Abstract
Several 2,5-disubstituted-1,3,4-oxadiazoles (4a–f) and 3,6-disubstituted-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazoles (7a–f) were synthesized and characterized by elemental analyses and spectral data. These compounds were screened for their anti-inflammatory, analgesic, ulcerogenic and lipid peroxidation activities. Compound 7c showed excellent anti-inflammatory and remarkable analgesic activity with reduced ulcerogenic and lipid peroxidation activity when compared with ibuprofen.
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are the most frequently prescribed drugs worldwide in musculoskeletal disordersCitation1. They have long been used as anti-inflammatory, antipyretic and analgesic agents. They reduce pain significantly in patients with arthritis, low back pain and soft tissue painCitation2. However, NSAIDs have important adverse effects, including gastrointestinal (GI) bleeding, peptic ulcer disease, hypertension, oedema and renal disease; more recently, some NSAIDs have also been associated with an increased risk of myocardial infarctionCitation3,Citation4. Therefore, development of novel compounds having anti-inflammatory and analgesic activity with improved safety profile is still a necessity.
During literature survey, we found that compounds having heterocyclic ring substituted with halogenated phenyl ring in their structure are reported as anti-inflammatory and analgesic agents, e.g. zomepirac,Citation5 fluproquazone,Citation6 SC-558Citation7 and DuP-697Citation8 (). It is reported that the incorporation of fluorine into a molecule provides a compound with enhanced biological activityCitation9. Accumulation of fluorine in carbon increases its oxidative and thermal stability. Thus fluorinated drugs are useful because of them being metabolically non-degradableCitation10. Also, inclusion of fluorine may lead to improved lipid solubility thereby enhancing the rate of absorption and transport of drug in vivoCitation11. Additionally, fluorinated drug candidates increase binding affinity to target proteinCitation12. Likewise, bromine-substituted analogues also show better anti-inflammatoryCitation13 activity.
Figure 1. Chemical structures of some halogenated anti-inflammatory and analgesic agents.
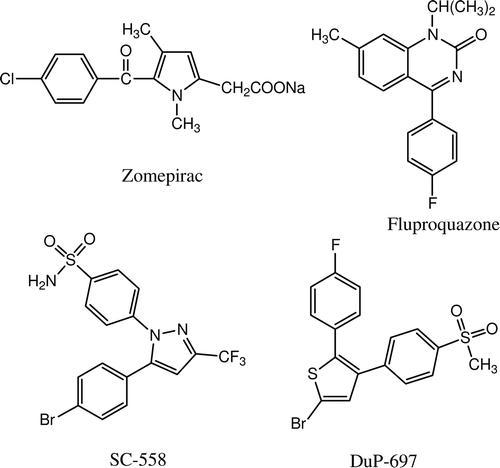
There are reports that dihalogen-substituted compounds exhibit superior biological activityCitation10,Citation14 and only one heterocyclic compound substituted with 3-bromo-4-fluoro phenyl ring is reported as potent analgesic used in neuropathic and inflammatory painCitation15.
It is reported that 1,3,4-oxadiazoles possess a broad spectrum of biological properties including anti-inflammatoryCitation16, antifungalCitation17, anti-mycobacterialCitation18, anticancerCitation19 and antibacterialCitation20 activities. Similarly, 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazoles are also reported to have anti-inflammatoryCitation21, anticancerCitation22, and antimicrobialCitation23 activities.
A survey of literature revealed that many researchers have synthesized 1,3,4- oxadiazoleCitation24 or 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazoleCitation21 derivatives possessing anti-inflammatory activity with reduced ulcerogenic effect.
The above reportings inspired us to synthesize 1,3,4- oxadiazole and 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazole derivatives bearing 3-bromo-4-fluoro phenyl moiety with the aim of getting potential anti-inflammatory and analgesic compounds with reduced GI side effects. The study of the effect of various substituents in the aromatic ring at 5 position of 1,3,4- oxadiazolesCitation25,Citation26 and 6 position of 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazolesCitation27 is based on various reportings.
We hereby report the syntheses of some new 2-(3-bromo-4-fluorophenyl)-5-substituted-1,3,4-oxadiazoles and 3-(3-bromo-4-fluorophenyl)-6-substituted-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazoles. These compounds were screened for their anti-inflammatory, analgesic, ulcerogenic and lipid peroxidation activities.
For the study, ibuprofen was chosen as reference drug. This selection was based on intensive literature survey. Many researchers have already used this drug for comparison purpose to study anti-inflammatory and related activities of different azolesCitation28,Citation29.
Ibuprofen is a traditional NSAID largely employed for its analgesic, anti-inflammatory and antipyretic properties. It is used both as prescription and non-prescription drugCitation3. It has large number of users and so provides the most stable baseline for comparisonsCitation30. Its use, however, may result in ulceration and/or bleedingCitation3. Therefore, by using ibuprofen as reference drug, we could compare both the desirable and non-desirable effects in the tested compounds.
