Abstract
Novel phenoxyalkylcarboxylic acid derivatives based on the natural scaffolds, flavonoids, or resveratrol were designed, synthesized, and evaluated for hypolipidaemic activity. Among the compounds, 30b lowered the triglycerides by 48.5% (P < 0.05) and total cholesterol by 44.2% (P < 0.05), respectively, and was more effective than the reference drug fenofibric acid in a Triton WR-1339-induced hyperlipidaemic mice model orally (300 mg/kg body weight). 30b also showed 59.4% triglycerides lowering in an alloxan-induced diabetic mice model orally (150 mg/kg body weight). Receptor docking studies revealed that compound 30b could interact with the amino acid residues in the ligand-binding domain essential for the activation of the PPARα. The results indicate that resveratrol should be a better scaffold to derive a new class of hypolipidaemic agents in comparison with a flavonoid scaffold.
Introduction
Hyperlipidaemia is the major risk factor for coronary heart disease (CHD), ischaemic cerebrovascular diseases, and peripheral vascular diseases.Citation1–3 Phenoxyalkylcarboxylic acid derivatives (fibrates), such as fenofibrate (2-[4-(4-chlorobenzoyl)phenoxy]-2-methylpropanoic acid isopropyl ester, 1a), gemfibrozil (5-(2,5-dimethylphenoxy)-2,2-dimethyl-pentanoic acid, 2) (), have been widely used in the clinic as an important class of lipid-modifying agents by lowering serum triglycerides and raising HDL cholesterol (HDLc) and remain the first choice for the treatment of severe hypertriglyceridaemia.Citation4–6 The effects of fibrates are attributed to the activation of the peroxisome proliferator-activated receptor alpha (PPARα), which is a known target for the treatment of dyslipidaemia.Citation7–9 However, most fibrates can cause myopathy (muscle pain with creatine phosphokinase elevations). Moreover, in combination with statin drugs, fibrates can cause an increased risk of rhabdomyolysis, idiosyncratic destruction of muscle tissue, leading to renal failure.Citation10–12 Elevated serum transaminase concentrations may occur with fenofibrate.Citation13 Fenofibrate retains some hepatotoxicity, especially in the rodents.Citation14 As a consequence, novel molecular scaffolds are expected to provide a superior clinical profile compared with that of existing fibrates for therapeutic intervention in dyslipidaemia and other metabolic disorders.Citation15
Figure 1. Synthetic triglyceride-lowering drug (fenofibrate, fenofibric acid, and gemfibrozil); naturally occurring flavone (chrysin), isoflavonoids (daidzein, formononetin, genistein), chalcone (isoliquirtigenin), flavanone (naringenin), and resveratrol.
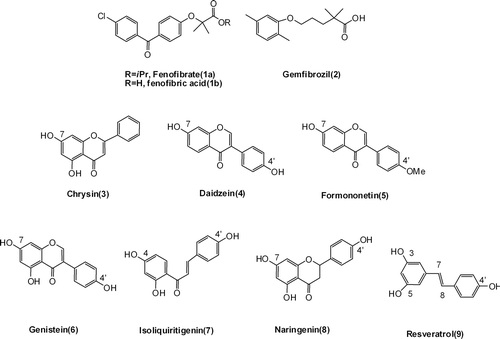
Flavonoids are naturally occurring phenolic compounds that are widely distributed in plants fulfilling many functions, such as antioxidant, antiallergic, anti-inflammatory, antimicrobial, and anticancer activities.Citation16–18 Consumers and food manufacturers have become interested in flavonoids for their medicinal properties, especially their potential role in the prevention of cancers and cardiovascular diseases.Citation19 They are relatively of low toxicity compared with other active plant compounds (for instance, alkaloids). The chemical structure of flavonoids are based on a C15 skeleton with a chromone ring bearing a second aromatic ring B in position 2 or 3 (). Various subgroups of flavonoids including flavones, isoflavonoids, chalcones, and flavanones are classified according to the substitution patterns of ring C ().
Figure 2. Chemical structures of flavonoids.
