Abstract
Tetrahydro-β-carboline derivatives (THBCs) have been identified as a class of potent Type-5 Phosphodiesterase (PDE5) inhibitors, showing benefits for the treatment of erectile dysfunction and also bearing anticancer properties. A computational strategy based on molecular docking studies, followed by docking-based Comparative Molecular Fields Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA), has been used to elucidate the atomic details of the PDE5/THBC interactions and to identify the most important features impacting the THBC PDE5 inhibitory activity. The final CoMSIA model resulted to be the more predictive, showing rncv2 = 0.96, rcv2 = 0.688, SEE = 0.248, F = 104.800, and r2pred = 0.78. The results allowed us to obtain useful information for the design of new THBC analogues, potentially acting as PDE5 inhibitors, and to predict their potency prior to synthesis.
Introduction
Cyclic nucleotide phosphodiesterases (PDEs) belong to a large family of enzymes able to catalyse the hydrolysis of second messengers cAMP and cGMP into the corresponding 5′-.nucleotide monophosphates. On the basis of their pharmacological properties and substrate affinity, eleven PDE sub-families (further divided in more than 60 isoforms and splice variants) have been identified to date. As the PDE4 family of enzymes is selective for cAMP, the PDE5 enzyme is selective for cGMPCitation1. The PDE5 isoform is expressed in smooth muscle tissue, importantly, in the corpus cavernosum2, and lung platelets. Thus, up to now PDE5 inhibitors have received considerable attention for having been successfully developed as potential drug for erectile dysfunction. Among them, three PDE5 inhibitors, tadalafil, sildenafil, and vardenafil, have reached the market, having received Food and Drug Administration (FDA) approval.
Notably, recent studies have highlighted that PDE5 is also overexpressed in various carcinomas like colon, pancreatic, lung and bladder cancersCitation3,Citation4, making this enzyme an interesting target for development of new PDE5 inhibitors, potentially bearing anticancer properties.
In particular, certain tadalafil analogues have shown tumor cell growth inhibitory effects, which appeared to be associated with PDE5 inhibitionCitation5.
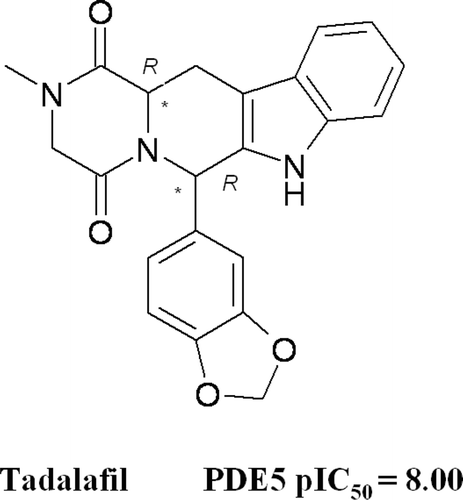
Chemically, tadalafil () is a tetrahydro-β-carboline (THBC) piperazinedione with two chiral carbons, both in R configuration, a methyl substituent on the piperazinedione nitrogen, and with a 1,3-benzodioxol substituent linked to C-6. In 2003, Maw and co-workers published a series of tadalafil analogues, especially exploring the effects caused by the introduction of flexible and/or bulky groups at the piperazinedione nitrogen atom (instead of the tadalafil methyl substituent), and by the replacement of the 1,3-benzodioxol ring with a phenyl one6. More recently, Abadi and collaborators discussed a series of new THBC hydantoines and piperazinediones as novel PDE5 inhibitorsCitation7.
Figure 1. Chemical structure of PDE5 inhibitor tadalafil.
Despite the utility of these compounds, one potential drawback could be cross-reactivity with the closely related PDE6 and PDE11 enzymes. A better understanding of the mode of function of PDE5/inhibitor complexes can be greatly enriched through analyses and docking procedures on the available PDE5 catalytic domain crystal structures, in tandem with quantitative-structure activity relationship analyses, performed on known PDE5 inhibitor seriesCitation8.
In particular, up to now, several computational and statistical studies have been performed on PDE5 inhibitors, bearing the imidazolo- or the pyrazolopirimidone scaffoldCitation9–11.
In this work, with the aim to elucidate the most important PDE5/THBC interactions and to elaborate a quantitative structure-activity relationship (QSAR) model, for enabling prediction of the THBC PDE5 inhibitory behaviour prior to synthesis, a computational protocol was applied. On the basis of the crystal structure of PDE5 in complex with tadalafil (pdb code:1XOZCitation12), a flexible docking simulation was performed on a series of 68 THBCs. The most probable docking poses, which resulted to be aligned into the enzyme catalytic site, were selected and submitted to the following docking-based 3D-QSAR studies, involving Comparative Molecular Fields Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA). The results allowed us to obtain useful suggestions for the design of new derivatives potentially acting as PDE5 inhibitors.