Materials and methods
Chemistry
Melting points were determined in open capillaries and are uncorrected. Chemicals were purchased from Merck Chemical Company, CDH (India), SD Fine (India) and Qualigens (India). Infrared (IR) spectra were recorded on Shimadzu 8400S IR spectrometer. The 1H and 13C NMR spectra were recorded on Bruker AV 300 MHz spectrophotometer using CDCl3 or DMSO-d6 as nuclear magnetic resonance (NMR) solvent. Mass spectra were recorded on a Waters UPLC/MS quadrupole system equipped with electron spray ionization (ESI) source and elemental analyses were carried out on a Vario-EL III CHNOS-Elemental analyzer. Thin layer chromatography (TLC) was performed to monitor progress of the reaction and purity of the compounds, spot being located under iodine vapours or UV light.
Synthesis of 3-bromo-4-fluorobenzoic acid (1)
The mixture of water (17 mL), 4-fluorobenzoic acid (3.50 g, 0.025 mol) and potassium bromate (3.93 g, 0.024 mol) was heated to 80°C and sulphuric acid (16 mL) was added slowly over a period of 3 h. The contents were then heated further at 90°C–100°C for 2 h. To this reaction mixture, water (50 mL) was added and the mixture was extracted with diethyl ether (3 × 30 mL). The organic layer extracts were combined, treated with sodium bisulphite solution and dried over anhydrous magnesium sulphate. The ether was removed to give the yellowish white solid. Yield (62%), m. p. 132°C–134°C (litCitation31, 137°C–139°C).
Synthesis of methyl 3-bromo-4-fluorobenzoate (2)
Compound 1 (2.19 g, 0.01 mol) was refluxed with methanol (30 mL) in presence of few drops of sulphuric acid for 4 h. The reaction mixture was poured onto crushed ice. A yellow oily liquid so obtained was extracted with diethyl ether (2 × 20 mL) and dried over magnesium sulphate. Ether was removed and light yellow oily liquid so obtained was used for the next step. Yield (78%).
Synthesis of 3-bromo-4-fluorobenzohydrazide (3)
Compound 2 (2.33 g, 0.01 mol) was refluxed with hydrazine hydrate (0.5 mL, 0.01 mol) in absolute ethanol (40 mL) for 6 h. The mixture was concentrated, cooled and poured onto crushed ice. The solid thus separated out was filtered, dried and recrystallised from ethanol to give light yellow crystals. Yield (64%); m. p. 120°C–122°C.
General method for the synthesis of 2-(3-bromo-4-fluorophenyl)-5-substituted-1,3,4-oxadiazoles (4a–f)
Compound 3 (0.23 g, 0.001 mol) was refluxed with equimolar quantity of appropriate aromatic acid in presence of phosphorous oxychloride (10 mL) for 8–12 h. After cooling, it was poured onto crushed ice and kept overnight. The solid thus separated out was filtered, washed with water, dried, and recrystallised from ethanol to give 4a–f.
2-(3-Bromo-4-fluorophenyl)-5-phenyl-1,3,4-oxadiazole (4a)
FTIR (KBr pellet) cm−1: 3124 (aromatic C-H stretch); 1627 (C = N stretch); 1532, 1500, 1407 (C = C ring stretch); 1280 (N-N = C); 1211 (asymmetric C-O-C stretch); 1018 (symmetric C-O-C stretch); 1149 (C-F stretch); 543 (C-Br stretch); 1H NMR (DMSO-d6) δ (ppm): 7.30 (dd, 1H, J= 8.4 Hz, J = 8.1 Hz, 5-H), 7.48 (m, 1H, ArH), 7.92 (m, 2H, ArH), 7.98 (ddd, 1H, J = 5.1 Hz, J = 4.5 Hz, J = 2.1 Hz, 6-H), 8.01 (m, 2H, ArH) 8.16 (dd, 1H, J = 6.3 Hz, J = 2.1 Hz, 2-H); 13C NMR (DMSO-d6) δ (ppm): 165.2 (C), 157.4 (C), 156.3 (C), 133.8 (C), 132.4 (C), 131.5 (C), 128.8 (C), 127.3 (2C), 126.1 (2C), 122.3 (C), 114.6 (C), 112.3 (C); MS: m/z 319 (M+); Anal. Calcd for C14H8BrFN2O: C, 52.69; H, 2.53; N, 8.78; Found C, 52.71; H, 2.54; N, 8.77%.