The stilbene-based components, or stilbenoids, have been suggested to have many health benefits including antioxidative, anti-inflammatory, antileukemic, antibacterial, antifungal, antiplatelet aggregation, vasodilator, and antitumour activities.Citation20–27 Resveratrol (trans-3,5,4′-trihydroxystilbene), the most widely studied stilbene, is a phytoalexin produced naturally by several plants when attacked by pathogens such as bacteria or fungi. Resveratrol is found in the skin of red grapes and is a constituent of red wine, and some scientists believe it is one of the factors behind the French Paradox.Citation28,Citation29 In mouse and rat experiments, anticancer, anti-inflammatory, blood sugar-lowering and other beneficial cardiovascular effects of resveratrol have been reported.Citation30 Resveratrol protects our heart and blood vessels by directly scavenging oxidants, which could cause oxidation of lipids, and by preventing oxidative stress-induced apoptosis of endothelial cells.Citation31,Citation32 It may also help to prevent heart damage after a cardiac arrest. Reduced platelet aggregation by resveratrol can reduce the risk of atherosclerosis.Citation33 Resveratrol has also been demonstrated to reduce blood lipid levels in animals.Citation34 Some naturally occurring or synthetic resveratrol analogues have shown to lower plasma lipid levels when fed to hamsters or to significantly activate PPARα.Citation35
These beneficial effects of flavonoids and resveratrol and their diverse chemical structures have attracted much attention, and they may provide novel scaffolds as therapeutic drugs. In this article, novel hypolipidaemic agents based on the flavonoid and resveratrol scaffolds were studied. The structure–activity relationship (SAR) on the fibrates shows that they consist of an acidic head group and a large lipophilic backbone.Citation36 The classical acidic head group could be 2,2-dimethyloxyvaleric acid or oxyisobutyric acid.Citation37 Here, we described the design and synthesis of novel hypolipidaemic agents by the combination of the fibrate head groups and the flavonoid or resveratrol scaffolds, which probably would provide synergistic pharmacological effects. The flavone, isoflavonoid, chalcone, flavanone, and resveratrol backbones were selected as the scaffolds in this study.
Materials and methods
Chemistry
All common laboratory chemicals were purchased from commercial sources and used without further purification. Melting points (uncorrected) were obtained by using an XT4 MP apparatus (Taike Corp., Beijing, China). 1H NMR and 13C NMR were measured on a JNM-ECA-400 NMR spectrometer (at 400 MHz for 1H and 100 MHz for 13C) using TMS as an internal standard. Coupling constants (J) are given in hertz. Electrospray mass spectra (ESI-MS) were recorded on an API3000 spectrometer. Thin-layer chromatography (TLC) was carried out on silica gel plates with a fluorescence indicator of F254 (0.2 mm, Qingdao Haiyang), and the spots were visualized in UV light. TOF-HRMS spectra were determined on an Agilent 1100 instrument.
The following illustrated the synthesis and characteristics of three representative target compounds (11, 17a, 17b), and the details of the synthesis of the other compounds, compound characterization data are available in the Supporting Materials.
5-Hydroxy-7-(1-methyl-1-ethoxycarboxylethoxy)flavone (10)
Ethyl 2-bromo-2-methylpropanate (2.21 mL, 15 mmol) was added to a solution of 5,7-dihydroxyflavone (1.90 g, 7.5 mmol) with anhydrous K2CO3 (3.14 g, 22.5 mmol) and KI (0.25 g, 1 mmol) in anhydrous dimethylformamide (DMF) (40 mL), and stirring was continued for 12 h at 120°C. The reaction mixture was cooled and filtered. The filtered solvent was removed under reduced pressure and the residue was purified by silica gel chromatography (PE/EtOAc 85:15) to afford 10 (2.07 g, 75% yield) as a yellow solid. Mp. 231–233°C; 1H NMR (DMSO-d6) δ 1.18 (3H, m, CH3), 1.66 (6H, m, 2CH3), 4.22 (2H, m, CH2), 6.14 (1H, d, J = 2 Hz, Ar-H), 6.55 (1H, d, J = 2 Hz, Ar-H), 7.04 (1H, s, Ar-H), 7.60 (3H, m, Ar-H), 8.10 (2H, m, Ar-H), 12.73 (1H, br.s, 5-OH); C21H20O6, MW calcd. 368.38; ESI-MS (MeOH): 369 [M+H]+.
5-Hydroxy-7-(1-methyl-1-carboxylethoxy) flavone (11)
K2CO3 (0.41 g, 3.00 mmol) was added to a solution of 10 (0.37 g, 1.00 mmol) in MeOH/H2O (1:1, 20 mL) and the solution was stirred and refluxed for 12 h. The mixture was concentrated under reduced pressure. The residue was poured into ice water (10 mL), acidified with a 3N HCl aqueous solution to pH 3–4 and extracted with EtOAc (3 × 20 mL). The organic extracts were washed with brine, dried over Na2SO4, and filtered. The solvent was removed under reduced pressure and the residue was purified by silica gel chromatography (PE/EtOAc 7:3) to afford 11 (0.22 g, 65% yield) as a white solid. Mp. 264–266°C; 1H NMR (400 MHz, DMSO-d6) δ 1.65(6H, m, 2CH3), 6.17(1H, d, J = 2 Hz, Ar-H), 6.51 (1H, d, J = 2 Hz, Ar-H), 7.02 (1H, s, Ar-H), 7.57–7.63 (3H, m, Ar-H), 8.09 (2H, m, Ar-H), 12.71 (1H, br.s, 5-OH), 13.21 (1H, br.s, COOH); C19H16O6, MW calcd. 340.33; ESI-MS (MeOH): 341[M+H]+; TOF-HRMS m/z = 341.1023 [M+H]+ (C19H17O6 requires 341.1025).