Methods
Data set
A dataset of 68 tetrahydro-β-carbolines were selected from two sample data setsCitation6,Citation7. The PDE5 inhibitory potency of compounds 1–42 (7) and of 43–68 (6), were measured by means of Immobilized Metal Affinity for Phosphochemicals (IMAP)-based Fluorescence Polarization (FP) phosphodiesterase evaluation assays, and using a Scintillation Proximity Assay (SPA)-based method, respectively. Since 1–68 pIC50 values are comparable (Tadalafil has been used as biological assays reference compound on both the two methods, displaying results within the same order of magnitude), all the compounds were submitted to CoMFA and CoMSIA analyses.
Table 1. Molecular structure of PDE5 inhibitors 1–6.
Table 2. Molecular structure of PDE5 inhibitors 6–22.
Table 3. Molecular structure of PDE5 inhibitors 23–42.
Table 4. Molecular structure of PDE5 inhibitors 43–68.
All the compounds were built, parameterized (Gasteiger-Hückel method) and energy minimized within MOE using MMFF94 force fieldCitation13.
Docking-based ligand alignment
To locate the appropriate binding orientations and conformations of carboline derivatives within the PDE5 catalytic site, a computational searching method was applied. Starting from a database including all the 68 compounds, a docking procedure was performed. Thus, on the basis of the three-dimensional structure co-ordinates of the PDE5/tadalafil complex (PDB entry 1XOZCitation12) (tadalafil in the bioactive conformation), each inhibitor was docked into the PDE5 catalytic site using the flexible docking module implemented in Surflex.
As reported by CardCitation12, tadalafil makes neither direct nor water-mediated interactions with the metal ions, belonging to the catalytic site. On the contrary, the inhibitor makes many hydrophobic interactions that strongly contribute to its high potency.
Thus, for docking simulations of tadalafil analogues, waters were not taken into account.
Surflex-Dock scores are expressed in –log10(Kd) units to represent binding affinitiesCitation14.
Since for all compounds the best-docked geometries were in agreement with the crystallographic data of the PDE5/tadalafil complex (and thus already aligned), they were directly submitted to CoMFACitation15 and CoMSIACitation16 studies by Sybyl-X 1.0 softwareCitation17.
3D-QSAR analyses
Training set and test set
All the compounds were grouped into a training set, for model generation, and a test set, for model validation, containing 53 and 15 compounds respectively. Both the training and the test set were divided manually according to a representative range of biological activities and structural variations. For QSAR analysis, IC50 values have been transformed into pIC50 values and then used as response variables. Compound pIC50 range covered 4 log orders of magnitude.
CoMFA and CoMSIA interaction energies
CoMFA methodCitation15 is a widely used 3D-QSAR technique to relate the biological activity of a series of molecules to their steric and electrostatic fields, which are calculated placing the aligned molecules, one by one, into a 3D cubic lattice with a 2 Å grid spacing. The van der Waals potential and Coulombic terms, which represent steric and electrostatic fields, respectively, were calculated using the standard Tripos force field method. The column-filtering threshold value was set to 2.0 kcal/mol to improve the signal-noise ratio. A methyl probe with a +1 charge was used to calculate the CoMFA steric and electrostatic fields. A 30 Kcal/mol energy cut-off was applied to avoid infinity of energy values inside the molecule. The CoMSIA methodCitation16 calculates five descriptors, namely steric, electrostatic and hydrophobic parameters, and the H-bond donor and H-bond acceptor properties. The similarity index descriptors were calculated using the same lattice box employed for the CoMFA calculations and a sp3 carbon as probe atom with a +1 charge, +1 hydrophobicity and +1 H-bond donor and +1 H-bond acceptor properties.
Partial least square (PLS) analysis and models validation
The partial least-squares (PLS) approach, an extension of the multiple regression analysis, was used to derive the 3D-QSAR models. CoMFA and CoMSIA descriptors were used as independent variables and pIC50 values were used as dependent variables. The leave one out (LOO) cross-validation method was used to check the predictivity of the derived model and to identify the optimal number of components (ONC) leading to the highest cross-validated r2 (r2cv). In the LOO methodology, one molecule is omitted from the dataset and a model is derived involving the rest of the compounds. Employing this model, the activity of the omitted molecule is then predicted.
The ONC obtained from cross-validation methodology was used in the subsequent regression model. Final CoMFA and CoMSIA models were generated using non-cross-validated PLS analysis. To further assess the statistical confidence and robustness of the derived models, a 100-cycle bootstrap analysis was performed. This is a procedure in which n random selections out of the original set of n objects are performed several times (100-times were required to obtain a good statistical information). In each run, some objects may not be included in the PLS analysis, whereas some others might be included more then once. The mean correlation coefficient is represented as bootstrap r2 (r2boot).
Predictive correlation coefficient (r2pred)
To further validate the CoMFA and CoMSIA derived model, the predictive ability for the test set of compounds (expressed as r2pred) was determined by using the following equation:
SD is the sum of the squared deviations between the biological activities of the test set molecules and the mean activity of the training set compounds and PRESS is the sum of the squared deviation between the observed and the predicted activities of the test set compounds.
