Abstract
Several non-peptide heterocyclic compounds reported as potent thrombin inhibitors in vitro were chosen to carry out a QSAR study upon them using MLR and ANN analysis. In order to identify the best QSAR models, the input for ANN consisted of those subsets of descriptors used in the MLR models. The best QSAR models contained the SIC0 descriptor as the main topological descriptor. To identify the physical and chemical properties involved in the ligand–thrombin complexes, an automated ligand-flexible docking procedure was used. The docking results suggest that the thrombin inhibition by these heterocyclic compounds is driven by π–π, hydrogen bonds and salt bridge interactions. The best Gibbs free energy of ligand binding was found at the thrombin sites S1 and D. We have shown that it is possible to build MLR models with geometries taken from two different sources (semi-empirical and MD geometries) and obtain similar results.
Introduction
Thrombin is a multifunctional serine protease that plays a central role in coordinating the processes of blood coagulationCitation1, inflammation and wound repair following tissue injuryCitation2–7. Recently, the search for a potent and orally administrated thrombin inhibitor for antithrombotic therapy has been one of the main challenges in medicinal chemistry since several problems were found in the early inhibitor types: for example, lack of selectivity, side effects or low oral bioavailabilityCitation8. These inhibitors possessed reactive functional groups such as aldehydes, activated ketones or boronic acidsCitation8.
The non-peptide thrombin inhibitors with a heterocyclic core working as molecules with oral bioavailability gained special attention during the late 1990s decadeCitation9–13. Recently, Ries et al.Citation14,Citation15 have designed and developed orally active coagulation inhibitors with remarkably low molecular weight containing heterocyclic core structures as potent thrombin inhibitors in vitro. This compound design was based on the benzimidazole lead structure (), by synthesis of alternative heterocyclic analogues and testing the activity. Through QSAR analysis, a wide variety of descriptors including constitutional, topological and quantum chemical have been reportedCitation16–20, which have aided to develop knowledge-based drug-design methodologies.
Figure 1. Benzimidazole lead structure.
Three recent papers proposed new methodologies that can be used to correlate the binding affinity data of several thrombin inhibitors with their molecular structure. Nicolotti et al.Citation21 reported an approach to rationally interface structure- and ligand-based drug design through the automated generation of molecular docking alignments for 3D-QSAR using a genetic algorithm in a large series of 3-amidinophenylalanine inhibitors of thrombin. Fukunishi et al.Citation22 proposed the hypothesis that “a model of active compound can be provided when integrating information of compounds high-ranked by docking simulation of a random compound library” where the inclusion of the true active compound in the high-ranked compounds is not necessary. Finally, Bonachéra and HorvathCitation23 reported the use of topological fuzzy pharmacophore triplets to establish and validate linear and non-linear QSAR models of a set of thrombin inhibitor models; in spite of the apparent success of the above mentioned methods, Nicolotti’s approach might be burdensome in terms of computational time, in the Fukunishi’s approach the QSAR model still provides higher performances than their pseudo structure–activity relationship (PSAR) models and in Bonachéra and Horvath’s approach their model revealed that some selected triplets make sense; however, their equations were unable to predict the activity of structurally different ligands.
Multiple linear regression (MLR) is still advantageous in building models with improved fit in the training set when the number of descriptors is smallCitation24; further improvement can be achieved using artificial neural networks (ANN) a non-parametric non-linear modelling technique. The flexibility of ANN models for discovering more complex relationships has found a wide application in QSAR studiesCitation25. Our working group has published some QSAR modelsCitation26–28 and also docking studiesCitation29. Given that docking simulations yield important structural details about the ligand recognition on protein targetCitation29, one can use the docking methodologyCitation30 to corroborate the QSAR analysis results and to identify the recognition details of the ligand–target complex such as in pharmacophore groupsCitation31. Furthermore, computational docking is an efficient tool that has been applied in studies for structure-based drug designCitation32,Citation33.
Generally speaking, the energy in docking studies is calculated as a function of five terms that include:
where the five ΔG terms on the right-hand side are coefficients empirically determined using linear regression analysis from a set of protein–ligand complexes with known binding constantsCitation30. The summations are performed over all pairs of ligand atoms, i, and protein atoms, j. The in vacuo contributions include three interaction energy terms: a Lennard-Jones 12-6 dispersion–repulsion term, a directional 12-10 hydrogen-bonding term and a screened Coulombic electrostatic potential. The four terms correspond to the number of sp3 bonds in the ligand whereas the last term corresponds to the solvatation parameters, which are very important due to ligand recognition on proteins, which is carried out in the water environmentCitation30.
In the present study, we have carried out a QSAR analysis over 50 thrombin inhibitor molecules ()Citation14,Citation15,Citation34 in order to derive a quantitative relationship between the thrombin inhibitory effects (IC50, µM) and their chemical structural properties by MLR, ANN and docking studies. For these compounds, we have calculated a wide variety of descriptors such as quantum chemical, topological, Galvez topological charge index, 2D autocorrelation, RDF, 3D-MoRSE, WHIM and GETAWAY. In MLR analysis, the Leave-One-Out (LOO) and Leave-Group-Out (LGO) tests were used to select the most relevant QSAR models. A chance correlation study was performed on the optimal model via randomization test. The input for the ANN consisted of those subsets of descriptors used in different MLR models. Finally, an automated ligand-flexible docking (Autodock 4.0.1) was used to identify the chemical properties involved in the ligand–thrombin complexes.