4′-Hydroxy-7-methoxymethoxyisoflavone (14a)
Chloromethyl methyl ether (3.17 mL, 39.4 mmol) was added dropwise to a solution of 4′,7-dihydroxyisoflavone (10 g, 39.4 mmol) with anhydrous K2CO3 (4.35 g, 31.5 mmol) in anhydrous acetone (250 mL), and the solution was stirred and refluxed for 12 h. The reaction mixture was filtered and the filtrate was concentrated. The residue was purified by silica gel chromatography (PE/EtOAc 85:15) to afford 14a (3.2 g, 27.4% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 3.81 (3H, s, OCH3), 5.27 (2H, s, OCH2O), 6.80–6.84 (3H, m, Ar-H), 6.95 (1H, dd, J = 9.0, 2.2 Hz, Ar-H), 7.36 (2H, d, J = 8.4 Hz, Ar-H), 7.98 (1H, d, J = 8.6 Hz, Ar-H), 8.32 (1H, s, Ar-H), 9.50 (1H, s, 4′-OH); C17H14O5, MW calcd. 298.29; ESI-MS (MeOH): 299 [M+H]+.
4′,5-Dihydroxy-7-methoxymethoxyisoflavone (14b)
The titled compound was prepared from 4′,5,7-trihydroxyisoflavone and ethyl 2-bromo-2-methylpropanate according to the method of compound 14a with the following yield: 44.8 %; yellow powder; 1H NMR (400 MHz, DMSO-d6) δ 3.78 (3H, s, OCH3), 5.23 (2H, s, OCH2O), 6.21 (1H, d, J = 2 Hz, Ar-H), 6.39 (1H, d, J = 2 Hz, Ar-H), 6.85 (2H, m, Ar-H), 7.49 (2H, m, Ar-H), 8.41 (1H, s, Ar-H), 9.54 (1H, s, 4′-OH), 12.89 (1H, s, 5-OH); C17H14O6, MW calcd. 314.29; ESI-MS (MeOH): 315 [M+H]+.
4′-(1-Methyl-1-ethoxycarboxylethoxy)-7-methoxymethoxyisoflavone (15a)
The titled compound was prepared from 14a and ethyl 2-bromo-2-methylpropanate according to the method of compound 10 with the following yield: 60%; white solid; 1H NMR (400 MHz, DMSO-d6) δ 1.16 (3H, m, CH3), 1.64 (6H, s, 2CH3), 3.83 (3H, s, OCH3), 4.25 (2H, m, CH2), 5.33 (2H, s, OCH2O), 6.81–6.84 (3H, m, Ar-H), 6.97 (1H, dd, J = 9.0, 2.2 Hz, Ar-H), 7.37 (2H, d, J = 8.4 Hz, Ar-H), 8.02 (1H, d, J = 8.6 Hz, Ar-H), 8.34 (1H, s, Ar-H); C23H24O7, MW calcd. 412.43; ESI-MS (MeOH): 413 [M+H]+.
4′-(1-Methyl-1-ethoxycarboxylethoxy)-5-hydroxy-7-methoxymethoxyisoflavone (15b)
The titled compound was prepared from 14b and ethyl 2-bromo-2-methylpropanate according to the method of compound 10 with the following yield: 50.3%; yellow powder; 1H NMR (400 MHz, DMSO-d6) δ 1.17 (3H, m, CH3), 1.63 (6H, s, 2CH3), 4.24 (2H, m, CH2), 3.78 (3H, s, OCH3), 5.24 (2H, s, OCH2O), 6.22 (1H, d, J = 2 Hz, Ar-H), 6.40 (1H, d, J = 2 Hz, Ar-H), 6.84 (2H, m, Ar-H), 7.45 (2H, m, Ar-H), 8.42 (1H, s, Ar-H), 12.90 (1H, s, 5-OH); C23H24O8, MW calcd. 428.43; ESI-MS (MeOH): 429 [M+H]+.
4′-(1-Methyl-1-ethoxycarboxylethoxy)-7-hydroxyisoflavone (16a)
Six millilitres of 10% aqueous HCl was added to a solution of 15a (1 g, 2.4 mmol) in MeOH (30 mL) and the mixture was stirred and refluxed for 30 min. The solution was poured onto water (40 mL), filtered, washed with water (100 mL), and dried in vacuum to give 16a as a white solid (0.75 g, 85% yield) without further purification.
