Abstract
A series of bis-N,N-(2-hydroxyethyl)glycine (bicine) derivatives, conjugated with an inhibitor of glucosamine-6-phosphate synthase, have been synthesized and their lipophilic and antifungal properties have been tested. The obtained compounds demonstrated higher lipophilicity than free inhibitor (FMDP) and, in consequence, an increased potential to cross the cytoplasmic membrane. All the tested compounds show better antifungal activity than parent compound.
Keywords::
Introduction
Infections caused mainly by human pathogenic fungi are regarded as one of the most important problems to be solved in modern chemotherapy. Only a very limited number of antifungal chemotherapeutics are in clinical use.Citation1 One of the possible solutions to overcome this problem is to consider an exploitation of new antifungal targets, for example enzymes involved in the biosynthesis pathway of the fungal cell wall.Citation2 Glucosamine-6-phosphate synthase (GlcN-6-P synthase, EC 2.6.1.16)Citation3 catalyses the formation of D-glucosamine-6-phosphate and therefore is one of the enzymes required for the biosynthesis of glucosamine-containing cell wall macromolecules:lipopolysaccharides and peptidoglycan in bacteria and mannoproteins and chitin in fungi.Citation4 Inhibition of this enzyme in fungal cells leads to morphological changes and lysis,Citation5,Citation6 whereas in mammalian cells temporary depletion of enzyme activity is not lethal due to long half-life of GlcN-6-P synthase and rapid expression of the genes encoding it.Citation7 Thus, GlcN-6-P synthase has been proposed as a potential target for designing new antimicrobial agents.Citation8 There are many inhibitors of GlcN-6-P synthase that are analogues of glutamine. Representative for this group of compounds is N3-(4-methoxyfumaroyl)-l-2,3-diaminopropanoic acid (FMDP), one of the most potent and selective inhibitors of the enzyme.Citation9,Citation10 FMDP is a non-peptide amino acid, therefore is poorly transported into fungal cells by rather specific amino acid permeases and exhibits only moderate antifungal activity. High polarity of the inhibitor effectively hampered it transport inside the cells. In order to overcome this problem, we have modified FMDP molecule, aimed at the construction latent and lipophilic derivatives that might be able to penetrate into the cells by free diffusion. Following uptake, the modifying group could be removed intracellularly. Modification of the carboxyl group involves the formation of latent esters, such as acetoxymethyl ester.Citation11 That approach is very common in penicillin group.Citation12 On the other hand, formation of prodrugs by modification of the amino group is relatively rare and difficult.Citation13 In our approach, we have tried to apply bicine, that is, bis-N,N-(2-hydroxyethyl)glycine as an acylating agent of FMDP molecule. Bicine amide underwent, at physiological pH, a facilitated cyclization (with t1/2 = 3 h) to a 4-(2-hydroxyethyl)morpholin-2-one with the releasing of ammonia molecule. Formation of the six-membered ring is the driving force in this reaction. The t1/2 for hydrolysis of glycinamide at physiological pH was determined to be ~7 years.Citation14 Therefore, the amide bond between bicine and FMDP should be cleaved as a result of intramolecular alcoholysis, thus leading to the formation of FMDP and 4-(2-hydroxyethyl)morpholin-2-one. To increase stability and lipophilicity of prodrug, esterification of hydroxyl groups in bicine was carried out, according to the published procedure.Citation15 The fully modified FMDP prodrug should penetrate into the fungal cells by free diffusion, and inside, the ester bonds should be cleaved by intracellular enzymes followed by cyclization of the bicine residue to a 4-(2-hydroxyethyl)morpholin-2-one and generation of free FMDP (). As a consequence of that reaction, inhibition of fungal cells should be observed. Moreover, we have also evaluated lipophilicity of the novel prodrugs by measuring their affinity to biological membranes.
Experimental
Chemistry
All reagents were purchased from Aldrich Chemical Co. Thin layer chromatography (TLC) was performed using Merck aluminum backed plates (Kieselgel 60 F254) and visualized by ultraviolet (UV) light. Separations by column chromatography were achieved using silica gel (0.063–0.200 mm). MS spectrum was recorded on a Quadrupolic Mass Spectrophotometer Trio-3 (FAB technique). 1H NMR spectra were recorded on a Varian Unity Plus spectrometer operating at 500 MHz using deuterated solvent (CDCl3). Chemical shifts are given in part per million (ppm) relative to internal standard tetramethylsilane.
