Abstract
The carbonic anhydrases (CAs, EC 4.2.1.1) constitute interesting targets for the design of pharmacological agents useful in the treatment or prevention of a variety of disorders such as, glaucoma, acid-base disequilibria, epilepsy, and other neuromuscular diseases, altitude sickness, edema, and obesity. A quite new and unexpected application of the CA inhibitors (CAIs) is with regard to their potential use in the management (imaging and treatment) of hypoxic tumors. A series of sulfonamides, including some clinically used derivatives like acetazolamide, methazolamide, ethoxzolamide, dichlorophenamide, dorzolamide, brinzolamide, benzolamide, and sulpiride, or indisulam, a compound in clinical development as antitumor drug, as well as the sulfamate antiepileptic drug topiramate have been reported to inhibit various human carbonic anhydrase isozyme. Various heterocyclic sulfonamides have been reported in this review with their potency to inhibit different carbonic anhydrases isozymes.
Introduction
The carbonic anhydrases (CAs) are a family of metalloenzymes involved in the catalysis of reversible hydration CO2 (CO2 + H2O ↔ HCO3- + H+) and, as a consequence, play an important part in a number of physiological processes such as pH regulation and electrolyte secretion, and in a number of biosynthetic pathways. From the viewpoint of cancer-drug discovery, perhaps the most interesting CA isoform is CA IX; because CA IX has been reported to be prevalently expressed in several human cancer cells but not in their ‘normal’ counterparts and high expression levels are related to poor patient prognosisCitation1.
They are involved in crucial physiological processes connected with respiration and transport of CO2/bicarbonate between metabolizing tissues and the lungs, pH and CO2 homeostasis, electrolyte secretion in a variety of tissues/organs, biosynthetic reactions (such as the gluconeogenesis, lipogenesis, and ureagenesis), bone resorption, calcification, tumorigenicity, and many other physiologic or pathologic processes. This enzyme has 16 different isoenzymes presently known in human. Basically, there are several cytosolic forms (CA I, CA II, CA III, and CA VII), four membrane bound forms (CA IV, CA IX, CA XII, and CA XIV), one mitochondrial form (CA V), as well as a secreted CA form (CA VI2). There are three additional “catalytic” CA isoforms (CA VIII, CA X, and CA XI) whose functions remain unclearCitation3. The isozymes CA VA and VB which are present in mitochondria are involved in several biosynthetic processes, such as ureagenesis, gluconeogenesis, and lipogenesisCitation4,Citation5. The isoenzymes CA IX and XII, are predominantly found in tumor cells and absent (or are present in very limited amount) in normal tissues. Several of these isoenzymes (CA II and CA IV) are expressed in human eyes. The most recently characterized members of this family are cytosolic CA XIII and membrane-bound CA XV. Although the activity of CA XIII is the second lowest reported thus far for any of the human CAs, it may have a role in maintaining the acid-base balance in the kidney and the gastrointestinal and reproductive tracts. CA XV is an exceptional enzyme, as it seems to be active in numerous species, such as rodents, birds and fish, but is absent from humans and chimpanzees. Mouse CA XV is a moderately active enzyme, suggesting that it may play a physiological role at least in the kidney. It is likely that other isozymes have substituted for this protein in humansCitation6.
The carbonic anhydrases constitute interesting targets for the design of pharmacological agents useful in the treatment or prevention of a variety of disorders such as, glaucoma, acid-base disequilibria, epilepsy, and other neuromuscular diseases, altitude sickness, edema, and obesityCitation7,Citation8. A quite new and unexpected application of the CA inhibitors (CAIs) is with regard to their potential use in the management (imaging and treatment) of hypoxic tumorsCitation9–16.