Table 1. Molecules considered in this study.
Methodology
Activity data and descriptor generation
There are at least 50 compounds that have shown thrombin inhibitory effect on factor XA (IC50) in vitro, the reported data were taken from refs. (Citation14,Citation15,Citation34). These compounds are characterized for containing several functional groups like quinoline, quinoxalinone, sulphonamides, carboxamide, aromatic rings, both hydrogen bond donating/accepting groups and an amine group.
All molecular structures were built as neutral species with the HYPERCHEM softwareCitation35. The molecular geometries were optimized under the PM3 semi-empirical approachCitation36 coupled to the Eigenvector following methodCitation37 included in MOPAC 06 packageCitation38. A conformational analysis was carried out to localize the global minimum of each molecule. Frequency calculations were performed to confirm the nature of the stationary point. Vibrational frequencies were unscaled.
In this study, a total of 1488 molecular descriptors were calculated, 1481 molecular descriptors belonging to 18 different types of theoretical descriptors were calculated for each molecule using the DRAGON softwareCitation39, and the rest seven quantum chemical descriptors were calculated using PM3 semi-empirical approach.
Data reduction, MLR and ANN modelling
The calculated descriptors were analysed for the existence of constant or near constant variables in about 85% of their elementsCitation40, and those found were eliminated. After this process, 1195 descriptors were selected. In addition, for each one of the analysed data sets (vide infra) the correlation among the descriptors was investigated. Those descriptor pairs with high collinear relationship (r ≥ 0.90) were eliminated, leaving 366 descriptors to work with. This process was performed with a subroutine written in MATLAB (Version 5.3)Footnotea.
The training models were built using the stepwise MLR subroutine of the SPSS for Windows 13.0 (2004)Footnoteb. The optimality of the linear models was determined by considering the t-statistical (models with all absolute values >4.0 were accepted) as well as the overall F-statistical test. The accepted linear models were subjected to a cross-validation (CV) analysis by LOO and LGO procedures to ensure that the models are not over-fitted or under-fitted. The LOO-CV and LGO-CV algorithms were written in MATLAB.
To demonstrate the absence of chance correlation on the best models obtained with the above procedure, we performed a Y-scrambling testCitation41,Citation42 where the output value (the biological response) of the compounds was shuffled randomly and the scrambled data set was re-examined by MLR method against real (unscrambled) input descriptors to determine correlation and productivity of the resulting ‘model’. This procedure was repeated 30 times on many different scrambled data sets. R2CC and LOO cross-validation (R2CV-CC) of the resulting models were averaged and compared with the real modelCitation43. The corresponding algorithm was written in MATLAB.
The accepted MLR models were further optimized using a feed-forward neural network (NN) with back propagation of error algorithm. This ANN subroutine was written in MATLAB (version 7.2.0)Footnotec. After several tests, the best results were obtained with a (N × 2N × 1) NN with one hidden and one output layer, where N is the number of descriptors obtained from the best MLR models.
Molecular modelling procedure
The 50 known thrombin inhibitors previously mentioned were also studied by docking simulation employing AutoDock 4.0.1Citation30. The 3D structure of the ligands in their minimum-energy conformation was obtained by PM3 semi-empirical approachCitation36 coupled to the Eigenvector following methodCitation37 included in MOPAC 06 packageCitation38.
To identify the ligand–thrombin recognition-binding sites, all the possible rotatable bonds, the torsional degrees of freedom, the atomic partial charges (Gasteiger) and merge-non-polar hydrogens of the ligands were assigned using AutoDockTools 1.5.4, which is included in the AutoDock programCitation30.
The 3D structural model for human thrombin was obtained from the Protein Data Bank (PDB code: 1A4W). Afterwards, hydrogens of human thrombin at pH ~7 were added. The Kollman charges for all atoms of the enzyme were assigned with AutoDockTools 1.5.4. The ligands were docked under the blind docking procedure (grid box of 124 × 124 × 124 Å with the grid points separated by 0.375 Å) on the enzyme by means of AutoDock software under the hybrid Lamarckian Genetic Algorithm, with an initial population of 100 randomly placed individuals and a maximum number of energy evaluations of 1 × 107. The resulting docked orientations within a root-mean square deviation of 0.5 Å were clustered together. The lowest energy cluster returned by AutoDock for each compound was used for further analysis. All other parameters were maintained at their default settings. All ligand–enzyme complexes were visualized using the Visual Molecular Dynamics v.1.8.6 (VMD) programCitation44.
Data analysis
The selected 50 thrombin inhibitors were arranged in four different data sets, first to test their independence on the biological data source and second to test the prediction strength of MLR analysis versus ANN analysis upon a general quinoline, quinoxalinone derivative data set.