Tert-butyl ester of bis-N,N-(2-hydroxyethyl)glycine (1)
A solution of tert-butyl bromoacetate (5.40 g, 27.70 mmol) and diethanolamine (11.60 g, 110.80 mmol) in anhydrous methylene chloride (DCM, 250 mL) was stirred at room temperature for 18 h. The reaction mixture was washed with water (4 × 50 mL), and the organic layer dried over anhydrous sodium sulphate, followed by filtration and removal of the solvent in vacuo to yield 1 (4.05 g, 67%). 1H NMR (500 MHz, CDCl3): δ 1.45 (s, 9H, C(CH3)3), 2.89 (t, 4H, J = 4.4 Hz, 2x(CH2N)). MS (FAB) m/z: 220 (MH+).
General method for the preparation of bis-N,N-(2-acyloxyethyl)glycine trifluoroacetate (3a–d)
A solution of compound 1 (0.20 g, 0.91 mmol), 4,4′-(dimethylamino)pyridine (DMAP, 0.11 g, 0.91 mmol) and appropriate carboxylic acid (0.91 mmol) in anhydrous DCM (15 mL) was cooled to 0°C. Dicyclohexylcarbodiimide (DCC, 0.206 g, 1.00 mmol) was added and the reaction mixture was stirred at 0°C for 30 min and then at room temperature for 12 h. The reaction mixture was filtered, the filtrate was washed with 0.5 N NaHCO3 (3 × 10 mL), water (10 mL) and dried (MgSO4). The solvent was evaporated in vacuo. Tert-butyl ester of bis-N,N-(2-acyloxyethyl)glycine (2a–d) was further purified by silica gel column chromatography. Compounds 2a–d were treated with TFA for 4 h at room temperature. Excess TFA was evaporated in vacuo, the residue was triturated with diethyl ether and the precipitate was filtered off and dried in vacuo over KOH pellets.
Bis-N,N-(2-acetoxyethyl)glycine tifluoroacetate (3a)
Tert-butyl ester of bis-N,N-(2-acetoxyethyl)glycine (2a) was purified by column chromatography on silica gel (ethyl acetate:petroleum ether, 1:7) to give 2a (0.25 g, 91%). Compound 3a was obtained as waxy solid (0.29 g, 97%). 1H NMR (200 MHz, CDCl3): δ 2.05 (s, 6H, 2x(CH3C(O))); 3.01 (t, 4H, J = 5.8 Hz, 2x(CH2N)); 3.41 (s, 2H, NH2C(O)); 4.15 (t, 4H, J = 5.8 Hz, 2x(OCH2)). MS(FAB) m/z: 248 (MH+).
Bis-N,N-(2-propionyloxyethyl)glycine tifluoroacetate (3b)
Tert-butyl ester of bis-N,N-(2-propionyl)glycine (2b) was purified by column chromatography on silica gel (ethyl acetate:petroleum ether, 1:8) to give 2b (0.23 g, 79%). Compound 3b was obtained as waxy solid (0.265 g, 95%).Citation1H NMR (500 MHz, CDCl3): δ 1.13 (t, 6H, J = 7.5 Hz, 2x(CH3)); 2.33 (q, 6 H, J = 7.5 Hz, 2x(CH2)); 3.00 (m, 4H, 2x(CH2N)); 3.42 (s, 2H, NH2C(O)); 4.17 (m, 4H, 2x(OCH2)). MS(FAB) m/z: 276 (MH+).
Bis-N,N-(2-butyryloxyethyl)glycine tifluoroacetate (3c)
Tert-butyl ester of bis-N,N-(2-butyryloxyethyl)glycine (2c) was purified by column chromatography on silica gel (ethyl acetate:petroleum ether, 1:10) to give 2c (0.23 g, 73%). Compound 3c was obtained as waxy solid (0.269 g, 97%). 1H NMR (500 MHz, CDCl3): δ 0.94 (t, 6H, J = 7.4 Hz, 2x(CH3)); 1.66 (q, 4H, J = 7.4 Hz, 2x(CH2)); 2.28 (t, 4 H, J = 7.4 Hz, 2x(CH2C(O))); 2.98 (t, 4H, J = 6 Hz, 2x(CH2N)); 3.39 (s, 2H, NH2C(O)); 4.13 (t, 4H, J = 6 Hz, 2x(C(O)CH2)). MS (FAB) m/z: 304 (MH+).