All calculations were carried out using a PC running the Windows XP operating system and an SGI O2 Silicon Graphics.
Results and discussion
Docking-based ligand alignment
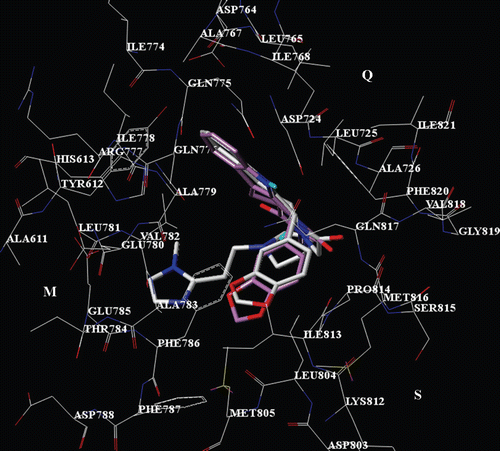
In this work, for the molecular modeling analysis we used the X-ray co-ordinates of the crystal structure of PDE5/tadalafil complex (pdb code:1XOZ). According to the experimental data reported by Card, the catalytic site of all PDEs can be subdivided in a metal binding pocket (M pocket); a solvent-filled side pocket (S pocket); and in a pocket containing a purine-selective glutamine and an hydrophobic clamp (Q pocketCitation12). As concern the PDE5 enzyme, the three pockets include: (i) His613, His617, His653, Asp654, His657, Asn662, Met681, Glu682, Asp724, Leu725, Asp764 (M pocket); (ii) Tyr612, Leu765, Ala767, Ile766, Gln775, Ile778, Ala779, Val782, Ala783. Leu804, Ile813, Met816, Gln817, Phe820 (Q pocket); Gly659, Asn661, Glu785, Phe786, Gln789, Thr802, Met805 (S pocket). Notably, the M pocket His613, His617, His653, Asp654, His657, Glu682, Asp764 residues, the Q cavity Gln817 and the S region Gly659 and Glu785, are absolutely conserved in all PDEs.
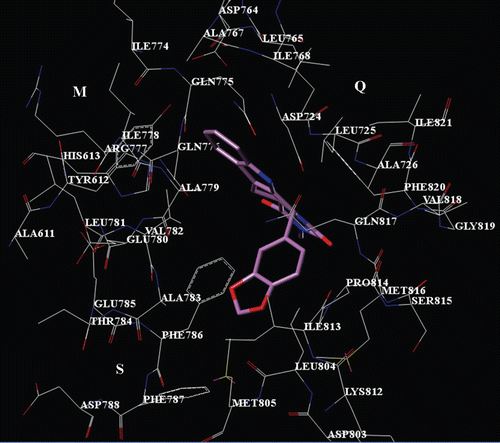
The 1XOZ X-ray tadalafil bioactive conformation (), displays one H-bond contact with Gln817 through the indole core nitrogen atom, being on the contrary especially engaged in contacts with Q pocket residues. Accordingly, the inhibitor shows a lot of hydrophobic contacts between: (i) the indole fragment and Tyr612, Leu765, Leu768, Val782, Phe786 and Phe820; (ii) the piperazinedione ring and Phe820, Ile821; (iii) the 1,3-benzodioxol substituent and Ala783, Phe786, Phe787, Ile813.
Figure 2. X-ray pose of compound tadalafil into the PDE5 catalytic site. The inhibitor is depicted in stick (C atom: violet).
Regarding the THBC derivatives, according to our calculations, all the inhibitors display one H-bond between the indole core nitrogen atom and the Gln817 side chain, while several van der Waals and π−π interactions especially with Q pocket residues are detected. In details, for all the compounds showing the R,R or R,S configuration, the R1 substituent is involved in contacts with Ala783, Phe786, Phe787, Ile813, the R2 substituent (or the corresponding hydantoine or piperazinedione ring) interacts with Leu725, Phe786, Leu804 and Phe820, while the indole core is properly oriented toward Tyr612, Leu765, Leu768 and Val782 (). On the other hand, compounds showing the S,R or S,S configuration do not properly interact with the Q pocket, since the R1 group results to be located on the opposite side respect to the tadalafil 1,3 benzodioxol ring (). These results are in agreement with the lower pIC50 values of these compounds, in comparison with those of the corresponding R,R or R,S isomeres.
Figure 3. Selected docking pose of THBC 23 into the PDE5 catalytic site. The inhibitor is depicted in stick (C atom: white). The tadalafil X-ray pose is also reported (C atom: violet).