2-(3-Bromo-4-fluorophenyl)-5-(4-chlorophenyl)-1,3,4-oxadiazole (4b)
2-(3-Bromo-4-fluorophenyl)-5-(4-fluorophenyl)-1,3,4-oxadiazole (4c)
2-(3-Bromo-4-fluorophenyl)-5-(4-methylphenyl)-1,3,4-oxadiazole (4d)
2-(3-Bromo-4-fluorophenyl)-5-(4-methoxyphenyl)-1,3,4-oxadiazole (4e)
2-(3-Bromo-4-fluorophenyl)-5-(1-naphthylmethyl)-1,3,4-oxadiazole (4f)
Synthesis of potassium salt of 3-bromo-4-fluorobenzohydrazide (5)
A mixture of compound 3 (2.33 g, 0.01 mol), carbon disulfide (0.9 mL, 0.015 mol) and potassium hydroxide (0.84 g, 0.015 mol) in absolute ethanol (50 mL) was refluxed for 14 h. It was cooled to room temperature and diluted with dry ether (25 mL). The solid so obtained was filtered, washed with ether and vacuum dried at 65°C. The salt so obtained was used as such for next step. Yield (67%).
Synthesis of 4-amino-5-(3-bromo-4-fluorophenyl)-4H-1,2,4-triazole-3-thiol (6)
A mixture of compound 5 (3.35 g, 0.01 mol), hydrazine hydrate (1.0 mL, 0.02 mol) and water (15 mL) was refluxed for 2 h. The contents turned green, hydrogen sulphide gas was evolved and a homogenous mixture was obtained. The contents were diluted with cold water (100 mL) and acidified with hydrochloric acid; a white solid so obtained was filtered, washed with cold water and recrystallised from aqueous ethanol to give compound 6. Yield (65%); m. p. 138°C–140°C.
FTIR (KBr pellet) cm−1: 3417 (N-H stretch); 3078 (aromatic C-H stretch); 2673 (S-H stretch) 1685 (C = N stretch); 1539, 1508, 1427 (C = C ring stretch); 1292 (N-N = C); 1049 (C-F stretch); 497 (C-Br stretch); 1H NMR (DMSO-d6) δ (ppm): 2.51 (s, 1H, S-H), 3.82 (s, 2H, NH2), 7.04 (dd, J = 8.4 Hz, J = 7.8 Hz, 1H, 5-H), 7.91 (ddd, 1H, J = 5.1 Hz, J = 4.2 Hz, J = 1.8 Hz, 6-H), 8.14 (dd, 1H, J = 6.6 Hz, J = 1.8 Hz, 2-H); 13C NMR (DMSO-d6) δ (ppm): 165.7 (C), 160.4 (C), 158.3 (C), 136.1 (C), 134.3 (C), 126.4 (C), 118.1 (C), 112.3 (C); MS: m/z 288 (M+); Anal. Calcd for C8H6BrFN4S: C, 33.23; H, 2.09; N, 19.38; Found C, 33.29; H, 2.09; N, 19.36%.
General method for the synthesis of 3-(3-bromo-4-fluorophenyl)-6-substituted-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazoles (7a–f)
Compound 6 (0.29 g, 0.001 mol) was refluxed with equimolar quantity of appropriate aromatic acid in presence of phosphorous oxychloride (10 mL) for 3–5 h. The reaction mixture was cooled to room temperature and poured onto crushed ice with constant stirring. The mixture was allowed to stand overnight and the solid thus separated out was filtered, treated with 5% of dilute sodium hydroxide solution, washed with water and recrystallised from ethanol to give 7a–f.
3-(3-Bromo-4-fluorophenyl)-6-phenyl-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazole (7a)
FTIR (KBr pellet) cm−1: 3039 (aromatic C-H stretch); 1704 (C = N stretch); 1550, 1527, 1442 (C = C ring stretch); 1384 (N = C-S); 1292 (N-N = C); 1053 (C-F stretch); 563 (C-Br stretch); 1H NMR (DMSO-d6) δ (ppm): 7.18 (dd, 1H, J = 8.7 Hz, J = 8.1 Hz, 5-H), 7.48 (m, 1H, ArH), 7.89 (m, 2H, ArH), 7.97 (ddd, 1H, J = 4.8 Hz, J = 4.5 Hz, J = 1.8 Hz, 6-H), 8.01 (m, 2H, ArH) 8.16 (dd, 1H, J = 6.9 Hz, J = 2.1 Hz, 2-H); 13C NMR (DMSO-d6) δ (ppm): 164.2 (C), 160.7 (C), 157.1 (C), 154.3 (C), 134.3 (C), 132.6 (C), 131.4 (C), 128.3 (C), 126.8 (2C), 126.2 (2C), 116.3 (C), 113.8 (C), 112.4 (C); MS: m/z 375 (M+); Anal. Calcd for C15H8BrFN4S: C, 48.01; H, 2.15; N, 14.93; Found C, 47.96; H, 2.15; N, 14.96%.