4′-(1-Methyl-1-ethoxycarboxylethoxy)-5,7-dihydroxyisoflavone (16b)
The titled compound was prepared from 15b according to the method of compound 16a with 90% yield as a yellow powder without further purification.
4′-(1-Methyl-1-carboxylethoxy)-7-hydroxyisoflavone (17a)
The titled compound was prepared from 16a according to the method of compound 11 with the following yield: 72 %; white solid; Mp. 246–248°C; 1H NMR (400 MHz, DMSO-d6) δ 1.54 (6H, m, 2CH3), 6.85–6.88 (3H, m, Ar-H), 6.94 (1H, dd, J = 8.8, 2.4 Hz, Ar-H), 7.48 (2H, m, Ar-H), 7.97 (1H, d, J = 8.8 Hz, Ar-H), 8.35 (1H, s, Ar-H), 10.80 (1H, s, 7-OH), 13.02 (1H, br.s, COOH); C19H16O6, MW calcd. 340.33; ESI-MS (MeOH): 341 [M+H]+, 363 [M+Na]+; TOF-HRMS m/z = 363.0843 [M+Na]+ (C19H16NaO6 requires 363.0845).
4′-(1-Methyl-1-carboxylethoxy)-5,7-dihydroxy isoflavone (17b)
The titled compound was prepared from 16b according to the method of compound 11 with the following yield: 72%; yellow powder; Mp. 222–224°C; 1H NMR (400 MHz, DMSO-d6) δ 1.54 (6H, m, 2CH3), 6.23 (1H, d, J = 2 Hz, Ar-H), 6.40 (1H, d, J = 2 Hz, Ar-H), 6.87 (2H, m, Ar-H), 7.46 (2H, m, Ar-H), 8.39 (1H, s, Ar-H), 10.94 (1H, s, 7-OH), 12.91 (1H, s, 5-OH), 13.14 (1H, s, COOH); C19H16O7, MW calcd. 356.33; ESI-MS (MeOH):357 [M+H]+, 379 [M+Na]+; TOF-HRMS m/z = 379.0792 [M+Na]+ (C19H16NaO7 requires 379.0794).
Biology
In vivo hypolipidaemic activity evaluation in Triton WR-1339-induced hyperlipidaemic mice
Male Kunming mice (18–22 g) bred in the animal house of the institute were divided into Triton, Triton plus drug-treated groups containing 10 mice in each. Overnight fasted mice of experimental groups were administered suspension of the desired test samples (made in 0.5% CMC-Na) at 300 mg/kg (i.g.) or 100 mg/kg (i.p.) dose. After 30 min, Triton WR-1339 (Sigma, St. Louis, MO) was administered (400 mg/kg body weight) by injecting via tail vein. Blood was withdrawn 22 h later from retro-orbital plexus and serum was separated by centrifugation at low speed and assayed for total cholesterol and triglycerides. The analysis of serum biochemical parameters was performed on a Semi-automatic Vital Microlab 300 Biochemistry Analyzer by standard enzymatic methods. The assay kits were purchased from BioSino Biotechnology and Science Inc., Changping, China. Differences between two groups were analysed using unpaired Student’s t-test and one-way ANOVA for multiple comparisons. P < 0.05 was considered significant.
In vivo hypolipidaemic activity evaluation in alloxan-induced diabetic mice
Male Kunming mice (18–22 g) bred in the animal house of the institute were divided into diabetic control, diabetic plus drug-treated groups containing 10 mice in each. Mice of experimental groups were administered suspension of the desired test samples orally (made in 0.5% CMC-Na) at 150 mg/kg dose daily from Day 1 to 3. Animals of control group were given an equal amount of 0.5% CMC-Na. On the second day, after 30 min of administration of overnight fasted mice, alloxan was administered (110 mg/kg body weight) by injecting via tail vein. The blood was collected on Day 4, 48 h after alloxan administration and serum was separated by centrifugation at low speed and assayed for triglycerides and glucose. The analysis of serum biochemical parameters was performed on a Semi-automatic Vital Microlab 300 Biochemistry Analyzer by standard enzymatic methods. The assay kits were purchased from BioSino Biotechnology and Science Inc.. Differences between two groups were analysed using unpaired Student’s t-test and one-way ANOVA for multiple comparisons. P < 0.05 was considered significant.
Molecular modelling
The crystal structure of PPARα in complex with GW409544 (1K7L) recovered from the Brookhaven Protein Data Bank was used as the target for molecular docking. The docking calculations of compounds with PPARα were performed with the AutoDock Vina1.0 program (Oleg Trott et al., The Scripps Research Institute, La Jolla, CA). These images were generated using the Discovery Studio 2.5 Visualizer program (Accelrys Software Inc.).