Bis-N,N-(2-benzoyloxyethyl)glycine tifluoroacetate (3d)
Tert-butyl ester of bis-N,N-(2-benzoyloxyethyl)glycine (2d) was purified by column chromatography on silica gel (ethyl acetate:petroleum ether, 1:12) to give 2d (0.225 g, 58%). Compound 3d was obtained as waxy solid (0.23 g, 92%). 1H NMR (500 MHz, CDCl3): δ 3.25 (t, 4H, J = 5.8 Hz, 2x(CH2N)); 3.58 (s, 2H, NH2C(O)); 4.46 (t, 4H, J = 5.8 Hz, 2x(C(O)CH2)); 7.36–7.58 (m, 6H,); 7.99–8.05 (m, 4H). MS (FAB) m/z: 372 (MH+).
General method for the preparation of FMDP acyloxyalkyl esters (6a–f)
To a solution of BocFMDP (4) (2.00 g, 6.33 mmol), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 0.96 g, 6.33 mmol) and sodium iodide (0.35 g, 1.90 mmol) in anhydrous acetonitrile (25 mL), the corresponding 1-chloroalkyl ester (5a–f) was added (7.60 mmol) and the mixture was refluxed with stirring for 5 h. The solvent was evaporated, the residue was diluted with ethyl acetate, washed with 0.5 N NaHCO3 (2 × 20 mL), water (20 mL) and dried (MgSO4). The solvent was evaporated in vacuo and the residue was purified by silica gel column chromatography (ethyl acetate:petroleum ether, 1:2). The product was treated with TFA for 2 h at room temperature. Excess TFA was evaporated in vacuo, the residue was triturated with diethyl ether and the precipitate was filtered off, dried in vacuo over KOH pellets.
Acetoxymethyl ester of N3-(4-methoxyfumaroyl)-(L)-2,3-diaminopropanoic acid trifluoroacetate (6a)
Yield 1.42 g (56%). 1H NMR (500 MHz, CDCl3): δ 2.15 (s, 3H, (O)CCH3); 3.82 (s, 5H, OCH3, NCH2); 4.45 (m, 1H, CH); 5.81 (dd, 2H, J = 5 Hz, OCH2O); 6.90 (ABq, 2H, J = 15 Hz, CH=CH). MS(FAB) m/z: 389 (MH+).
(Isobutyryloxy)methyl ester of N3-(4-methoxyfumaroyl)-(L)-2,3-diaminopropanoic acid trifluoroacetate (6b)
Yield 1.58 g (58%). 1H NMR (500 MHz, CDCl3): δ 1.18 (d, 6H, J = 6.5 Hz, 2x(CH3)); 2.64 (m, 1H, CH); 3.80 (s, 5H, OCH3, NCH2); 5.75 (dd, 2H, J = 5 Hz, OCH2O); 6.86 (ABq, 2H, J = 15 Hz, CH=CH). MS (FAB) m/z: 417 (MH+).
(2-Phenylacetoxy)methyl ester of N3-(4-methoxyfumaroyl)-(L)-2,3-diaminopropanoic acid trifluoroacetate (6c)
Yield 1.91 g (63%). 1H NMR (500 MHz, CDCl3): δ 3.71 (s, 2H, CH2); 3.80 (s, 5H, OCH3, NCH2); 4.42 (m, 1H, CH); 5.75 (dd, 2H, J = 5 Hz, OCH2O); 6.86 (ABq, 2H, J = 15 Hz, CH=CH); 7.32 (m, 5H, Ar). MS (FAB) m/z: 465 (MH+).
[(2-Ethylbutanoyl)oxy]methyl ester of N3-(4-methoxyfumaroyl)-(L)-2,3-diaminopropanoic acid trifluoroacetate (6d)
Yield 2.17 g (75%).Citation1H NMR (500 MHz, CDCl3): δ 0.86 (t, 6H, J = 7.4 Hz, 2x(CH3)); 1.62 (m, 4H, 2x(CH2)); 2.35 (m, 1H, CH); 3.79 (s, 5H, OCH3, NCH2); 4.42 (m, 1H, CH); 5.78 (dd, 2H, J = 5.6 Hz, OCH2O); 6.88 (ABq, 2H, J = 15 Hz, CH=CH). MS (FAB) m/z: 445 (MH+).
1-Acetoxypropyl ester of N3-(4-methoxyfumaroyl)-(L)-2,3-diaminopropanoic acid trifluoroacetate (6e)
Yield 1.44 g (53%).Citation1H NMR (500 MHz, CDCl3): δ 0.99 (m, 3H, CH3); 1.84 (m, 2H, CH2); 2.11 (s, 3H, CH3C(O)); 3.80 (s, 5H, OCH3, NCH2); 4.45 (m, 1H, CH); 5.65 (m, 1H, OCHO); 6.86 (m, 2H, CH=CH). MS (FAB) m/z: 417 (MH+).