3-(3-Bromo-4-fluorophenyl)-6-(4-chlorophenyl)-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazole (7b)
3-(3-Bromo-4-fluorophenyl)-6-(4-fluorophenyl)-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazole (7c)
3-(3-Bromo-4-fluorophenyl)-6-(4-methylphenyl)-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazole (7d)
3-(3-Bromo-4-fluorophenyl)-6-(4-methoxyphenyl)-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazole (7e)
3-(3-Bromo-4-fluorophenyl)-6-(1-naphthylmethyl)-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazole (7f)
Animals
Adult Wistar albino rats of either sex, weighing between 160 and 200 g were used for anti-inflammatory, ulcerogenic and lipid peroxidation activities, whereas Swiss albino mice of either sex, weighing between 18 and 25 g were used to evaluate analgesic activity. The animals were randomly distributed into groups at the beginning of all the experiments. In each group six animals were kept. The animals were allowed food and water ad libitum except during the experiments. All the test compounds and the reference drug were administered orally, suspended in 1% carboxymethylcellulose (CMC) with one drop of twin-80 solution wherever required. The experimental protocol was approved by the animal ethics committee of Jamia Hamdard.
Anti-inflammatory activity
Anti-inflammatory activity was evaluated by carrageenan-induced hind paw oedema methodCitation32. Into the subplantar region of the right hind paw of each rat, 0.1 mL of 1% carrageenan solution in saline was injected subcutaneously, 1 h after administration of the test compounds and standard drug. Control group received only solution of 10 mL/kg 1% CMC with one drop of twin-80. Standard drug ibuprofen was administered orally at a dose of 20 mg/kg. The test compounds were administered orally at same dose of 20 mg/kg. The right hind paw volume was measured with a digital plethysmometer (Panlab LE 7500) before and after 1, 3 and 4 h of carrageenan treatment. Percentage of anti-inflammatory activity was calculated according to the following formula:
where Vc represents the mean increase in paw volume in control group of rats and Vt represents the mean increase in paw volume in rats treated with test compounds and standard drug.
Analgesic activity
Analgesic activity was evaluated by acetic acid-induced writhing methodCitation33. It was performed by an Intraperitoneal (IP) injection of 1% aqueous acetic acid solution in a volume of 0.1 mL. Screening of analgesic activity was performed after PO (oral) administration of test compounds at a dose of 20 mg/kg. All the compounds were dissolved in 1% CMC. One group was kept as control and received PO administration of 1% CMC. Ibuprofen 20 mg/kg was used as reference drug. After 1 h of drug administration, 0.1 mL of 1% acetic acid solution was given to mice intraperitoneally. The total number of writhes was recorded for 10 min beginning 5 min after the acetic acid injection. The analgesic activity was expressed in terms of percentage inhibition of the number of writhes.
where Nc represents the mean number of writhes of control group of mice and Nt represents the mean number of writhes of test group of mice.
Acute ulcerogenicity studies
Acute ulcerogenicity screening was performed according to the method reported by Cioli et al.Citation34 Ulcerogenic activity was evaluated after PO administration of test compounds or ibuprofen at a dose of 20 mg/kg. Control group received PO administration of vehicle (suspension of 1% CMC). Food, but not water, was removed 24 h before administration of the test compounds and standard drug. After the drug treatment, the rats were fed with normal diet for 17 h and then sacrificed under light ether anaesthesia. The stomach was then removed and opened along the greater curvature, washed with distil water and cleaned gently by dipping in saline. The mucosal damage was examined by means of a magnifying glass. For each stomach, the mucosal damage was assessed according to the following scoring system:
0.5 Redness
1.0 Spot ulcers
1.5 Hemorrhagic streaks
2.0 Ulcers < 3, but ≤ 5
3.0 Ulcers > 5
The mean score of each treated group minus the mean score of control group was regarded as severity index of gastric mucosal damage.
Lipid peroxidation
Lipid peroxidation in the gastric mucosa was evaluated according to the method reported by Ohkawa et al.Citation35 After screening for ulcerogenic effect of synthesized compounds the gastric mucosa of animals was scraped with two glass slides, weighed (100 mg) and homogenized in 1.8 mL of 1.15% ice-cold KCl solution. The homogenate was supplemented with 0.2 mL of 8.1% sodium dodecyl sulphate (SDS), 1.5 mL of acetate buffer (pH 3.5) and 1.5 mL of 0.8% thiobarbituric acid (TBA). The mixture was heated at 95°C for 60 min. After cooling, the contents were shaken vigorously for 1 min and centrifuged for 10 min at 4000 rpm after supplementing with 5 mL of the mixture of n-butanol and pyridine (15:1 v/v). The supernatant organic layer was separated and absorbance was measured at 532 nm on UV spectrophotometer. The results are expressed as nmols of MDA per 100 mg tissue, using extinction coefficient 1.56 × 105 cm−1M−1.
Acute toxicity study
Approximate 50% lethal dose (ALD50) of the promising compounds was determined in albino mice by the reported methodCitation36. Swiss albino mice of either sex weighing 18–25 g were used. The test compounds were dissolved in DMSO in 500, 750, and 1000 mg/kg body weight doses and were injected intraperitoneally. The toxic symptoms and mortality rates in each group were recorded 24 h after drug administration.