Having in mind that the Ockham’s razorCitation45 suggests that the larger the ratio of number of molecules to the number of properties, the better expected predictive power of the model. In the following procedure, we applied the commonly accepted lower limit of 5 for this valueCitation46.
The first data set considered all molecules tested (50 molecules) as the training sample. This data set was called M50. To construct the next three data sets, five molecules were eliminated each time at random from the M50 set and allowed them to form part of the external prediction set. The remaining molecules were used to construct three different sets by taking at random 36, 32 and 27 compounds, respectively, as the training sample; the remaining 9, 13 and 18 molecules in each set were part of the prediction sample. These sets were called M36, M32 and M27, respectively.
Despite that many authors consider a high q2 (for instance, q2 > 0.5) as an indicator or even as the ultimate proof that the model is highly predictiveCitation47, to define the relative importance of the QSAR models, we have also performed several other tests beyond the q2. These include the Akaike’s information criterion (AIC)Citation48, the Kubinyi fitness function (FIT)Citation49 and the calculation of the correlation coefficient for regression through the origin (RTO, R20) were performed for the training and predicted activity setsCitation50.
Finally, we have also taken into consideration the suggestion made by Guha and JursCitation40 who have shown that compounds that are very similar in the training set, generally exhibit smaller residuals and standard errors of prediction. These authors suggested that the cutoff value must be selected so that the outliers are approximately 20–30% of the whole data set.
Results
Pool of descriptors
The first step was to select the best descriptors for data set M50. After application of the stepwise MLR subroutine from the SPSS software, 21 descriptors were chosen from 10 different groups (see Table SI1 in the supporting information). The best model obtained was built with 17 variables from the reduced descriptor pool; however, the relation molecule number:descriptor is 2.94, which is lower than the ideal value of 5Citation46, in this sense the maximum number of variables should be 10 (see Equation 1). In the resulting equation, the main descriptors correspond to one topological descriptor and several chemical descriptors weighted by atomic van der Waals volumes.
or
R = 0.9526, SD = 0.3275, P < 0.0001, N = 50, SEE = 0.3816, q2LOO = 0.6502, q2LGO = 0.6650, FIT = 2.5725, AIC = 0.1420, F = 38.218.
The main descriptor in this equation is SIC0, a topological descriptor developed by Basak et al.Citation51; this descriptor takes into account all atoms in the constitutional formula (included hydrogen atoms) and considers the information content provided by various classes of atoms based on their topological neighbourhood. The SICr descriptors (r is the order level and could be 0 to 6) are not intercorrelated with other topological indices. Particularly, at the zero-order level the atom set is partitioned solely on the basis of its chemical nature. The SIC0 descriptor has a regression coefficient of R = 0.693 with respect to the experimental biological activity of the 50 compounds.
The correlation matrix of this model showed no colinearity among the chosen descriptors. The largest colinearity value is found between SIC0 and ATS6v descriptors (R = −0.715). The ATS6v descriptor is a 2D autocorrelation descriptor; the computation of this descriptor involves the summations of different autocorrelation functions corresponding to different fragment lengths and leads to different autocorrelation vectors corresponding to the lengths of the structural fragmentsCitation52. As a result, this descriptor addresses the topology of the structure or parts thereof in association with a selected physicochemical property.
For a cutoff = 1 used in all experiments performed in this study, only MOL19 was present as an outlier (see Figure SI1 in the supporting information). No apparent geometrical characteristic is responsible for its behaviour. The structure of MOL19 is very similar to that of MOL18, which includes an ester group that is more voluminous than the acid group of MOL19. It is important to mention that the IC50 of MOL18 and MOL19 are 0.027 and 3.3 µM, respectively; this means that in terms of inhibition of thrombin, the ester group is more important than the acid group.
Finding the best thrombin inhibitor model
To determine the optimum number of descriptors arising from data set M50, several training:prediction ratios were studied (see ‘Data analysis’ section). The compounds included in these ratios were taken randomly. For each data set, three different subsets were built; in total, nine subsets were studied by MLR methodology. The best inhibitor of thrombin MLR models were selected according to the best statistical parameters. Finally, the ANN analysis was applied to the best MLR models.
MLR models
The maximum number of descriptors for data set M36, M32 and M27 will be 7, 6 and 5, respectively; on the other hand, the best models of each data set were built using 4 (M36-MLR), 3 (M32-MLR) and 3 (M27-MLR) descriptors (see Table SI2 in the supporting information). These three models present the same structure on the descriptor type, that is, the SIC0 descriptor is the main variable as in M50 MLR model (see ‘Pool of descriptors’ section). The RBF constitutional descriptorCitation19 is taken to form part of these three models. This descriptor corresponds to the rotatable bond fraction in the molecule; the negative term associated with the descriptor indicates that the fractional decrease in the rotatable bond in the molecule is conductive for the thrombin inhibitory activities of the derivatives; for example, molecule MOL06 is the most potent compound whose X-ray structure with thrombin revealed an unexpected binding mode of this lead novel coagulation inhibitor structureCitation14, where the ester group interacts with the S4 subsite. Two of the cyclopentyl methylene groups point into the bulk solvent indicating that they do not contribute to the enzyme affinity. The rigidity of the cyclopentyl group is an important factor of this enzyme affinity.