Results and discussion
Chemistry
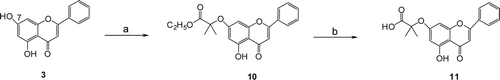
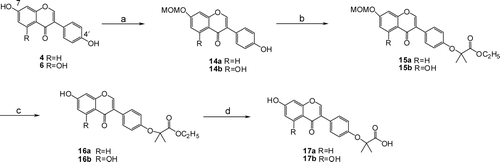
The flavone and isoflavonoid derivatives were synthesized starting with the natural scaffolds, chrysin (5,7-dihydroxyflavone, 3), daidzein (4′,7-dihydroxyisoflavone, 4), formononetin (7-hydroxy-4′-methoxyisoflavone, 5), genistein (4′,5,7-trihydroxyisoflavone, 6), one common natural flavone and three common natural isoflavonoids. The target compounds were prepared by the alkylation of the phenols with ethyl 2-bromo-2-methylpropanate, and then hydrolysis of the esters as shown in and and S1 (in Supplementary Materials). As the 7-hydroxyl group is more reactive than 4′-hydroxyl group, the latter could be selectively alkylated following MOM protection of the 7-hydroxyl group. MOM deprotection and then hydrolysis of the esters provided the isoflavonoid derivatives 17a and 17b.
Scheme 1. (a) BrC(CH3)2COOC2H5, K2CO3, DMF and (b) (i) K2CO3, MeOH/H2O, reflux, (ii) aqueous HCl.
Scheme 2. (a) MOMCl, acetone, K2CO3, reflux; (b) BrC(CH3)2COOC2H5, K2CO3, DMF; (c) 10% HCl/MeOH, reflux, 0.5 h; and (d) (i) K2CO3, MeOH/H2O, reflux, (ii) aqueous HCl.
The method for the synthesis of chalcone derivatives is shown in S2 (in Supplementary Materials). After aldol condensation and MOM deprotection, the chalcone scaffolds were obtained and the phenols were alkylated with ethyl 2-bromo-2-methylpropanate. Alkaline hydrolysis of the esters gave the target compounds 23a and 23b.
Cyclization of 21b by treatment with sodium acetate and then MOM deprotection gave the flavanone scaffold. The subsequent alkylation and hydrolysis afforded the target compound 26 as shown in S3 (in Supplementary Materials).
In previous studies, simple groups were introduced at 4′ position and the methoxy groups were kept at the 3 and 5 positions of resveratrol.Citation38 In our target compounds, the classical fibrate head groups were introduced at the 3, 5, and/or 4′ positions of resveratrol by the alkylation of the phenols with either ethyl 2-bromo-2-methylpropanate or isobutyl 5-chloro-2,2-dimethylvalerate, and the remaining phenolic hydroxyl groups could be methylated by iodomethane or not. The compounds with two or three fibrate head groups were also obtained by the one-pot synthesis method in order to better understand the SAR. Furthermore, as the reactivity of the 4′-hydroxyl group of resveratrol was comparable with the 3-hydroxyl group, the resveratrol-4′-O- and 3-O-monosubstitutes were synthesized by the one-pot synthetic approach. The synthesis routes are shown in S4 and S5 (in Supplementary Materials). All the target compounds above were confirmed by spectroscopy. Like resveratrol, the trans configuration of the target compounds were confirmed as the relatively large coupling constant (~16 Hz) between H-7 and H-8.
Biological activity
The ability of the compounds to affect lipid/cholesterol homeostasis was first examined on Triton WR-1339-induced hyperlipidaemia in mice. shows the activity of flavonoid derivatives at the dose of 300 mg/kg body weight orally. Out of these eight derivatives, isoflavonoid derivative 17a and chalcone derivative 23a were the two most potent compounds. 17a lowered the triglycerides by 20.2% (P < 0.05), total cholesterol by 16.6% (P < 0.05), and 23a lowered 19.8% (P < 0.05) in triglycerides, 16.3% (P < 0.05) in total cholesterol quantity. Moreover, the isoflavonoid derivatives 17a and 17b showed more potent than their parent compounds daidzein and genistein, respectively. These results suggested that the enhanced hypolipidaemic activities of 17a and 17b can be attributed to the presence of the fibrate head moieties and the modification can lead to increased activity. But none of the synthesized compounds was more potent than the reference drug fenofibric acid (2-[4-(4-chlorobenzoyl)phenoxy]-2-methylpropanoic acid, 1b), the active metabolite of fenofibrate.
Table 1. In vivo hypolipidaemic activity of flavonoid derivatives in Triton WR-1339-induced hyperlipidaemic mice.