Statistical analysis
Statistical analysis was carried out by using Graphpad Prism 3.0 (Graphpad software; San Diego, California, USA). All results were expressed by mean ± SD. Graphs of data were compared with an analysis of variance (ANOVA) followed by Dunnett’s multiple comparison test. Values were considered statistically significant as p < 0.05.
Results and discussion
Chemistry
The reaction sequences involved in the synthesis of target compounds are shown in .
Scheme 1. Reagents and conditions: Synthetic route to target compounds 4a–f and 7a–f. (i) KBrO3, 80°C; (ii) H2SO4 add over a period of 3 h; (iii) heat, 90°C−100°C, 2 h; (iv) CH3OH, H2SO4, reflux for 4 h; (v) NH2NH2.H2O, absolute C2H5OH, reflux for 6 h; (vi) POCl3, Ar-COOH, reflux for 8–12 h; (vii) KOH, CS2, absolute C2H5OH, reflux for 14 h, (viii) NH2NH2.H2O, reflux for 2 h; (ix) POCl3, Ar-COOH, reflux for 3–5 h. Where a, Ar= C6H5; b, Ar= 4-Cl-C6H4; c, Ar= 4-F-C6H4; d, Ar= 4-CH3-C6H4; e, Ar= 4-OCH3-C6H4; f, Ar= C11H9;
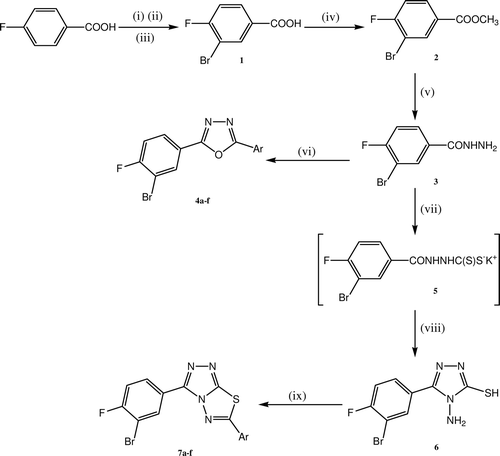
The starting material 3-bromo-4-fluorobenzoic acid 1 was successfully synthesized by the bromination of 4-fluoro-benzoic acid on the basis of the synthesis of 3-bromobenzoic acidCitation37 and halogenation reactionsCitation31. Compound 1 on esterification with methanol in presence of catalytic amounts of sulphuric acid gave methyl 3-bromo-4-fluorobenzoate 2. Further, compound 2 was converted into 3-bromo-4-fluorobenzohydrazide 3, by reacting it with hydrazine hydrate in presence of absolute ethanol. The 2-(3-bromo-4-fluorophenyl)-5-substituted-1,3,4-oxadiazoles 4a–f were successfully obtained by reacting 3 with appropriate aromatic acid in presence of phosphorous oxychloride. However, refluxing the compound 3 with carbon disulphide and potassium hydroxide in absolute alcohol formed potassium salt of 3-bromo-4-fluorobenzohydrazide 5. This, potassium salt on treatment with hydrazine hydrate gave 4-amino-5-(3-bromo-4-fluorophenyl)-4H-1,2,4-triazole-3-thiol 6. Compound 6 on reacting with appropriate aromatic acid in presence of phosphorous oxychloride yielded 3-(3-bromo-4-fluorophenyl)-6-substituted-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazoles 7a–f.
Anti-inflammatory activity
All the newly synthesized compounds 4a–f and 7a–f were evaluated for their in vivo anti-inflammatory activity by carrageenan-induced rat paw oedema method of Winter et al.Citation32 The effect of various substituents in the aromatic ring was studied. The compounds were tested at 20 mg/kg oral dose and response was compared with the standard drug ibuprofen at the same oral dose. The percentage inhibition was calculated after 1, 3 and 4 h of carrageenan treatment. Since maximum inhibition was observed after 4 h, this was made the basis of discussion. The tested compounds showed anti-inflammatory activity ranging from 23.75%–51.86% (), whereas the standard drug ibuprofen showed 60.06% inhibition after 4 h. The 1,3,4-oxadiazole derivatives 4a–f exhibited anti-inflammatory activity in the range from 23.75% to 35.36%. The 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazole derivatives 7a–f displayed better activity ranging from 29.95% to 51.86%.
Table 1. Anti-inflammatory activity of the synthesized compounds after oral administration (20 mg/kg).