Statistically, the M27-MLR model is the finest model that has a correlation coefficient of 0.8725, q2LOO value of 0.6737, FIT parameter of 2.2784 and AIC parameter of 0.2528; therefore, the M36- and M32-MLR models show statistical values close to M27-MLR values (see Table SI2 in the supporting information).
In the three models, the cutoff was defined at ±1.0 logarithmic units from the ideal line; this value is less than the common cutoff value that is three times the standard deviation (in these cases, this value should be between 1.395 and 1.512 units). A small number of outliers were found in each MLR model. The M36-MLR model shows two outliers from the Training set (MOL40 and MOL04), whereas the M32-MLR model also shows two outliers, one from the training set (MOL37) and one from the prediction set (MOL31), and finally the M27-MLR model presents four outliers: one from the training set (MOL37) and three from the predicted set (MOL13, MOL14 and MOL31). In these three models, validation sets molecules are always inside of the cutoff range.
To obtain the best statistic representation and to decide on the best model, the ANN techniques were employed.
The Y-scrambling test showed that the average for R2CC and R2CV-CC are 0.1220, 0.1287, 0.1165 and 0.3375, 0.1836, 0.1198 for M36-MLR, M32-MLR and M27-MLR, respectively. The lower values in the predicted set in M36-MLR decrease the probability of chance correlation effect. From these results, we concluded that increasing the number of molecules in the training set or decreasing the number of molecules in the prediction set increases the probability of chance correlation.
ANN models
To seek improvement on the MLR results, the ANN methodology was applied to M36-MLR, M32-MLR and M27-MLR models. We used a feed-forward NN with a back propagation of error algorithm. After several trial and error tests, the best models were obtained for a N × 2N × 1 network, with one hidden and one output layer, where N is the number of descriptors of the best MLR models.
shows the equations and statistical parameters of the three models obtained using the ANN methodology. The statistical parameters for the three models were improved when using the ANN methodology where the correlation coefficients were increased to values around 0.90 units. Moreover, the q2 values were increased.
Table 2. The three best models obtained by the ANN methodology.
In terms of outliers, the behaviour is very similar. The M36-ANN and M32-ANN models continue with two outliers, whereas the M27-ANN model shows a decrease in this number. The outliers in ANN models are almost the same found in the MLR models; in M36-ANN model, only the MOL39 molecule is a new outlier in place of MOL04 molecule.
To establish which one of these models is the best model, RTO calculations were performed upon the training and predicted sets (), the best model will be the one that shows the correlation coefficients close to 1 (R0 and R0′ for training and predicted set) and slopes close to 1 too. Three models show correlation coefficients and slopes close to 1 and therefore it is not possible to determine with these parameters which model is the best. It is clear that the molecule randomization does not have influence in the different models; at this point, the selection of the best model should be by the number of descriptors and the number of molecules in the model. After performing the MLR, ANN, RTO and descriptor number analysis, we concluded that the best model is M27-ANN (see Figure SI2 in the supporting information).
Table 3. The three best models obtained by the RTO methodology.
Docking results
This protein is characterized by having sites named D-pocket, P-pocket and the most important binding site the Ser195 amino acid, this is a site S or S1. In this work, we identified that the vast majority of the ligands tested reached the binding site S1 by making several hydrogen bonds, electrostatic interactions and π−π interaction as has been reported by Frédérick et al.Citation53 Most ligands can reach the three sites at the same time being more common site S1 and the D pocket (see Table SI3 in the supporting information). This is because these two sites are very close to each other and the ligands have a volume that cover the entire cavity and they also share the same pharmacophore group. In addition, some ligands do not reach the reported sites as lowest energy complexes (see Table SI3 in the supporting information).
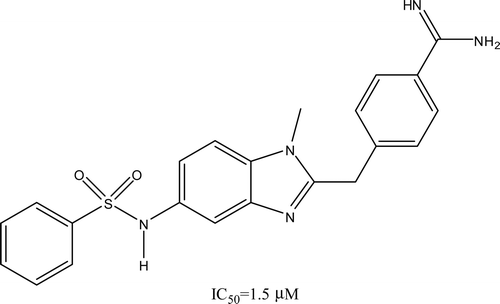
The binding energy in the enzyme–ligand complex is calculated from the addition of the next five energies (see Table SI4 in the supporting information): the ΔGIntermol (final intermolecular energy); ΔGvdW (van der Waals energy + hydrogen bond energy + desolvation energy); ΔGElect (electrostatic energy); ΔGinternal (final total internal energy); ΔGTorsional (torsional free energy)Citation30. Due to the lack of several biological properties, it is very common not to find relationships between the theoretical and experimental data. However, after studying each energy contribution, a relationship between four descriptors taken from the M50-MLR and ΔGinternal were found as suitable, this model is called M50-MLR-docked (, Equation 2). It is important to mention that the main descriptor in the M50-MLR model is the same descriptor as in the M50-MLR-docked model, that is, the QSAR models from semi-empirical and docking calculations are similar. The equation obtained for the M50-MLR-Docked model is:
Figure 2. Training set of the M50-MLR-docked data set. Calculated equation by multiple linear regression (solid line), ideal QSAR line (dotted line) and residual line limits (dashed line, cutoff = |1.0|). Outliers are also depicted.