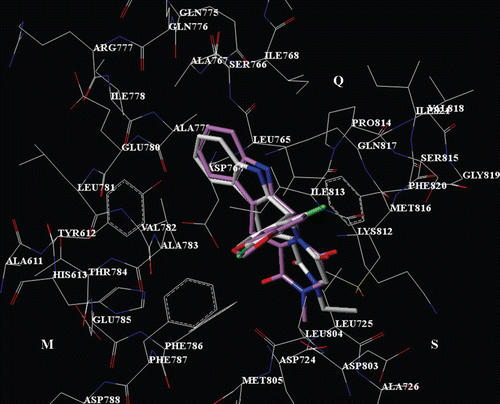
Figure 4. Selected docking pose of THBC 29 into the PDE5 catalytic site. The inhibitor is depicted in stick (C atom: yellow). The tadalafil X-ray pose is also reported (C atom: violet).
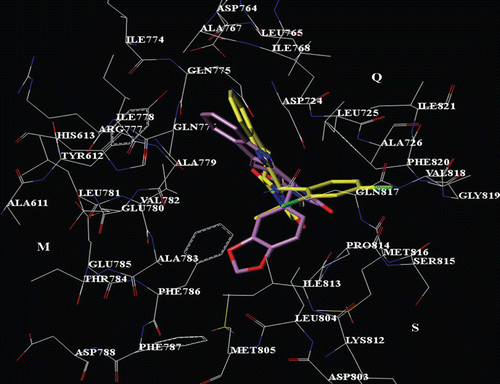
Interestingly, compound 62, which bears a bulky R3 substituent, is involved in much more interactions with the M and S pockets, by means of contacts with His613, L725, Glu785 and Phe786 (). Furthermore, the 1,3-benzodioxol group properly displays the same interactions we have previously discussed for tadalafil. Accordingly, compounds 43–68 (bearing a bulky R3 group and/or a 1,3-benzodioxol ring at the R1 substituent; mean pIC50 value = 8.00) show higher PDE5 inhibitory potency values than those of THBC 1–42 (bearing a short-alkyl R3 group and a phenyl ring at the R1 substituent; mean pIC50 value = 6.33) and, in particular 48–68 (bearing a bulky R3 group and a 1,3-benzodioxol ring at the R1 substituent; mean pIC50 value = 8.09) are the most potent inhibitors of the two series.
Figure 5. Selected docking pose of THBC 27 into the PDE5 catalytic site. The inhibitor is depicted in stick (C atom: white). The tadalafil X-ray pose is also reported (C atom: violet).
3D-QSAR analyses
CoMFA and CoMSIA analyses were performed dividing compounds 1–68 into a training set (1, 3, 5–11, 13–19, 21–23, 27–29, 32–38, 40, 42–47, 49–55, 57–60, 62–67) for model generation and into a test set (2, 4, 12, 20, 24–26, 30, 31, 39, 41, 48, 56, 61, 68) for model validation. CoMFA and CoMSIA analyses were developed using, respectively, CoMFA steric and electrostatic fields and CoMSIA steric, electrostatic, hydrophobic, H-bond donor and H-bond acceptor properties, as independent variables, and the ligand pIC50 as dependent variable.
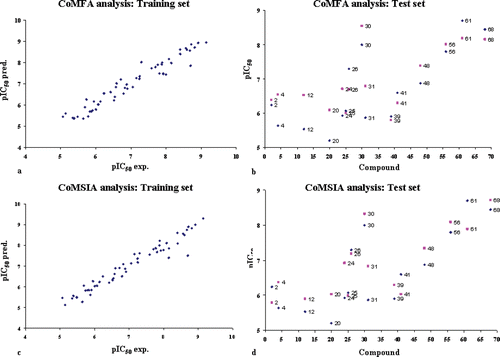
The final CoMFA model was generated employing non-cross-validated PLS analysis with the optimum number of components (ONC = 6) to give a non-cross validated r2 (r2ncv) = 0.94, a test set r2 (r2pred) = 0.72, Standard Error of Estimate (SEE) = 0.288, steric contribution = 0.507 and electrostatic contribution = 0.493. The model reliability thus generated was supported by bootstrapping results. All statistical parameters supporting CoMFA model are reported in . The final CoMSIA model consisting of steric, electrostatic, hydrophobic, H-bond donor and H-bond acceptor fields with a r2ncv = 0.96, r2pred = 0.78, SEE = 0.248, steric contribution = 0.157, electrostatic contribution = 0.190, hydrophobic contribution = 0.323, H-bond acceptor contribution = 0.216 and H-bond donor contribution = 0.114 was derived. All statistical parameters supporting CoMSIA model are reported in .
Experimental and predicted pIC50 values for the training set and test set are reported in . Distribution of experimental and predicted pIC50 values for training set and for test set compounds according CoMFA and CoMSIA analyses are reported in .
Figure 6. Distribution of experimental and predicted pIC50 values for training set compounds according to CoMFA analysis (a), for test set compounds according to CoMFA analysis (b), for training set compounds according to CoMSIA analysis (c), and for test set compounds according to CoMSIA analysis (d).