It was interesting to note that the compounds 4a and 7a having unsubstituted aryl ring showed the lowest anti-inflammatory activity in their respective series (23.75% and 29.95%, respectively). It was observed that introduction of halogen atom in the aryl ring increased the activity in both the series of compounds. Compound 7c, a 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazole derivative, the aryl ring of which is substituted with fluorine atom showed the highest activity (51.86%) which is comparable with ibuprofen (60.06%). When this fluorine atom was replaced by chlorine atom the obtained compound 7b showed 46.61% inhibition. The same pattern of anti-inflammatory activity was also observed with 1,3,4-oxadiazole derivatives 4a–f. Compound 4c containing fluorine atom at para-position in the aryl ring present at 5 position of 1,3,4-oxadiazole nucleus showed an increase in anti-inflammatory activity from 23.75% to 34.66% compared with unsubstituted aryl ring. When the fluorine atom was replaced by chlorine atom, the obtained compound 4b showed 33.03% anti-inflammatory activity. It was noted when the aryl ring was replaced by naphthyl ring, the obtained compound 4f showed the highest activity 35.36% among 1,3,4-oxadiazole derivatives. It was also observed that changing electronic parameter adversely affected the activity, a decrease in activity was observed by replacing halogen atom with methyl (4d and 7d; 26.97% and 32.17%, respectively) or methoxy (4e and 7e; 28.19% and 34.25%, respectively) group. Statistically significant testing using ANOVA followed by Dunnett’s multiple comparison test showed that the anti-inflammatory activity of all the tested compounds except 7c were in the range of p < 0.01 when compared with ibuprofen. Anti-inflammatory activity of compound 7c was found comparable to ibuprofen. Potency of synthesized compounds was determined in comparison to ibuprofen. Compounds having potency > 0.50 were further selected for determining their ulcerogenic activity.
Analgesic activity
The analgesic activity of the synthesized compounds was evaluated by acetic acid-induced writhing methodCitation33. The tested compounds showed analgesic activity ranging from 15.65% to 48.48%, whereas ibuprofen showed 60.48% inhibition (). The 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazole derivatives 7a–f showed better analgesic activity (25.61%–48.48%) compared with 1,3,4-oxadiazole derivatives 4a–f (15.65%–27.71%). It was observed that compound 7c possessing excellent anti-inflammatory activity also showed highest analgesic activity 48.18%. In compound 7c, aryl ring is substituted with fluorine atom. When the fluorine atom of this compound was replaced by chlorine atom, the obtained compound 7b was also found to have a good analgesic activity 44.48%. Compounds 7e and 7f showed moderate analgesic activity (36.95% and 35.83%, respectively).
Table 2. Analgesic, ulcerogenic and lipid peroxidation activities of the synthesized compounds.
Acute ulcerogenicity studies
The ulcerogenic activity was done according to Cioli et al.Citation34 The compounds showed ulcerogenic activity ranging from 0.750 to 0.925. The standard drug ibuprofen showed high severity index of 1.738 (). It was clear that the ulcerogenic effect of all the test compounds was appreciably less than ibuprofen. Five compounds 4b, 4c, 7b, 7c and 7d showed severity index having half the value of ibuprofen. It is interesting to note that all the 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazole derivatives substituted with halogenated phenyl ring at 6 position have very less ulcerogenic activity.
Lipid peroxidation
Compounds with low ulcerogenic activity are also reported to have reduced malondialdehyde (MDA) content, a by-product of lipid peroxidationCitation35 Therefore by determining the MDA level, an attempt was made to correlate the reduction in ulcerogenic activity of the compounds with that of lipid peroxidation. The lipid peroxidation was measured as nmols of MDA/100 mg of tissue. All the compounds which were evaluated for ulcerogenic activity were also subjected to lipid peroxidation studies. The animals treated with ibuprofen showed lipid peroxidation 7.375, whereas the control group exhibited 3.978 and the groups treated with the synthesized compounds showed it to be ranging from 5.008 to 5.615 (). These results further confirmed that the synthesized compounds have less ulcerogenic effects compared with the standard drug ibuprofen.
Acute toxicity
Compounds 7b and 7c exhibiting prominent anti-inflammatory and analgesic activity were further selected for evaluating their approximate lethal dose (ALD50) in mice as described in the literatureCitation36. However, no toxic symptoms or mortality rates were observed 24 h post-administration even at the dose of 1000 mg/kg body weight suggesting their wide margin of safety.
Conclusion
We report herein that the syntheses of 2-(3-bromo-4-fluorophenyl)-5-substituted-1,2,4-oxadiazoles 4a–f and 3-(3-bromo-4-fluorophenyl)-6-substituted-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazoles 7a–f. The synthesized compounds were studied for their anti-inflammatory, analgesic, ulcerogenic and lipid peroxidation properties. The biological results showed that, compound 7c possessed highest anti-inflammatory (51.86%) and analgesic (48.18%) activity. Also, there was marked reduction in ulcerogenic and lipid peroxidation activity (0.763 and 5.108, respectively) compared with the standard drug (1.738 and 7.375, respectively). Similarly, compound 7b exhibited moderate anti-inflammatory and analgesic activity with less ulceration and lipid peroxidation (46.61%, 44.48%, 0.788 and 5.008, respectively). Compounds 7b and 7c were further selected for toxicity studies and were found to have a wide margin of safety.