or
R = 0.883, SD = 0.469, P < 0.0001, N = 50, SEE = 0.5544, q2LOO = 0.7175, q2LGO = 0.6580, FIT = 2.0762, AIC = 0.3006, F = 31.100.
In this model, three molecules are considered as outliers (), again MOL19 is an outlier as in M50-MLR model.
Discussion
Brandstetter et al.Citation54 and Banner and HadváryCitation55 studied the interaction between the bovine–thrombin complex with the benzamidine-based inhibitor NAPAP, an indicator of what molecule type could present some biological activity. This analysis was performed by comparison of several molecular interactions, for instance, the amidine group forms a bidentate salt bridge with the carboxylate of Asp189, hydrophobic interactions are formed by the piperidine ring and the naphthyl moiety with the proximal and the distal pocket of the thrombin active site. Additionally, the bridging glycine moiety of NAPAP, in analogy to peptidic serine protease substrates, forms the canonical hydrogen-bonding patterns with residue Trp215 and Gly216 at the rim of the S1 pocket. On the other hand, MOL16 and human α-thrombin form a complexCitation34 with the same salt bridge between Asp189 and MOL16 as found in the NAPAP complex, where the terminal phenyl ring is bound by a hydrophobic interaction to the D-pocket and the N-methyl group fits nicely into the P-pocket. The hydrogen bond between the inhibitor and the enzyme is not present; this last interaction should be very important in the inhibitor activity because the NAPAP and MOL16 show IC50 = 0.2 µM and IC50 = 1.5 µM, respectively.
In the case of NAPAP–MOL06, the first hydrophobic interaction is formed by the quinoline ring and its methyl substitution and the second hydrophobic interaction is formed by ethyl moiety or cyclopentyl moiety with the proximal and the distal pocket of the thrombin active site. These types of interactions could be very important in setting up the biological activity. On the other hand, NAPAP–MOL35 shows that p-CF3 (and o-CF3) substitution in the phenyl moiety is important to decrease the biological activity, that is, the higher polar substitution in the hydrophobic interaction, the less biological activity (see Figure SI3 in supporting information).
In M27-MLR model, the leader compound (MOL06) presents the smaller value of the SIC0 descriptor, whereas the MOL35 compound presents the worse biological activity that shows a higher value of the SIC0 descriptor, that is, the SIC0 descriptor is the main descriptor in the biological activity correlation. The linear regression between the biological activity and the SIC0 descriptor, gives a correlation coefficient of 0.748.
The leader compound is located in the prediction set and the worse biological activity compound is located in the training set. In the linear regression, residual values are |0.6172| and |0.4648|, respectively, whereas in the M27-MLR model, these values change to |0.7559| and |0.2662|, in both cases, the residuals are inside the outlier region. The best residual of leader compound is found in the M27-ANN model with |0.4406|.
Docking
Several types of interaction drive the ligand–protein recognition in the thrombin system. We discussed what happens when MOL01 is the ligand (see ) for which the binding energy is ΔG = −7.6 kcal/mol. In this figure, several ligand–protein interactions shown explain the best affinity; for example, there are several π−π interactions with Tyr228, Trp60D, Tyr60A, and the R of the compounds (that is an aromatic ring) also makes π−π interactions with Trp215. This confirms that the π−π interactions are very important for stabilizing these molecular complexes as has been reported by othersCitation56. In addition, a cluster of hydrogen bonds is made with the backbone of Gly216, Gly219 and Phe227. Finally, as have been reported elsewhere, electrostatic interaction of salt bridge is carried out between the amine of the compound with carboxylic acid of the Asp189. This means that aromatic substituents with few hindrance effects could be necessary to increase the affinity, chemical characteristics that MOL01 has. The same is observed for ligands from MOL01 to MOL15.
Figure 3. Ligand interactions of the compounds with best predicted affinity on thrombin (pdb code: 1A4W) classified according to their chemical cores, (A) sample belonging to MOL01–MOL15 subset, (B) sample belonging to MOL17–MOL28 subset, (C) sample belonging to MOL29–MOL44 subset and (D) sample belonging to MOL45–MOL50.