1-[(Ethoxycarbonyl)oxy]ethyl ester of N3-(4-methoxyfumaroyl)-(L)-2,3-diaminopropanoic acid trifluoroacetate (6f)
Yield 1.44 g (51%). 1H NMR (500 MHz, CDCl3): δ 1.35 (m, 3H, CH3); 1.58 (m, 2H, CH2); 3.80 (s, 5H, OCH3, NCH2); 4.28 (m, 2H, CH2); 4.45 (m, 1H, CH); 5.78 (m, 1H, OCHO); 6.86 (m, 2H, CH=CH). MS (FAB) m/z: 433 (MH+).
General method for the preparation of acyloxyalkyl ester of N2-(bis-N,N-(2-acyloxyethyl)glycinyl)-N3-(4-methoxyfumaroyl)-(L)-2,3-diaminopropanoic acid hydrochloride (8a–i)
A solution of bis-N,N-(2-acyloxyethyl)glycine trifluoroacetate (3a–d) (1 mmol), triethylamine (0.20 g, 2 mmol) and N-hydroxysuccinimide (0.114 g, 1 mmol) in tetrahydrofuran (THF) (10 mL) was cooled to 0°C and DCC (0.216 g, 1.05 mmol) was added. After 12 h, the dicyclohexylurea was filtered off and the filtrate was added drop wise to solution of corresponding acyloxyalkyl ester of FMDP (1 mmol) and triethylamine (0.20 g, 2 mmol) in THF (7 mL). The mixture was stirred at 0°C for 30 min, and then at room temperature for 14 h. The solvent was evaporated, the residue was dissolved in ethyl acetate (20 mL), washed with 0.5 N NaHCO3 (2 × 15 mL), water (10 mL) and dried (MgSO4). The solvent was evaporated in vacuo and the residue was purified by silica gel column chromatography (ethyl acetate:petroleum ether, 3:1). The product was dissolved in ethyl acetate and equimolar amount of 2.00 M HCl in diethyl ether was added. The precipitate was filtered off and dried in vacuo over KOH pellets.
Acetoxymethyl ester of N2-(bis-N,N-(2-acetoxyethyl)glycinyl)-N3-(4-methoxyfumaroyl)-(L)-2,3-diaminopropanoic acid hydrochloride (8a)
Yield 0.23 g (41%). 1H NMR (500 MHz, CDCl3): δ 2.09 (s, 6H, 2x(CH3C(O)); 2.12 (s, 3H, CH3C(O)); 2.88 (m, 4H, 2x(CH2N)); 3.31 (d, 2H, J = 5.4 Hz, NCH2C(O)); 3.78 (s, 5H, OCH3, HNCH2); 4.10–4.35 (m, 4H, 2x((CH2OC(O)); 4.70 (m, 1H, NHCH); 5.75 (dd, 2H, J = 5.6 Hz, OCH2O); 6.90 (ABq, 2H, J = 15.4 Hz, CH=CH). MS (FAB) m/z: 518 (MH+).
Acetoxymethyl ester of N2-(bis-N,N-(2-propionyloxyethyl)glycinyl)-N3-(4-methoxyfumaroyl)-(L)-2,3-diaminopropanoic acid hydrochloride (8b)
Yield 0.28 g (48%). 1H NMR (500 MHz, CDCl3): δ 1.13 (t, 6H, J = 7.5 Hz, 2x(CH3); 2.12 (s, 3H, CH3C(O)); 2.36 (q, 4H, J = 7.5 Hz, 2x(CH2C(O))); 2.88 (m, 4H, 2x(CH2N)); 3.29 (s, 2H, NCH2C(O)); 3.78 (s, 5H, OCH3, HNCH2); 4.15–4.35 (m, 4H, 2x((CH2OC(O)); 4.70 (m, 1H, NHCH); 5.75 (dd, 2H, J = 5.6 Hz, OCH2O); 6.90 (ABq, 2H, J = 15.4 Hz, CH=CH). MS (FAB) m/z: 546 (MH+).