The 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazoles derivative having fluorine-substituted aryl ring at 6 position was identified as most potent anti-inflammatory and analgesic agent with reduced ulcerogenic effect.
Acknowledgements
The authors are thankful to the Jamia Hamdard, New Delhi for providing research facilities. They are also thankful to IIT, New Delhi for providing the 1H NMR and 13C NMRspectra. Special thanks to Prof. M.S.Y. Khan, Prof. Emeritus, Jamia Hamdard, New Delhi for his encouragement and blessings.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Ray WA, Stein CM, Byrd V, Shorr R, Pichert JW, Gideon P et al. Educational program for physicians to reduce use of non-steroidal anti-inflammatory drugs among community-dwelling elderly persons: A randomized controlled trial. Med Care 2001;39:425–435.
- Rang HP, Dale MM, Ritter JM, Flower RJ. Anti-inflammatory and immunosuppressant drugs. Rang and dale’s pharmacology. Elsevier Science Health Science div; 2007, pp. 227.
- Cocco MT, Congiu C, Onnis V, Morelli M, Cauli O. Synthesis of ibuprofen heterocyclic amides and investigation of their analgesic and toxicological properties. Eur j Med Chem 2003;38:513–518.
- Dannhardt G, Kiefer W. Cyclooxygenase inhibitors–current status and future prospects. Eur j Med Chem 2001;36:109–126.
- Marnett LJ, Kalgutkar AS. Design of selective inhibitors of cyclooxygenase-2 as nonulcerogenic anti-inflammatory agents. Curr Opin Chem Biol 1998;2:482–490.
- Herrmann U, Laube W, Roth F, Schultheiss HR, Berger M, Schürmann P et al. Analgesic effect of fluproquazone in postoperative patients. Clin Pharmacol Ther 1980;27:379–385.
- Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, Pak JY et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996;384:644–648.
- Prasit P, Wang Z, Brideau C, Chan CC, Charleson S, Cromlish W et al. The discovery of rofecoxib, [MK 966, Vioxx, 4-(4’-methylsulfonylphenyl)-3-phenyl-2(5H)-furanone], an orally active cyclooxygenase-2-inhibitor. Bioorg Med Chem Lett 1999;9:1773–1778.
- Isanbor C, O’Hagan D. Fluorine in medicinal chemistry: A review of anti-cancer agents. J Fluorine Chem 2006;127:303–319.
- Karthikeyan MS, Holla BS, Kumari NS. Synthesis and antimicrobial studies on novel chloro-fluorine containing hydroxy pyrazolines. Eur j Med Chem 2007;42:30–36.
- Karthikeyan MS, Prasad DJ, Mahalinga M, Holla BS, Kumari NS. Antimicrobial studies of 2,4-dichloro-5-fluorophenyl containing oxadiazoles. Eur j Med Chem 2008;43:25–31.
- Shah P, Westwell AD. The role of fluorine in medicinal chemistry. j Enzyme Inhib Med Chem 2007;22:527–540.
- Goel B, Ram T, Tyagi R, Bansal E, Kumar A, Mukherjee D, Sinha JN. 2-Substituted-3-(4-bromo-2-carboxyphenyl)-5-methyl-4-thiazolidinones as potential anti-inflammatory agents. Eur J Med Chem 1999;34:265–269.
- Zheng X, Li Z, Wang Y, Chen W, Huang Q, Liu C, Song G. Syntheses and insecticidal activities of novel 2,5-disubstituted 1,3,4-oxadiazoles. J Fluorine Chem 2003;123:163–169.
- Knutsen LJ, Hobbs CJ, Earnshaw CG, Fiumana A, Gilbert J, Mellor SL et al. Synthesis and SAR of novel 2-arylthiazolidinones as selective analgesic N-type calcium channel blockers. Bioorg Med Chem Lett 2007;17:662–667.
- Bhandari SV, Bothara KG, Raut MK, Patil AA, Sarkate AP, Mokale VJ. Design, synthesis and evaluation of antiinflammatory, analgesic and ulcerogenicity studies of novel S-substituted phenacyl-1,3,4-oxadiazole-2-thiol and Schiff bases of diclofenac acid as nonulcerogenic derivatives. Bioorg Med Chem 2008;16:1822–1831.
- Chen CJ, Song BA, Yang S, Xu GF, Bhadury PS, Jin LH et al. Synthesis and antifungal activities of 5-(3,4,5-trimethoxyphenyl)-2-sulfonyl-1,3,4-thiadiazole and 5-(3,4,5-trimethoxyphenyl)-2-sulfonyl-1,3,4-oxadiazole derivatives. Bioorg Med Chem 2007;15:3981–3989.