CoMFA steric and electrostatic regions
As shown in (for simplicity, only the structure of compounds 23 and 27 are depicted), the steric contour map predicts (for compounds 1–42) favourable interaction polyhedra (green) in proximity of the para and one of the two ortho positions of the R1 phenyl moiety, around the R2 substituent (or in the proximity of the corresponding hydantoin and piperazinedione core fragment) and in the area located between the R1 and the R2 groups. On the contrary, unfavourable polyhedra (yellow) are positioned near one of the two meta positions of the R1 phenyl ring.
Figure 7. Contour maps of CoMFA steric regions (green, favoured; yellow, disfavoured) depicted around THBC 23 and 27 (a) and around the ihibitor 62 (b) Inhibitors are depicted in stick mode and coloured by atom type.
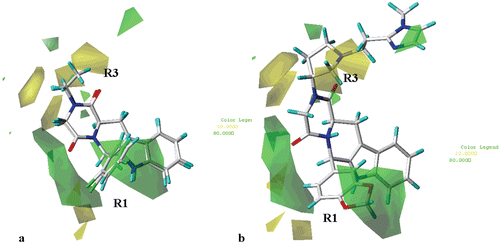
Notably, compounds showing the S,S or S,R configuration do not orient the R1 and the R3 substituents toward the favourable steric map regions previously discussed, being on the contrary sorrounded by yellow areas. Accordingly, these compounds are lower pIC50 values in comparison with those of the corresponding R,R or R,S isomeres, respectively.
In addition, the steric contour maps also show green polyhedra around the 1,3-benzodioxol group of the compound 62 R1 substituent (, 62 is taken as representative of the second series of compounds), near the pyrrolidine position 5 and onto the imidazolic ring. Accordingly, compounds 62 (pIC50 = 9.15) and 63 (pIC50 = 8.92) show high potency levels.
The reliability of the CoMFA steric map is also verified by the higher pIC50 values of those compounds bearing a 2,4 di-chloro phenyl ring at the R1 substituent, in comparison with those inhibitors bearing a 3,4 di-chloro phenyl ring at the same position [compare: (i) 7 (R3 = ethyl; pIC50 = 8.00), 8 (R3 = ethyl; pIC50 = 7.05), 9 (R3 = ethyl; pIC50 = 6.07), 10 (R3 = ethyl; pIC50 = 8.15), with compounds 11 (R3 = ethyl; pIC50 = 6.82), 12 (R3 = ethyl; pIC50 = 5.54), 13 (R3 = ethyl; pIC50 = 5.06) and 14 (R3 = ethyl; pIC50 = 6.52), (ii) 27 (R3 = ethyl; pIC50 = 8.70), 29 (R3 = ethyl; pIC50 = 6.00) and 30 (R3 = ethyl; pIC50 = 8.00), with compounds 31 (R3 = ethyl; pIC50 = 5.87), 32 (R3 = ethyl; pIC50 = 5.48) and 33 (R3 = ethyl; pIC50 = 5.69)].
On the basis of the electrostatic fields contour map of the CoMFA analysis plotted in (for simplicity, only the structure of compound 23 and 27 are depicted as representative), less positive moieties would be favoured (red polyhedra) in proximity of the carbonyl oxygen atom of the R2 substituent (or of the corresponding hydantoin and piperazinedione core), near the para position and one of the two ortho positions of the R1 phenyl ring, and in the area located between the R1 and the R2 substituents. On the other hand, more electropositive substituent are predicted to be beneficial (blue area) in proximity of the R3 substituent and near one of the two meta positions of the R1 phenyl ring. Accordingly, compounds bearing the 2,4-dichlorophenyl ring as R1 substituent (showing the two chlorine atoms electron-withdrawing properties), are more potent than the corresponding 3,4-dichloro phenyl ring derivatives.
Figure 8. CoMFA electrostatic areas are displayed around compounds 23 and 27 (a) and around THBC 62 (b). Blue regions are favourable for more positively charged groups; red regions are favourable for less positively charged groups. Inhibitors are depicted in stick mode and coloured by atom type.
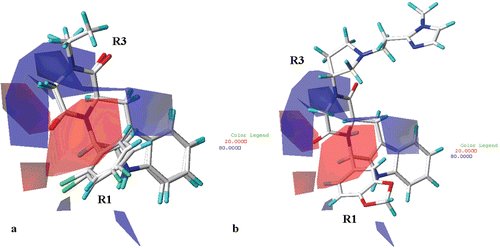
Interestingly, compounds showing the S,S or S,R configuration (which display lower pIC50 values in comparison with those of the corresponding R,R or R,S isomers) do not properly occupy the electrostatic maps. In fact, the two ortho positions of the R1 phenyl ring are sorrounded by blue areas, while the R3 substituent is partially located outside the blue area cited above.