The in vivo hypolipidaemic activity of resveratrol derivatives in a Triton WR-1339-induced hyperlipidaemic mice model is shown in . The synthesized compounds, employing the 2,2-dimethyloxyvaleric acid group, or the oxyisobutyric acid group, showed more potent than the parent compound, resveratrol (9) by intraperitoneal injection (100 mg/kg body weight) or orally (300 mg/kg body weight). The reference drug fenofibric acid was the most potent followed by 30b at the dose of 100 mg/kg when administration by intraperitoneal injection. But at the dose of 300 mg/kg orally, 30b was identified as the most potent and lowered the triglycerides by 48.5% (P < 0.05), total cholesterol by 44.2% (P < 0.05), whereas fenofibric acid lowered the triglycerides by 38.2% (P < 0.05), total cholesterol by 30.8% (P < 0.05). Moreover, interestingly, methylation of the remaining phenolic hydroxyl groups led to increased activities: 30a was more effective than 28a, and 34 was more effective than 28c, and 33b was more effective than 28g. The compounds, employing the 2,2-dimethyloxyvaleric acid group, showed more potent than the compounds employing the oxyisobutyric acid group at the corresponding positions of the compounds (for instance, 28e > 28b, 28g > 28c). Furthermore, 34 exhibited more effective than 30a suggesting the substitution position of the fibrate head group could influence the activity. 28b bearing two oxyisobutyric acid moieties showed more potent than the compounds 28a (i.g., 300 mg/kg) and 28c (i.v., 100 mg/kg) which have only one oxyisobutyric acid moiety in their molecules.
Table 2. In vivo hypolipidaemic activity of resveratrol derivatives in Triton WR-1339-induced hyperlipidaemic mice.
Compounds 28e, 28f, 30a, and 30b were further tested for their hypolipidaemic activity in an alloxan-induced diabetic mice model at a dose of 150 mg/kg body weight orally. In this experiment, the model mice exhibited hypertriglyceridaemia and hyperglycaemic. These compounds showed good lowering in triglycerides, and 30b was the most potent among them. 30b lowered 59.4% (P < 0.01) in triglycerides, which was slightly more active than fenofibric acid (). In addition, 28e (53.5%), 30a (52.6%), and fenofibric acid (54.1%) showed similar triglyceride-lowering activity. Like fenofibric acid, none of the compounds exhibited any blood glucose-lowering activity in this model.
Table 3. In vivo hypolipidaemic activity of compounds in alloxan-induced diabetic mice.
Docking studies
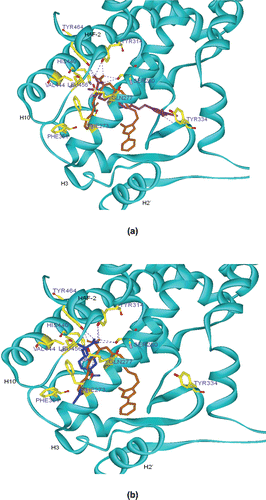
Molecular modelling (docking) studies were carried out to gain an insight into the likely binding interactions between the most potent compound 30b and PPARα. Our proposed binding pose of 30b superimposed on the co-crystal structure of GW409544 inside the PPARα ligand-binding domain (PDB:1K7L) is illustrated in . GW409544 is a full PPARα and PPARγ agonist.Citation39 Docking studies revealed that one of the carboxylate oxygens of 30b could form a bifurcated H-bond with the Tyr464 OH and the Tyr314 OH groups of the PPARα. The other carboxylate oxygen of 30b was at H-bonding distance from Ser280 OH. Ser280, Tyr314, Tyr464 are known to be the key interaction residues for the activation of the PPARα.Citation39 The gem-dimethyl substituents of 30b were directed into a lipophilic pocket bounded by Phe273, Gln277, Val444, and Leu456, a region at the top end of the so-called “benzophenone” pocket. The remaining two phenyl rings were docked into the hydrophobic pocket formed by the helices 2′, 3, and β sheet, and one methoxy group at the 3 or 5 position of 30b could produce a hydrogen bond interaction with Tyr334 OH. In in vivo studies, 30b showed good hypolipidaemic activity, so it can be predicted that the activity may be due to the activation of the PPARα. However, unlike compound 30b, docking of fenofibric acid, the marketed activator of PPARα, showed the two phenyl rings were docked into the hydrophobic pocket formed by the helices 3, 6, and 10 adjacent to the AF-2 helix, also known as “benzophenone” pocket (). These docking models revealed that 30b may bind to the receptor in a different mode from fenofibric acid.