Acetoxymethyl ester of N2-(bis-N,N-(2-butyryloethyl)glycinyl)-N3-(4-methoxyfumaroyl)-(L)-2,3-diaminopropanoic acid hydrochloride (8c)
Yield 0.28 g (46%). 1H NMR (500 MHz, CDCl3): δ 0.94 (t, 6H, J = 7.3 Hz, 2x(CH3)); 1.64 (m, 4H, 2x(CH2)); 2.12 (s, 3H, CH3C(O)); 2.32 (t, 4 H, J = 7.5 Hz, 2x(CH2C(O))); 2.89 (t, 4H, J = 5.6 Hz 2x(CH2N)); 3.31 (s, 2H, NCH2C(O)); 3.78 (s, 5H, OCH3, HNCH2); 4.14–4.30 (m, 4H, 2x((CH2OC(O)); 4.72 (m, 1H, NHCH); 5.76 (dd, 2H, J = 5.6 Hz, OCH2O); 6.88 (ABq, 2H, J = 15.4 Hz, CH=CH). MS (FAB) m/z: 574 (MH+).
Acetoxymethyl ester of N2-(bis-N,N-(2-benzoyloxyethyl)glycinyl)-N3-(4-methoxyfumaroyl)-(L)-2,3-diaminopropanoic acid hydrochloride (8d)
Yield 0.28 g (41%). 1H NMR (500 MHz, CDCl3): δ 2.09 (s, 3H, CH3C(O)); 3.18 (t, J = 5.4 Hz, 4H, 2x(CH2N)); 3.53 (s, 2H, NCH2C(O)); 3.72 (s, 5H, OCH3, HNCH2); 4.53 (m, 4H, 2x((CH2OC(O)); 4.58 (m, 1H, NHCH); 5.83 (dd, 2H, J = 5.6 Hz, OCH2O); 6.82 (ABq, 2H, J = 15.4 Hz, CH=CH); 7.35–7.90 (m, 10H). MS (FAB) m/z: 642 (MH+).
(Isobutyryloxy)methyl ester of N2-(bis-N,N-(2-acetoxyethyl)glycinyl)-N3-(4-methoxyfumaroyl)-(L)-2,3-diaminopropanoic acid hydrochloride (8e)
Yield 0.23 g (40%). 1H NMR (500 MHz, CDCl3): δ 1.16 (d, 6H, J = 6.5 Hz, 2x(CH3)); 2.09 (s, 6H, 2x(CH3C(O))), 2.69 (m, 1H, CH); 2.98 (m, 4H, 2x(CH2N)); 3.75 (s, 5H, OCH3, HNCH2) 4.10–4.40 (m, 4H, 2x((CH2OC(O)); 4.69 (m, 1H, NHCH); 5.78 (dd, 2H, J = 5.4 Hz, OCH2O); 6.89 (ABq, 2H, J = 15.4 Hz, CH=CH). MS (FAB) m/z: 546 (MH+).
(2-Phenylacetoxy)methyl ester of N2-(bis-N,N-(2-acetoxyethyl)glycinyl)-N3-(4-methoxyfumaroyl)-(L)-2,3-diaminopropanoic acid hydrochloride (8f)
Yield 0.23 g (37%). 1H NMR (500 MHz, CDCl3): δ 2.09 (s, 6H, 2x(CH3C(O))); 3.05 (m, 4H, 2x(CH2N)), 3.71 (s, 2H, CH2); 3.78 (s, 5H, OCH3, HNCH2); 4.10–4.40 (m, 4H, 2x((CH2OC(O)); 4.65 (m, 1H, NHCH), 5.75 (dd, 2H, J = 5.4 Hz, OCH2O); 6.88 (ABq, 2H, J = 15.4 Hz, CH=CH). MS (FAB) m/z: 594 (MH+).
[(2-Ethylbutanoyl)oxy]methyl ester of N2-(bis-N,N-(2-acetoxyethyl)glycinyl)-N3-(4-methoxyfumaroyl)-(L)-2,3-diaminopropanoic acid hydrochloride (8g)
Yield 0.26 g (43%). 1H NMR (500 MHz, CDCl3): δ 0.87 (t, 6H, J = 7.4 Hz, 2x(CH3)); 1.57 (m, 6H, 2x(CH2)); 2.09 (s, 6H, 2x(CH3C(O)); 2.27 (m, 1H, CHC(O)); 2.89 (m, 4H, 2x(CH2N)); 3.30 (m, 2H, NCH2C(O)); 3.78 (s, 5H, OCH3, HNCH2); 4.10–4.40 (m, 4H, 2x((CH2OC(O)); 4.70 (m, 1H, NHCH); 5.80 (dd, 2H, J = 5.5 Hz, OCH2O); 6.90 (ABq, 2H, J = 15.6 Hz, CH=CH). MS (FAB) m/z: 574 (MH+).