Finally, red areas are also located near the two oxygen atoms of the 1,3-benzodioxol group of the compound 62 R1 substituent (), while a small blue region sorround the methylene located between the two oxygen atoms. These results are in agreement with the high pIC50 values of those THBC (48–68, pIC50 = 6.79–9.15) bearing an 1,3-benzodioxol group on the R1 substituent.
The CoMSIA steric and electrostatic regions are in agreement with the CoMFA steric and electrostatic areas.
CoMSIA hydrophobic, H-bond acceptor and H-bond donor regions
As shown in (for simplicity, only the structure of compound 23 and 27 are depicted as representative), the introduction of hydrophobic substituents in proximity of the para and one of the two ortho positions of the R1 phenyl moiety, around the R2 substituent (or around the corresponding hydantoin and piperazinedione core fragment) and in the area located between the R1 and the R2 groups, is favoured (yellow regions). On the contrary, unfavourable polyhedra (white) are positioned near one of the two meta positions of the R1 phenyl ring. These results are in agreement with the information derived from the CoMFA steric and electrostatic maps, previously discussed, especially regarding the R1 substituent.
Figure 9. Contour maps of CoMSIA hydrophobic regions (yellow, favoured; white, disfavoured) are depicted around compounds 23 and 27 (a) and around THBC 62 (b), shown in stick mode and coloured by atom type.
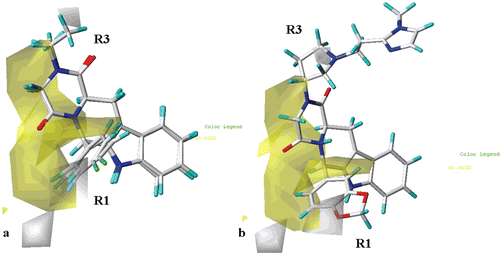
Furthermore, the presence of hydrophilic groups is also recommended (white polyhedra) onto one of the two oxygen atoms of the 1,3-benzodioxol group of the compound 62 R1 substituent (), and on the pyrrolidine nitrogen atom. These results are supported by the high pIC50 values of those THBC (48–68, pIC50 = 6.79–9.15) bearing an 1,3-benzodioxol group on the R1 substituent.
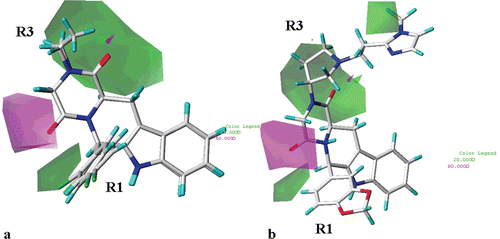
(for simplicity, only the structure of compound 23 and 27 are depicted as representative) shows that H-bond acceptor functions are predicted to be beneficial (magenta polyhedra) around the carbonyl oxygen atom of the R2 substituent and on both the twe two oxygen atoms of the hydantoin and piperazinedione derivatives. On the contrary, the introduction of H-bond acceptor groups are disfavoured (green areas) in proximity of one of the two ortho postions of the R1 substituent. As shown in , the presence of H-bond acceptor groups onto the methylene linked to the pyrrolidine ring of compound 62 is also disfavoured. Accordingly, compound 64 (pIC50 = 7.70) displays a lower potency value in comparison with those of 65–68 (pIC50 = 8.08–8.60).
Figure 10. CoMSIA hydrogen bond acceptor polihedra are shown around THBC 23 and 27 (a) and around the inhibitor 62 (b). H-bond acceptor groups: magenta, favoured; green, disfavoured. THBCs are reported in stick and coloured by atom type.
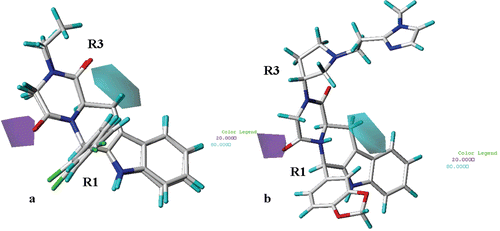
H-bond donors ( and , for simplicity, only the structure of compound 23 and 27 are depicted as representative) are predicted to be unfavourable (purple region) near the carbonyl oxygen atom of the R2 substituent (or of the corresponding hydantoin and piperazinedione core). The introduction of H-bond donor groups is favoured (cyan region) in the area located between the hydantoin (or piperazindione) ring and the tetrahydro-β-carboline scaffold.
Figure 11. CoMSIA hydrogen bond donor polihedra (b) are displayed around compounds 23 and 27 (a) and around THBC 62 (b). H-bond donor groups: cyan, favoured; purple, disfavoured. Compounds are depicted in stick mode and coloured by atom type.