Figure 3. (A) Proposed binding pose of 30b (purple) superimposed on the co-crystal structure of GW409544 (golden) inside PPARα ligand-binding domain. Blue dotted line represents hydrogen bonding. Hydrogen atoms are not shown for clarity. The protein backbone is shown as cyan ribbon. Important residues are shown in yellow. (B) Proposed binding pose of fenofibric acid (blue) superimposed on the co-crystal structure of GW409544 (golden) inside PPARα ligand-binding domain. Blue dotted line represents hydrogen bonding. Hydrogen atoms are not shown for clarity. The protein backbone is shown as cyan ribbon. Important residues are shown in yellow.
Conclusions
In conclusion, novel phenoxyalkylcarboxylic acid derivatives based on the natural scaffolds, flavonoids or resveratrol, were prepared and screened for their hypolipidaemic activity in vivo. Compound 30b was the most potent among the synthesized compounds and was orally more effective than the reference drug fenofibric acid in two animal models. Docking studies revealed that compound 30b could interact with the amino acid residues in the ligand-binding domain essential for the activation of the PPARα and may bind to the receptor in a different mode from fenofibric acid. The results of this study indicate that resveratrol can be used as a better scaffold to derive a new class of hypolipidaemic agents compared with a flavonoid scaffold. Further work is in progress to find out the mechanism of action of 30b in hypolipidaemic activity, and whether 30b can indeed activate the PPARα through interactions with the ligand-binding domain of the receptor as predicted by these modelling studies. Moreover, as we mentioned above, there should be further research to find whether the compounds could produce synergistic pharmacological effects by the combination of the pharmacophore moieties and the natural scaffolds. We hope this information might be helpful for the discovery of new hypolipidaemic agents.
Supplementary Material
Download PDF (194.8 KB)Acknowledgement
The authors are grateful to management of Bo-hua Zhong Group for encouragement, and the analytical department for support.
Declaration of interest
The authors report no conflict of interest, and the authors alone are responsible for the content of this article.
References
- Anum EA, Adera T. Hypercholesterolemia and coronary heart disease in the elderly: a meta-analysis. Ann Epidemiol 2004;14:705–721.
- Auboeuf D, Rieusset J, Fajas L, Vallier P, Frering V, Riou JP et al. Tissue distribution and quantification of the expression of mRNAs of peroxisome proliferator-activated receptors and liver X receptor-alpha in humans: no alteration in adipose tissue of obese and NIDDM patients. Diabetes 1997;46:1319–1327.
- Banner CD, Göttlicher M, Widmark E, Sjövall J, Rafter JJ, Gustafsson JA. A systematic analytical chemistry/cell assay approach to isolate activators of orphan nuclear receptors from biological extracts: characterization of peroxisome proliferator-activated receptor activators in plasma. J Lipid Res 1993;34:1583–1591.
- Chong PH, Bachenheimer BS. Current, new and future treatments in dyslipidaemia and atherosclerosis. Drugs 2000;60:55–93.
- Brown PJ, Winegar DA, Plunket KD, Moore LB, Lewis MC, Wilson JG et al. A ureido-thioisobutyric acid (GW9578) is a subtype-selective PPARalpha agonist with potent lipid-lowering activity. j Med Chem 1999;42:3785–3788.
- Bunnelle WH, Dart MJ, Schrimpf MR. Design of ligands for the nicotinic acetylcholine receptors: the quest for selectivity. Curr Top Med Chem 2004;4:299–334.
- Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature 2000;405:421–424.
- Willson TM, Brown PJ, Sternbach DD, Henke BR. The PPARs: from orphan receptors to drug discovery. J Med Chem 2000;43:527–550.
- Fruchart JC, Staels B, Duriez P. The role of fibric acids in atherosclerosis. Curr Atheroscler Rep 2001;3:83–92.
- Shek A, Ferrill MJ. Statin–fibrate combination therapy. Ann Pharmacother 2001;35:908–917.
- Adverse Drug Reactions Advisory Committee. Risk factors for myopathy and rhabdomyolysis with the statins. Aust Adv Drug React Bull 2004;23:2.
- Jones PH, Davidson MH. Reporting rate of rhabdomyolysis with fenofibrate + statin versus gemfibrozil + any statin. Am J Cardiol 2005;95:120–122.
- Laboratoires Fournier SA. Lipidil (Fenofibrate) Product Information, 13 October 2003.
- Peters JM, Cattley RC, Gonzalez FJ. Role of PPAR alpha in the mechanism of action of the nongenotoxic carcinogen and peroxisome proliferator Wy-14,643. Carcinogenesis 1997;18:2029–2033.
- Staels B, Dallongeville J, Auwerx J, Schoonjans K, Leitersdorf E, Fruchart JC. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 1998;98:2088–2093.