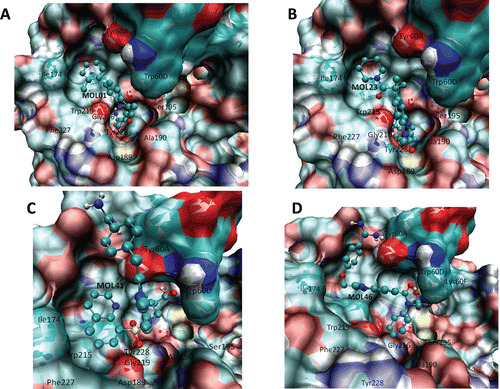
Other set of compounds that share the same core was the set of compounds from MOL17 to MOL28, being MOL23 the one with the best affinity (ΔGbinding = −7.89 kcal/mol). As it occurs with MOL01, there are several π−π interactions with aromatic residues such as Tyr60A and Trp60D and hydrogen bonds with backbone of Gly216, Gly219 and Phe227. Also a π–cation interaction with the amine group and the aromatic ring of the Tyr228 could take place. Additionally, hydrophobic interactions are found between the cyclohexane group of the compound and the aromatic ring of Trp215, since the carbonyl group is close to the cyclohexane it can make a hydrogen bond with the NH group of the aromatic ring of Trp215 and with the amine group of the Gly216 backbone. These ligand–protein interaction descriptions are shown in . With these results, it is possible to infer that the chemical core is necessary to fit in the thrombin protein, that the R group that normally is bound to Trp215 is required to be hydrophobic and not necessarily an aromatic ring; however, it is important that there must not be high hindrance effects.
depicts the best compound belonging to the set from MOL29 to MOL44; this is MOL41 with ΔGbinding = −8.14 kcal/mol, which shares the same chemical core while changing a six member cycle by a five member cycle (). In this compound, a binding mode occurred for which we did not find a previous report. As one can see, the aromatic system could make several interactions with Trp215, Trp60D and Tyr60A. Some hydrogen bonds are also observed between the carbonyl group from S and Gly216 backbone, and the amine group of five cycle member with the hydroxyl group of Tyr60A.
Finally, shows the ligand that had the best affinity on the thrombin-binding site; this is MOL46 (ΔGbinding = -8.18 kcal/mol), which belongs to the group of compounds from MOL45 to MOL50. These compounds have the same chemical core present in the above described compounds (MOL29 to MOL44); however, the set of ligands from MOL45 to MOL50 had more hindrance effects due to the aromatic ring. MOL46 makes several π−π interactions with Trp60D, Tyr60A and Trp215 and several hydrogen bonds with the hydroxyl group of Ser 195, Lys60F and the hydroxyl group of Tyr60A.
Conclusion
In this study, we have explored a set of 50 heterocyclic molecules under QSAR and docking studies. Several models were studied with MLR and ANN methodology where it is found that the chemical nature of some atoms (SIC0 descriptor) in the molecules was the most important descriptor. QSAR shows that the main interaction between the heterocyclic and the target protein is by hydrogen bond interactions, whereas docking studies show electrostatic and π−π interactions are important. These results suggest that it is possible that other hydrophobic substituents in the phenyl moiety could be very important in decreasing the biological activity.
The observed difference in the QSAR models obtained with MLR might be the effect of the strategy used in assigning the type and given number of molecules present in the training/predicted sets. Such effect has been reported by the Hemmateenejad groupCitation57.
Finally, we have shown that it is possible to build MLR models with geometries taken from two different sources, such as semi-empirical or MD methodologies, and obtain similar results. An option to improve the prediction is to separate the different contributions of the binding energy and to take them all as independent descriptors.
Supplementary Material
Download PDF (691.2 KB)Acknowledgements
RGJ acknowledges partial support from CONACyT grant 46061-R. JCB acknowledges partial support from CONACyT grant 62488, ICyTDF and Fundación Miguel Aleman AC, EDI/SIP-SIBE/COFAA-IPN.
Declaration of interest
The authors report no conflicts of interest.
Notes
a MATLAB Version 5.3 1.29215a (R11.1), The MathWorks Inc., Natick, MA, 1999.
b SPSS Version 13 for Windows, Chicago, IL, 2004.
c MATLAB Version 7.2.0.232 (R2006a). The MathWorks Inc., Natick, MA, 2006.
References
- Berndt M, Phillips D. Platelet membrane proteins: composition and receptor function. In: Gordon J, ed. Platelet in Biology and Pathology. North Holland, Amsterdam: Elsevier, 1981, pp. 43–74.
- Bar-Shavit R, Kahn A, Fenton JW 2nd, Wilner GD. Receptor-mediated chemotactic response of a macrophage cell line (J774) to thrombin. Lab Invest 1983;49:702–707.
- Harlan JM, Thompson PJ, Ross RR, Bowen-Pope DF. Alpha-thrombin induces release of platelet-derived growth factor-like molecule(s) by cultured human endothelial cells. J Cell Biol 1986;103:1129–1133.
- Jones A, Geczy CL. Thrombin and factor Xa enhance the production of interleukin-1. Immunology 1990;71:236–241.
- Okazaki H, Majesky MW, Harker LA, Schwartz SM. Regulation of platelet-derived growth factor ligand and receptor gene expression by alpha-thrombin in vascular smooth muscle cells. Circ Res 1992;71:1285–1293.