A comparison between the CoMFA and CoMSIA analyses and the PDE5/62 docking model
In order to verify the reliability of the 3D-QSAR analyses, CoMFA and CoMSIA maps have been compared with the results of docking analysis. For simplicity, only the CoMFA steric map and the CoMSIA hydrophobic and H-bond acceptor maps are reported in . Briefly, the CoMFA steric model proves to match with the PDE5 catalytic site topology described by CardCitation12 (), suggesting bulky substitution around the compound 62 1,3-benzodioxol ring and indole core (which interact especially with the Q pocket) and in proximity of the imidazolyl substituent, to optimize the contacts with the S pocket Glu785, Phe786 and Met805 residues. On the contrary, the pyrrolidine ring is sorrounded by a yellow area since a bulky group at this position could be in clash contacts with Leu725 (M pocket) or Leu804 (Q pocket).
Figure 12. The CoMFA steric map (green, favoured; yellow, disfavoured) is displayed around the compound 62 selected docking pose (in stick), located into the PDE5 catalytic site. The most important residues are labelled.
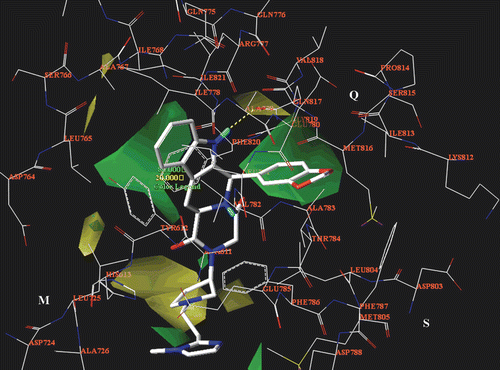
Figure 13. The CoMSIA hydrophobic map (yellow, favoured; white, disfavoured) is displayed around the compound 62 selected docking pose (in stick), located into the PDE5 catalytic site. The most important residues are labelled.
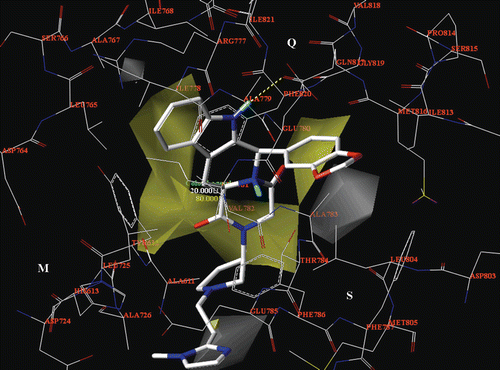
Figure 14. The CoMSIA H-bond acceptor map is displayed around the compound 62 selected docking pose (in stick), located into the PDE5 catalytic site. H-bond acceptor groups: magenta, favoured; green, disfavoured. The most important residues are labelled.
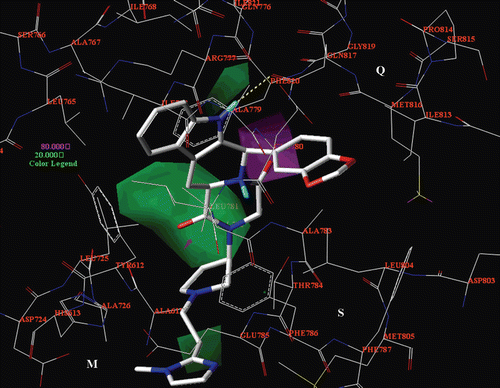
With this purpose, the introduction of a ethyl phthalimide group as R3 substituent or the substitution of the imidazolyl ring with a indole one (or N-methyl-indol one), could enhance the inhibitor potency. The CoMFA electrostatic map points out the beneficial presence of electropositive moieties in proximity of the R3 substituent, which is properly surrounded by negative charged residues (the M pocket Asp724 and the S cavity Glu785); and near the methylene located between the two 1,3-benzodioxol ring oxygen atoms, oriented toward Glu780. Thus, a piperazine ring could be beneficial as a portion of the R3 substituent, being protonated and sorrounded by Asp724 and Glu785. As shown in , the CoMSIA hydrophobic maps underline the importance of lipophilic group (yellow areas) around the indole core and near the area located between the R2 substituent (or at the corresponding hydantoine or piperazinedione ring) and the R1 group, being principally involved in π−π interactions or Van der Waals contacts with Q pocket residues. The CoMSIA H-bond acceptor and H-bond donor maps confirm the importance of the introduction of polar groups as a driving force for the proper disposition of the inhibitor into the PDE5 catalytic site (). In particular, the introduction of the 1,3-benzodioxol ring as R1 substituent (bearing two potential H-bond acceptor atoms) seems to be particularly favoured, much more than the presence of the phenyl ring (showing no H-bond acceptor atoms). Accordingly, the R1 substituent could be modified in a catechol ring or in an allyl-substituted moiety (bearing H-bond acceptor groups such as cyano or sulphonyl functions). Taking all the results into account, higher potency values toward the PDE5 enzyme could be achieve by enhancing inhibitor interactions with the S and Q pocket. Accordingly, compounds bearing a bulky R3 group and/or a 1,3-benzodioxol ring at the R1 substituent (mean pIC50 value = 8.00) are the most potent inhibitors of the two series.