1-Acetoxypropyl ester of N2-(bis-N,N-(2-acetoxyethyl)glycinyl)-N3-(4-methoxyfumaroyl)-(L)-2,3-diaminopropanoic acid hydrochloride (8h)
Yield 0.24 g (41%). 1H NMR (200 MHz, CDCl3): δ 0.97 (m, 3H, CH3); 1.82 (m, 2H, CH2); 2.11 (s, 3H, CH3C(O)); 2.13 (s, 6H, 2x(CH3C(O)); 2.92 (m, 4H, 2x(CH2N)); 3.40 (m, 2H, NCH2C(O)); 3.79 (s, 5H, OCH3, HNCH2); 4.10–4.35 (m, 4H, 2x((CH2OC(O)); 4.69 (m, 1H, NHCH); 6.70 (m, 2H, OCHO); 6.80 (m, 2H, CH=CH). MS (FAB) m/z: 546 (MH+).
1-[(Ethoxycarbonyl)oxy]ethyl ester of N2-(bis-N,N-(2-acetoxyethyl)glycinyl)-N3-(4-methoxyfumaroyl)-(L)-2,3-diaminopropanoic acid hydrochloride (8i)
Yield 0.23 g (38%). 1H NMR (500 MHz, CDCl3): δ 1.32 (m, 3H, CH3); 1.58 (m, 2H, CH2); 2.09 (s, 6H, 2x(CH3C(O)); 2.89 (m, 4H, 2x(CH2N)); 3.28 (m, 2H, NCH2C(O)); 3.75 (s, 5H, OCH3, HNCH2); 4.10–4.35 (m, 4H, 2x((CH2OC(O)); 4.22 (m, 2H, CH2); 4.71 (m, 1H, NHCH); 6.75 (m, 1H, OCHO); 6.90 (m, 2H, CH=CH). MS (FAB) m/z: 562 (MH+).
Determination of the affinity to the artificial biological membrane
Interactions between the tested compounds and immobilized artificial membrane were investigated using HPLC column IAM.PC.DD2 (Regis Technologies, Inc., Morton Grove, IL).Citation16 The column dimensions were 3 cm × 4.6 mm, particle diameter 10 μm and pore width 300 Å. The chromatographic system consisted of a UV/vis detector Model G1315B, Vacuum degasser Model G1322A, Quaternary pump Model G1311A (Agilent Technologies 1200 Series, Palo Alto, CA) . 0.1 M Sörensen buffer (K2HPO4/KH2PO4, pH 7.2)/acetonitrile was used in proportions 50:50, 60:40, 65:35, 70:30, 75:25, 80:20 (v/v) as mobile phase. The injection volume was 10 μL; the flow rate was 1 mL/min; the samples were detected at 254 nm. The dead volume of the column was determined by the retention time of citric acid (aqueous solution, 50 μg/mL) and was used to calculate capacity factors k′IAM. The dependence of logk′IAM versus concentration of acetonitrile was linear. The logk′IAM,0 was obtained by extrapolation of logarithmic capacity factor to concentration of acetonitrile equal to 0. A standard, commercially available statistical package for regression analysis was applied on a personal computer.
Antifungal susceptibility tests
Minimal inhibitory concentrations (MICs) of the examined compounds were determined by the serial 2-fold dilution microtitre plate method, in the minimal liquid yeast nitrogen base (YNB) medium without amino acids and ammonium sulphate containing 2% glucose and l-proline (4 mg/mL). Wells containing serially diluted test compounds and control were inoculated with 104 cells/mL of an overnight culture of fungal cells and the microtitre plates were incubated for 24 h at 30°C. Fungal growth was measured using the microplate reader (Labsystem Multiscan Biochromatic) Please provide city and state for Labsystem Multiscan Biochromatic at λ = 595 nm. The MIC was defined as the inhibitor concentration preventing at least 80% of fungal growth, as compared with the inhibitor-free control.