- Jeremy PES. Flavonoids: modulators of brain function? Br J Nutr 2008;99:ES60–ES77.
- Cushnie TP, Lamb AJ. Antimicrobial activity of flavonoids. Int J Antimicrob Agents 2005;26:343–356.
- “Studies force new view on biology of flavonoids”, by David Stauth, EurekAlert!. Adapted from a news release issued by Oregon State University. URL accessed.
- Alan L, Miller ND. Antioxidant flavonoids: structure, function and clinical usage. Altern Med Rev 1996;1:103–111.
- Stivala LA, Savio M, Carafoli F, Perucca P, Bianchi L, Maga G et al. Specific structural determinants are responsible for the antioxidant activity and the cell cycle effects of resveratrol. J Biol Chem 2001;276:22586–22594.
- Kimura Y, Okuda H, Arichi S. Effects of stilbenes on arachidonate metabolism in leukocytes. Biochim Biophys Acta 1985;834:275–278.
- Mannila E, Talvitie A, Kolehmainen E. Xanthone O-glycosides from Polygala tenuifolia. Phytochemistry 1993;33:813–816.
- Mahady GB, Pendland SL, Chadwick LR. Resveratrol and red wine extracts inhibit the growth of CagA+ strains of Helicobacter pylori in vitro. Am J Gastroenterol 2003;98:1440–1441.
- Creasy LL, Coffee M. Phytoalexin production potential of grape berries. J Am Soc Hortic Sci 1988;113:230–234.
- Chung MI, Teng CM, Cheng KL, Ko FN, Lin CN. An antiplatelet principle of Veratrum formosanum. Planta Med 1992;58:274–276.
- Inamori Y, Kubo M, Tsujibo H, Ogawa M, Saito Y, Miki Y et al. The ichthyotoxicity and coronary vasodilator action of 3,3′-dihydroxy-alpha,beta-diethylstilbene. Chem Pharm Bull 1987;35:887–890.
- Schneider Y, Duranton B, Gossé F, Schleiffer R, Seiler N, Raul F. Resveratrol inhibits intestinal tumorigenesis and modulates host-defense-related gene expression in an animal model of human familial adenomatous polyposis. Nutr Cancer 2001;39:102–107.
- Hegsted DM, Ausman LM. Diet, alcohol and coronary heart disease in men. J Nutr 1988;118:1184–1189.
- Renaud S, de Lorgeril M. Wine, alcohol, platelets, and the French paradox for coronary heart disease. Lancet 1992;339:1523–1526.
- Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov 2006;5:493–506.
- Frankel EN, Waterhouse AL, Kinsella JE. Inhibition of human LDL oxidation by resveratrol. Lancet 1993;341:1103–1104.
- Ungvari Z, Orosz Z, Rivera A, Labinskyy N, Xiangmin Z, Olson S et al. Resveratrol increases vascular oxidative stress resistance. Am J Physiol Heart Circ Physiol 2007;292:H2417–H2424.
- Wang Z, Zou J, Huang Y, Cao K, Xu Y, Wu JM. Effect of resveratrol on platelet aggregation in vivo and in vitro. Chin Med J 2002;115:378–380.
- Arichi H, Kimura Y, Okuda H, Baba K, Kozawa M, Arichi S. Effects of stilbene components of the roots of Polygonum cuspidatum Sieb. et Zucc. on lipid metabolism. Chem Pharm Bull 1982;30:1766–1770.
- Rimando AM, Nagmani R, Feller DR, Yokoyama W. Pterostilbene, a new agonist for the peroxisome proliferator-activated receptor alpha-isoform, lowers plasma lipoproteins and cholesterol in hypercholesterolemic hamsters. J Agric Food Chem 2005;53:3403–3407.
- Li Z, Liao C, Ko BC, Shan S, Tong EH, Yin Z et al. Design, synthesis, and evaluation of a new class of noncyclic 1,3-dicarbonyl compounds as PPARalpha selective activators. Bioorg Med Chem Lett 2004;14:3507–3511.
- Sierra ML, Beneton V, Boullay AB, Boyer T, Brewster AG, Donche F et al. Substituted 2-[(4-aminomethyl)phenoxy]-2-methylpropionic acid PPARalpha agonists. 1. Discovery of a novel series of potent HDLc raising agents. J Med Chem 2007;50:685–695.
- Mizuno CS, Ma G, Khan S, Patny A, Avery MA, Rimando AM. Design, synthesis, biological evaluation and docking studies of pterostilbene analogs inside PPARalpha. Bioorg Med Chem 2008;16:3800–3808.
- Xu HE, Lambert MH, Montana VG, Plunket KD, Moore LB, Collins JL et al. Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc Natl Acad Sci usa 2001;98:13919–13924.