On the basis of our results, some compounds (a-d) have been built in silico and their pIC50 values have been predicted by the 3D-QSAR analyses previously derived and discussed. The a-d chemical structure and the predicted inhibitory potency values have been reported in .
Table 5. Summary of CoMFA and CoMSIA results.
Table 6. Experimental and predicted pIC50 values of compounds 1–52.
Table 7. Chemical structure and the predicted pIC50 values for compounds a-d.
Conclusions
The computational studies here presented analyse the main interactions responsible for the different potency values of the dataset compounds and give useful suggestions for the synthesis of new analogues potentially active as PDE5 inhibitors. In particular, our studies highlight the importance of a bulky R3 group, which interacts with the enzyme pocket S. Notably, Phe786 and Met805 (S region) are unconserved among PDEs and therefore they could be recognized of strategic relevance for the design of new compounds potentially acting as selective PDE5 inhibitors. The 3D-QSAR models, which underline the beneficial presence of H-bond acceptor group and of hydrophobic moieties around the THBC scaffold, proved to be in agreement with the docking studies previously elaborated, and have been exploited to design new compounds and predict their potency prior to synthesis.
Declaration of interest
This work was financially supported by the University of Genova. The authors report no declarations of interest.
References
- Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem 2007;76:481–511.
- Wallis RM, Corbin JD, Francis SH, Ellis P. Tissue distribution of phosphodiesterase families and the effects of sildenafil on tissue cyclic nucleotides, platelet function, and the contractile responses of trabeculae carneae and aortic rings in vitro. Am j Cardiol 1999;83:3C–12C.
- Whitehead CM, Earle KA, Fetter J, Xu S, Hartman T, Chan DC et al. Exisulind-induced apoptosis in a non-small cell lung cancer orthotopic lung tumor model augments docetaxel treatment and contributes to increased survival. Mol Cancer Ther 2003;2:479–488.
- Tinsley HN, Gary BD, Keeton AB, Zhang W, Abadi AH, Reynolds RC et al. Sulindac sulfide selectively inhibits growth and induces apoptosis of human breast tumor cells by phosphodiesterase 5 inhibition, elevation of cyclic GMP, and activation of protein kinase G. Mol Cancer Ther 2009;8:3331–3340.
- Abadi AH, Abouel-Ella DA, Ahmed NS, Gary BD, Thaiparambil JT, Tinsley HN et al. Synthesis of novel tadalafil analogues and their evaluation as phosphodiesterase inhibitors and anticancer agents. Arzneimittelforschung 2009;59:415–421.
- Maw GN, Allerton CM, Gbekor E, Million WA. Design, synthesis and biological activity of beta-carboline-based type-5 phosphodiesterase inhibitors. Bioorg Med Chem Lett 2003;13:1425–1428.
- Mohamed HA, Girgis NM, Wilcken R, Bauer MR, Tinsley HN, Gary BD et al. Synthesis and molecular modeling of novel tetrahydro-ß-carboline derivatives with phosphodiesterase 5 inhibitory and anticancer properties. j Med Chem 2011;54:495–509.
- Yang GF, Huang X. Development of quantitative structure-activity relationships and its application in rational drug design. Curr Pharm Des 2006;12:4601–4611.
- Yang GF, Lu HT, Xiong Y, Zhan CG. Understanding the structure-activity and structure-selectivity correlation of cyclic guanine derivatives as phosphodiesterase-5 inhibitors by molecular docking, CoMFA and CoMSIA analyses. Bioorg Med Chem 2006;14:1462–1473.
- Chen G, Wang H, Robinson H, Cai J, Wan Y, Ke H. An insight into the pharmacophores of phosphodiesterase-5 inhibitors from synthetic and crystal structural studies. Biochem Pharmacol 2008;75:1717–1728.
- Chen CY, Chang YH, Bau DT, Huang HJ, Tsai FJ, Tsai CH et al. Discovery of potent inhibitors for phosphodiesterase 5 by virtual screening and pharmacophore analysis. Acta Pharmacol Sin 2009;30:1186–1194.
- Card GL, England BP, Suzuki Y, Fong D, Powell B, Lee B et al. Structural basis for the activity of drugs that inhibit phosphodiesterases. Structure 2004;12:2233–2247.
- MOE: Chemical Computing Group Inc. Montreal. H3A 2R7 Canada. http://www.chemcomp.com
- Jain AN. Scoring noncovalent protein-ligand interactions: a continuous differentiable function tuned to compute binding affinities. j Comput Aided Mol Des 1996;10:427–440.
- Cramer RD 3rd, Patterson DE, Bunce JD. Recent advances in comparative molecular field analysis (CoMFA). Prog Clin Biol Res 1989;291:161–165.
- Klebe G, Abraham U, Mietzner T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. j Med Chem 1994;37:4130–4146.
- Sybyl-X 1.0. Tripos Inc 1699 South Hanley Road. St Louis. Missouri. 63144. USA.