scoring function to estimate the binding constant for a protein–ligand complex of known three-dimensional structure. J Comput Aided Mol Des 1994, 8, 243–256.
- Muegge I. A knowledge-based scoring function for protein–ligand interactions: probing the reference state. Perspect Drug Discovery Des 2000, 20, 99–114.
- Muegge I, Martin YC. A general and fast scoring function for protein–ligand interactions: a simplified potential approach. J Med Chem 1999, 42, 791–804.
- Muegge I. Effect of ligand volume correction on PMF scoring. J Comput Chem 2001, 22, 418–425.
- Böhm HJ. Prediction of binding constants of protein ligands: a fast method for the prioritization of hits obtained from de novo design or 3D database search programs. J Comput Aided Mol Des 1998, 12, 309–323.
- Jain AN. Scoring noncovalent protein–ligand interactions: a continuous differentiable function tuned to compute binding affinities. J Comput Aided Mol Des 1996, 10, 427–440.
- Rowlands YM, Newbatt CP, Laurence HP, Paul W, Wynne A. High-throughput screening assay for inhibitors of heat-shock protein 90 ATPase activity. Analyt Biochem 2004, 327, 176–183.
- Matts RL, Manjarrez JR. Assays for identification of Hsp90 inhibitors and biochemical methods for discriminating their mechanism of action. Curr Top Med Chem 2009, 9, 1462–1478.
- Schulz-Gasch T, Stahl M. Scoring functions for protein–ligand interactions: a critical perspective. Drug Discov Today 2004, 1, 231–239.
- Tame JR. Scoring functions: a view from the bench. J Comput Aided Mol Des 1999, 13, 99–108.
- Krovat EM, Langer T. Impact of scoring functions on enrichment in docking-based virtual screening: an application study on renin inhibitors. J Chem Inf Comput Sci 2004, 44, 1123–1129.
- Clark RD, Strizhev A, Leonard JM, Blake JF, Matthew JB. Consensus scoring for ligand/protein interactions. J Mol Graph Model 2002, 20, 281–295.
- Miklos F. Consensus scoring for protein–ligand interactions. Drug Discov Today, 2006, 11, 421–428.
- Song CM, Lim SJ, Tong JC. Recent advances in computer-aided drug design. Brief Bioinformatics 2009, 10, 579–591.
- Jorgensen WL. Efficient drug lead discovery and optimization. Acc Chem Res 2009, 42, 724–733.
- Leach AR, Shoichet BK, Peishoff CE. Prediction of protein–ligand interactions. Docking and scoring: successes and gaps. J Med Chem 2006, 49, 5851–5855.
- Krovat EM, Langer T. Impact of scoring functions on enrichment in docking-based virtual screening: an application study on renin inhibitors. J Chem Inf Comput Sci 2004, 44, 1123–1129.
- Klebe G. Virtual ligand screening: strategies, perspectives and limitations. Drug Discov Today 2006, 11, 580–594.
- Krissinel, E. Crystal contacts as nature’s docking solutions. J Comput Chem 2009, 31, 133–143.
- Clarke’s Analysis of Drugs and Poisons, © Pharmaceutical Press, Monographs, 2005.
- Senem S A, Yüksel A, Nurullah S, Güleren A, Jose L B. Solvent Effects on pKa values of some substituted sulfonamides in acetonitrile–water binary mixtures by the UV-spectroscopy method. J Chem Eng Data 2009, 54, 3014–3021.