Results and discussion
Chemistry
The syntheses of compounds 3a–d () was started with preparation of tert-butyl ester of bis-N,N-(2-hydroxyethyl)glycine (1). Compound 1 was prepared by a displacement reaction using tert-butyl bromoacetate. Condensation of compound 1 with corresponding carboxylic acids was carried out in the presence of DCC/DMAPCitation17 to give appropriate tert-butyl ester of bis-N,N-(2-acyloxyethyl)glycine (2a–d). Deprotection of the carboxyl group was performed in anhydrous trifluoroacetic acid. The bicine derivatives 3a–d were prepared as trifluoroacetate salts. In the case of the FMDP acyloxyalkyl esters synthesis (), BocFMDP (4) was treated with an appropriate chloroalkyl estersCitation18 in the presence of DBU and NaI.Citation19 The reaction proceeds smoothly in a polar solvent such as acetonitrile. The products were purified by silica gel column chromatography with ethyl acetate:petroleum ether (1:2) as a mobile phase. After deprotection of the amino group using anhydrous TFA in CH2Cl2, compounds 6a–f were obtained as hygroscopic trifluoroacetate salts. The synthesis of compounds 8a–i was performed using standard synthetic procedures (). The compounds 3a–d were activated with N-hydroxysuccinimide (HOSu) and DCCCitation20 to give the active esters (7a–d) and condensed with appropriate acyloxyalkyl esters of FMDP (5a–f). The products were purified by silica gel column chromatography with ethyl acetate:petroleum ether (3:1) as a mobile phase. Using anhydrous hydrochloride in diethyl ether, compounds 8a–i were obtained as amorphous hydrochloride salts.
Scheme 1. Syntheses of bis-N,N-(2-acyloxyethyl)glycine trifluoroacetate (3a–d). (i) diethanolamine, CH2Cl2; (ii) RCOOH, DMAP, DCC and (iii) TFA.
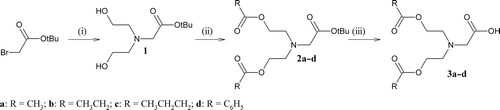
Scheme 2. Syntheses of FMDP acyloxyalkyl esters. (i) CH3CN, DBU, NaI. * denotes salt formation.
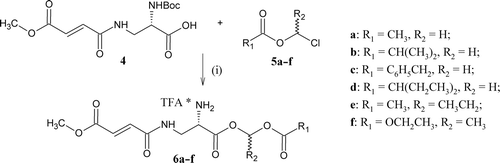
Scheme 3. Syntheses of acyloxyalkyl ester of N2-(bis-N,N-(2-acyloxyethyl)glycinyl)-N3-(4-methoxyfumaroyl)-(l)-2,3-diaminopropanoic acid hydrochloride (8a–i), (i) HOSu/DCC and (ii) HCl/Et2O and hypothetical degradation pathway. * denotes salt formation.
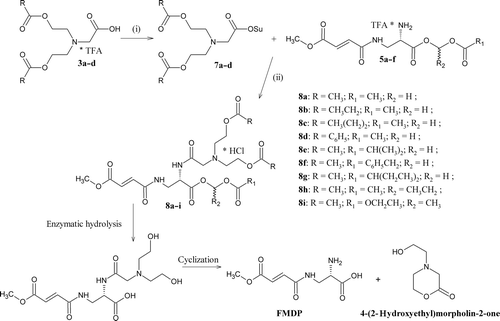
Membrane affinity
The affinity of a drug for biological membrane is an important factor facilitating its diffusion into fungal cell. For compounds 8a–i, their interaction with a biological membrane model was investigated using an HPLC chromatographic column immobilized artificial membrane (IAM) PC.DD 2 with stationary phase mimicking the cell membrane. The retention time determined for the examined compounds was expressed as logk′IAM and extrapolated to logk′IAM,0. The highest logk′IAM,0 value was determined for compound 8d (containing an aromatic and methyl substituents), whereas the lowest one was determined for compound 8a (with only methyl substituents). These values comprise not only the lipophilic properties but also other interactions with membrane such as hydrogen bond formation or electrostatic interactions. Therefore, the logk′IAM,0 values reflect rather membrane affinity than lipophilic properties.
Antifungal activity
MICs were determined in YNB medium with proline as carbon source. present activity against few species. Most of the synthesized compounds exhibited moderate antifungal activity in the range from 15.6 to 1000 μg/mL and their activity is not correlated with apparent lipophilicity. The lack of correlation between the logk′IAM,0 and MIC values may be attributed to the slow releasing of FMDP molecule from bicine conjugate. Compounds 8a–d displayed almost the same antifungal activity thus showing that the type of substituent in “R” position is not important for the activity. On the other hand, the change of latent ester of FMDP in compounds 8e–i (position “R1” and “R2”) improves antifungal activity of these compounds. This may be explained by different rates of intracellular hydrolysis of these compounds resulting in higher level of FMDP inside the cells. All the obtained compounds have better lipophilic properties than free inhibitor—FMDP. The logk′IAM,0 values between 0.91 and 2.11 show that compounds 8a–i may be able to cross the cell membrane by free diffusion.
Table 1. Antifungal activity of compounds 8a–i and their affinity to biological membrane.