- McNamara CA, Sarembock IJ, Gimple LW, Fenton JW 2nd, Coughlin SR, Owens GK. Thrombin stimulates proliferation of cultured rat aortic smooth muscle cells by a proteolytically activated receptor. J Clin Invest 1993;91:94–98.
- Carney DH, Glenn KC, Cunningham DD. Conditions which affect initiation of animal cell division by trypsin and thrombin. J Cell Physiol 1978;95:13–22.
- Wiley MR, Fisher MJ. Small molecules direct thrombin inhibitors. Exp Opin Ther Pat 1997;7:1265–1282.
- Sanderson PEJ, Dyer DL, Naylon-Olsen AM, Vacca JP, Gardell SJ, Lewis SD et al. L-373,890: an achiral, noncovalent, subnanomolar thrombin inhibitor. Bioorg Med Chem Lett 1997;7:1497–1500.
- Sanderson PE, Cutrona KJ, Dorsey BD, Dyer DL, McDonough CM, Naylor-Olsen AM et al. L-374,087, an efficacious, orally bioavailable, pyridinone acetamide thrombin inhibitor. Bioorg Med Chem Lett 1998;8:817–822.
- Sanderson PE, Lyle TA, Cutrona KJ, Dyer DL, Dorsey BD, McDonough CM et al. Efficacious, orally bioavailable thrombin inhibitors based on 3-aminopyridinone or 3-aminopyrazinone acetamide peptidomimetic templates. J Med Chem 1998;41:4466–4474.
- Tamura SY, Semple JE, Reiner JE, Goldman EA, Brunck TK, Lim-Wilby MS et al. Design and synthesis of a novel class of thrombin inhibitors incorporating heterocyclic dipeptide surrogates. Bioorg Med Chem Lett 1997;7:1543–1548.
- Iwanowicz EJ, Lau WF, Lin J, Roberts DGM, Seiler SM. Derivatives of 5-amide indole as inhibitor of thrombin catalytic activity. Bioorg Med Chem Lett 1996;6:1339–1344.
- Ries UJ, Priepke HW, Hauel NH, Haaksma EE, Stassen JM, Wienen W et al. Heterocyclic thrombin inhibitors. Part 1: design and synthesis of amidino-phenoxy quinoline derivatives. Bioorg Med Chem Lett 2003;13:2291–2295.
- Ries UJ, Priepke HW, Hauel NH, Handschuh S, Mihm G, Stassen JM et al. Heterocyclic thrombin inhibitors. Part 2: quinoxalinone derivatives as novel, potent antithrombotic agents. Bioorg Med Chem Lett 2003;13:2297–2302.
- Coop A, MacKerell AD. The future of opioid analgesics. Am J Pharm Educ 2002;66:153–156.
- Hansch C, Leo A. QSAR of nonspecific toxicity. In: Heller SR, ed. Exploring QSAR: Fundamentals and Applications in Chemistry and Biology. Washington, DC: American Chemical Society, 1995, pp. 169–221.
- Krogsgaard-Larsen P, Liljerfors T, Madsen U. Textbook of Drug Design and Discovery. New York, USA: Taylor & Francis, CRC Press, 2002.
- Todeschini R, Consonni V. Handbook of Molecular Descriptors. Weinheim, Germany: Wiley-VCH, 2000.
- Karelson M. Molecular Descriptors in QSAR/QSPR. New York, USA: Wiley-Interscience, 2000.
- Nicolotti O, Miscioscia TF, Carotti A, Leonetti F, Carotti A. An integrated approach to ligand- and structure-based drug design: development and application to a series of serine protease inhibitors. J Chem Inf Model 2008;48:1211–1226.
- Fukunishi H, Teramoto R, Shimada J. Hidden active information in a random compound library: extraction using a pseudo-structure–activity relationship model. J Chem Inf Model 2008;48:575–582.
- Bonachéra F, Horvath D. Fuzzy tricentric pharmacophore fingerprints. 2. Application of topological fuzzy pharmacophore triplets in quantitative structure–activity relationships. J Chem Inf Model 2008;48:409–425.
- Xu L, Zhang WJ. Comparison of different methods for variable selection. Anal Chem Acta 2001;446:475–481.
- Schneider G, Wrede P. Artificial neural networks for computer-based molecular design. Prog Biophys Mol Biol 1998;70:175–222.
- Ramírez-Galicia G, Garduño-Juárez R, Deeb O, Hemmateenejad B. MLR-ANN and RTO approach to mu-opioid receptor-binding affinity. Pooling data from different sources. Chem Biol Drug Des 2008;71:260–270.
- Ramírez-Galicia G, Garduño-Juarez R, Hemmateenejad B, Deeb O, Estrada-Soto S. QSAR study on the relaxant agents from some Mexican medicinal plants and synthetic related organic compounds. Chem Biol Drug Des 2007;70:143–153.
- Ramírez-Galicia G, Garduño-Juárez R, Hemmateenejad B, Deeb O, Deciga-Campos M, Moctezuma-Eugenio JC. QSAR study on the antinociceptive activity of some morphinans. Chem Biol Drug Des 2007;70:53–64.