- Ali MA, Shaharyar M. Oxadiazole mannich bases: Synthesis and antimycobacterial activity. Bioorg Med Chem Lett 2007;17:3314–3316.
- Aboraia AS, Abdel-Rahman HM, Mahfouz NM, El-Gendy MA. Novel 5-(2-hydroxyphenyl)-3-substituted-2,3-dihydro-1,3,4-oxadiazole-2-thione derivatives: Promising anticancer agents. Bioorg Med Chem 2006;14:1236–1246.
- Joshi SD, Vagdevi HM, Vaidya VP, Gadaginamath GS. Synthesis of new 4-pyrrol-1-yl benzoic acid hydrazide analogs and some derived oxadiazole, triazole and pyrrole ring systems: A novel class of potential antibacterial and antitubercular agents. Eur j Med Chem 2008;43:1989–1996.
- Amir M, Kumar H, Javed SA. Synthesis and pharmacological evaluation of condensed heterocyclic 6-substituted-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazole derivatives of naproxen. Bioorg Med Chem Lett 2007;17:4504–4508.
- Holla BS, Poojary KN, Rao BS, Shivananda MK. New bis-aminomercaptotriazoles and bis-triazolothiadiazoles as possible anticancer agents. Eur j Med Chem 2002;37:511–517.
- Methew V, Keshavayya J, Vaidya VP. Heterocyclic system containing bridgehead nitrogen atom: Synthesis and pharmacological activities of some substituted 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazoles. Eur J Med Chem 2006;41:1048–1058.
- Chandrakantha B, Shetty P, Nambiyar V, Isloor N, Isloor AM. Synthesis, characterization and biological activity of some new 1,3,4-oxadiazole bearing 2-flouro-4-methoxy phenyl moiety. Eur j Med Chem 2010;45:1206–1210.
- Husain A, Ahmad A, Alam MM, Ajmal M, Ahuja P. Fenbufen based 3-[5-(substituted aryl)-1,3,4-oxadiazol-2-yl]-1-(biphenyl-4-yl)propan-1-ones as safer antiinflammatory and analgesic agents. Eur j Med Chem 2009;44:3798–3804.
- Palaska E, Sahin G, Kelicen P, Durlu NT, Altinok G. Synthesis and anti-inflammatory activity of 1-acylthiosemicarbazides, 1,3,4-oxadiazoles, 1,3,4-thiadiazoles and 1,2,4-triazole-3-thiones. Farmaco 2002;57:101–107.
- Karegoudar P, Prasad DJ, Ashok M, Mahalinga M, Poojary B, Holla BS. Synthesis, antimicrobial and anti-inflammatory activities of some 1,2,4-triazolo[3,4-b][1,3,4]thiadiazoles and 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines bearing trichlorophenyl moiety. Eur j Med Chem 2008;43:808–815.
- Akhter M, Husain A, Azad B, Ajmal M. Aroylpropionic acid based 2,5-disubstituted-1,3,4-oxadiazoles: Synthesis and their anti-inflammatory and analgesic activities. Eur j Med Chem 2009;44:2372–2378.
- Sun XY, Hu C, Deng XQ, Wei CX, Sun ZG, Quan ZS. Synthesis and anti-inflammatory activity evaluation of some novel 6-alkoxy(phenoxy)-[1,2,4]triazolo[3,4-a]phthalazine-3-amine derivatives. Eur j Med Chem 2010;45:4807–4812.
- Carson JL, Strom BL, Morse ML, West SL, Soper KA, Stolley PD et al. The relative gastrointestinal toxicity of the nonsteroidal anti-inflammatory drugs. Arch Intern Med 1987;147:1054–1059.
- Chambers R, Skinner C, Atherton M, Moilliet J. Halogenation Reactions. US Patent, 667, 088 (2001).
- Winter CA, Risley EA, Nuss GW. Carrageenin-induced edema in hind paw of the rat as an assay for antiiflammatory drugs. Proc Soc Exp Biol Med 1962;111:544–547.
- Siegmund E, Cadmus R, Lu G. A method for evaluating both non-narcotic and narcotic analgesics. Proc Soc Exp Biol Med 1957;95:729–731.
- Cioli V, Putzolu S, Rossi V, Scorza Barcellona P, Corradino C. The role of direct tissue contact in the production of gastrointestinal ulcers by anti-inflammatory drugs in rats. Toxicol Appl Pharmacol 1979;50:283–289.
- Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem 1979;95:351–358.
- Bekhit AA, Fahmy HT. Design and synthesis of some substituted 1H-pyrazolyl-oxazolidines or 1H-pyrazolyl-thiazolidines as anti-inflammatory-antimicrobial agents. Arch Pharm (Weinheim) 2003;336:111–118.
- Harrison JJ, Pellegrini JP, Selwitz CM. Bromination of deactivated aromatics using potassium bromate. J Org Chem 1981;46:2169–2171.