Conclusion
We have synthesized a series of derivatives of bicine and glucosamine-6-phosphate synthase inhibitor. The modification of FMDP at the amino group improves lipophilic properties of obtained compounds. Compounds 8a–i are soluble in water and show better antifungal activity than the parent compound—FMDP.
Acknowledgments
This work was supported by the Ministry of Education and Science (Grant No. NN 405 1829 35). The HPLC instrument (which was used in this study) is a part of equipments of The Research Laboratory Center of Excellence “ChemBioFarm.” Its purchase was partially funded by the European Union Structural Funds under the Sectoral Operational Programme—Improvement of the Competitiveness of Enterprises.
Declaration of interest
The authors report no conflicts of interest.
References
- Sundriyal S, Sharma RK, Jain R. Current advances in antifungal targets and drug development. Curr Med Chem 2006;13:1321–1335.
- Andriole TA. Current and future antifungal therapy: new targets for antifungal agents. J Antimicrob Chemother 1999;44:151–162.
- Milewski S. Glucosamine-6-phosphate synthase—the multi-facets enzyme. Biochim Biophys Acta 2002;1597:173–192.
- Chmara H, Smulkowski M, Borowski E. Growth inhibitory effect of amidotransferase inhibition in Candida albicans by epoxy-peptides. Drugs Under Exptl Clin Res 1980;6:7–14.
- Milewski S, Chmara H, Borowski E. Antibiotic tetaine—a selective inhibitor of chitin and mannoprotein biosynthesis in Candida albicans. Arch Microbiol 1986;145:234–240.
- Badet B, Vermoote P, Le Goffic F. Glucosamine synthetase from Escherichia coli: kinetic mechanism and inhibition by N3-fumaroyl-l-2,3-diaminopropionic derivatives. Biochemistry 1988;27:2282–2287.
- Bates CJ, Adams WR, Handschumacher RE. Control of the formation of uridine diphospho-N-acetyl-hexosamine and glycoprotein synthesis in rat liver. J Biol Chem 1966;241:1705–1712.
- Borowski E. Novel approaches in the rational design of antifungal agents of low toxicity. Farmaco 2000;55:206–208.
- Andruszkiewicz R, Chmara H, Milewski S, Kasprzak L, Borowski E. Structural determinants of inhibitory activity of N3-(4-methoxyfumaroyl)-l-2,3-diaminopropanoic acid towards glucosamine-6-phosphate synthase. Pol J Chem 1993;67:673–683.
- Andruszkiewicz R, Chmara H, Milewski S, Borowski E. Synthesis of N3-fumaramoyl-l-2,3-diaminopropanoic acid analogues, the irreversible inhibitors of glucosamine synthetase. Int J Pept Protein Res 1986;27:449–453.
- Clayton JP, Cole M, Elson SW, Ferres H, Hanson JC, Mizen LW et al. Preparation, hydrolysis, and oral absorption of lactonyl esters of penicillins. J Med Chem 1976;19:1385–1391.
- Daehne W, Frederiksen E, Gundersen E, Lund F, Morch P, Petersen HJ et al. Acyloxymethyl esters of ampicillin. J Med Chem 1970;13:607–612.
- Greenwald RB, Zhao H, Yang K, Reddy P, Martinez A. A new aliphatic amino prodrug system for the delivery of small molecules and proteins utilizing novel PEG derivatives. J Med Chem 2004;47:726–734.
- Suggs JW, Pires RM. Facile hydrolysis and formation of amide bonds by N-hydroxyethylation of α-amino acids. Tetrahedron Lett 1997;38:2227–2230.
- Andruszkiewicz R, Koszel D. A simple access to O-acyl bicine. Pol J Chem 2007;81:295–298.
- Ong S, Liu H, Pidgeon C. Immobilized-artificial-membrane chromatography: measurements of membrane partition coefficient and predicting drug membrane permeability. J Chromatogr A 1996;728:113–128.
- Hassner A, Alexanian V. Direct room temperature esterification of carboxylic acids. Tetrahedron Lett 1978;46:4475–4478.
- Ulich LH, Adams R. The reaction between acid halides and aldehydes. J Am Chem Soc 1921;43:660–667.
- Ono N, Yamada T, Saito T, Tanaka K, Kaji A. A convenient procedure for esterification of carboxylic acids. Bull Chem Soc JPN 1978;51:2401–2404.
- Anderson GW, Zimmerman JE, Callahan FM. The use of esters of N-hydroxysuccinimide in peptide synthesis. J Am Chem Soc 1964;86:1839–1842.