- Deeb O, Rosales-Hernández MC, Gómez-Castro C, Garduño-Juárez R, Correa-Basurto J. Exploration of human serum albumin binding sites by docking and molecular dynamics flexible ligand–protein interactions. Biopolymers 2010;93:161–170.
- Morris GM, Goodsell DS, Halliday RS, Huey R, Hart E, Belew RK et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem 1998;19:1639–1662.
- Chen Z, Li HL, Zhang QJ, Bao XG, Yu KQ, Luo XM et al. Pharmacophore-based virtual screening versus docking-based virtual screening: a benchmark comparison against eight targets. Acta Pharmacol Sin 2009;30:1694–1708.
- Blaney JM, Dixon JS. A good ligand is hard to find: automated docking methods. Perspect Drug Discov Design 1993;1:301–319.
- Kuntz ID, Meng EC, Shoichet BK. Structure-based molecular design. Acc Chem Res 1994;27:117–123.
- Hauel NH, Nar H, Priepke H, Ries U, Stassen JM, Wienen W. Structure-based design of novel potent nonpeptide thrombin inhibitors. J Med Chem 2002;45:1757–1766.
- HyperChem™ [computer program]. Release 7.1 for Windows. Gainesville, FL: Hypercube, Inc., 2002.
- Stewart JJP. Optimization of parameters for semiempirical methods I. Method. J Comp Chem 1989;10:209–220.
- Stewart JPP. MOPAC 6.0 QCPE Program No 455. Chemistry Department, Indiana University, Bloomington, IN, 1989.
- Baker J. An algorithm for the location of transition states. J Compt Chem 1993;7:385–395.
- Todeschini R, Consonni V, Pavan M. Dragon Software version 2.1-2002 Pisani 13, Milano, Italy. Dragon Software and references therein, 2002.
- Guha R, Jurs PC. Determining the validity of a QSAR model—a classification approach. J Chem Inf Model 2005;45:65–73.
- Topliss JG, Costello RJ. Change correlations in structure–activity studies using multiple regression analysis. J Med Chem 1972;15:1066–1068.
- Topliss JG, Edwards RP. Chance factors in studies of quantitative structure–activity relationships. J Med Chem 1979;22:1238–1244.
- Fernández M, Caballero J. QSAR models for predicting the activity of non-peptide luteinizing hormone-releasing hormone (LHRH) antagonists derived from erythromycin A using quantum chemical properties. J Mol Model 2007;13:465–476.
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph 1996;14:33–8, 27.
- Hoffman R, Minkin VI, Carpenter BK. Ockham’s razor and chemistry. HYLE Int J Phil Chem 1997;3:3–28.
- Walker CA, Owasoyo JO. The influence of serotonin, GABA and dl-dopa on the circadian rhythm in the toxicity of picrotoxin, pentylenetetrazol and phenobarbital in mice. Int J Chronobiol 1974;2:125–130.
- Golbraikh A, Tropsha A. Beware of q2! J Mol Graph Model 2002;20:269–276.
- Akaike H. A new look at the statistical model identification. IEEE Trans Automat Contr AC-19 1974;716–723.
- Kubinyi H. Variable selection in QSAR studies 2: a highly efficient combination of systematic search and evolution. QSAR 1994;13:393–401.
- Anderson MJ, Legendre P. An empirical comparison of permutation methods for test of partial regression coefficients in a linear model. J Stat Comput Simul 1999;62:271–303.
- Basak SC, Balaban AT, Grunwald GD, Gute BD. Topological indices: their nature and mutual relatedness. J Chem Inf Comput Sci 2000;40:891–898.
- Broto P, Moreau G, Vandycke C. Molecular structures perception, autocorrelation descriptor and SAR studies. Eur J Med Chem 1984;19:66–70.
- Frédérick R, Robert S, Charlier C, Wouters J, Masereel B, Pochet L. Mechanism-based thrombin inhibitors: design, synthesis, and molecular docking of a new selective 2-oxo-2H-1-benzopyran derivative. J Med Chem 2007;50:3645–3650.
- Brandstetter H, Turk D, Hoeffken HW, Grosse D, Stürzebecher J, Martin PD et al. Refined 2.3 A X-ray crystal structure of bovine thrombin complexes formed with the benzamidine and arginine-based thrombin inhibitors NAPAP, 4-TAPAP and MQPA. A starting point for improving antithrombotics. J Mol Biol 1992;226:1085–1099.
- Banner DW, Hadváry P. Crystallographic analysis at 3.0-A resolution of the binding to human thrombin of four active site-directed inhibitors. J Biol Chem 1991;266:20085–20093.
- Nishi H, Ota M. Amino acid substitutions at protein–protein interfaces that modulate the oligomeric state. Proteins 2010;78:1563–1574.
- Hemmateenejad B, Javadnia K, Elyasi M. Quantitative structure–retention relationship for the Kovats retention indices of a large set of terpenes: a combined data splitting-feature selection strategy. Anal Chim Acta 2007;592:72–81.