Inhibitors of zinc metalloenzymes need precise structural requirements in order to tightly bind to the metal ion(s) present in their active sites. For example, primary sulfonamides (RSO2NH2), sulfamates (ROSO2NH2), and sulfamides (RNHSO2NH2) show high affinity for binding the Zn(II) ion present in α-carbonic anhydrases acting as potent inhibitors with clinical applications as antiglaucoma, diuretic, antiobesity or antitumor drugs. In all of them, the sulfonamide/sulfamate/sulfamide drug binds in deprotonated form to the catalytically critical Zn(II) ion, also participating in extensive hydrogen bond and van der Waals interactions with amino acid residues both in the hydrophobic and hydrophilic halves of the enzyme active site, as shown by X-ray crystallographic studies of enzyme-inhibitor complexesCitation17. The classical recognition motif for small molecules to bind the active site of CA is an aromatic sulfonamide moiety (ArSO2NH2). The deprotonated sulfonamide (ArSO2NH−) coordinates to the CA active site Zn2+, and so inhibits the binding of the endogenous substrates (CO2 and H2O), thereby reducing the catalytic ability of the enzymeCitation18,Citation19.
Heterocyclic moieties with carbonic anhydrase inhibitiory activity
Thiadiazole
A small series of 2-substituted-1,3,4-thiadiazole-5-sulfamides were synthesized and assayed for the inhibition of five physiologically relevant isozymes, the cytosolic CA I and II, the membrane-associated CA IV and the mitochondrial CA VA and VB. The new compounds showed weak inhibitory activity against hCA I (KIs of 102 nM–7.42 μM), hCA II (KIs of 0.54–7.42 μM) and hCA IV (KIs of 4.32–10.05 μM) but were low-nanomolar inhibitors of hCA VA and hCA VB, with inhibition constants in the range of 4.2–32 nM and 1.3–74 nM, respectivelyCitation19. As extension of their former work C. Temperini et al.Citation20 reported interaction of 2-N,N-Dimethylamino-1,3,4-thiadiazole-5-methanesulfonamide [1] with the 12 catalytically active mammalian carbonic anhydrase isozymes, CA I-XIV. The compound was found to be a potent inhibitor of CA IV, VII, IX, XII, and XIII (KIs of 0.61–39 nM), a medium potency inhibitor of CA II and VA (KIs of 121–438 nM), and a weak inhibitor against the other isoforms (CA III, VB, VI, and XIV) making it a very interesting candidate for situations in which a strong/ selective inhibition of certain isozymes is needed. They included that compound 1 is completely different from those of clinically used sulphonamides like acetazolamide. Indeed compound 1 is one of the most potent CA IV and CA VII inhibitors ever reported, while acting as a much weaker inhibitor of the ubiquitous isoform CA II which is often considered as housekeeping enzyme, whose inhibition is not desired.
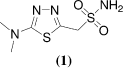
5-Amino-1,3,4-thiadiazole-2-sulfonamide has several biological activities and antibacterial properties. Several derivatives of this drug have been synthesized and used in treatment of glaucoma; one of them is acetazolamide which is used systematically (i.e. an oral medication) to reduce intraocular pressure by lowering the humor formation in the eye. A number of side effects of this drug are however experienced such as numbness and tingling in the fingers and toes, taste alterations, blurred vision, kidney stones, and an increase in urinationCitation21. To overcome these side effects Rahmi et al.Citation22 synthesized various novel pyrazole carboxylic acid derivatives of 5-amino-1,3,4-thiadiazole-2-sulfonamide, [2a-b, 3, 4a-g]. They also reported the inhibitory effects of all newly synthesized amides on hydratase and esterase activities of carbonic anhydrase isoenzymes (hCA-I and hCA-II) in vitro. The comparison of newly synthesized amides to inhibitor 5-amino-1,3,4-thiadiazole-2-sulfonamide and to acetazolamide, indicated that the new derivatives inhibit CA isoenzymes and they are more potent inhibitors than the parent inhibitor. The most effective compounds were 4g for hCA-I, 2a, 4f and 4g for hCA-II.
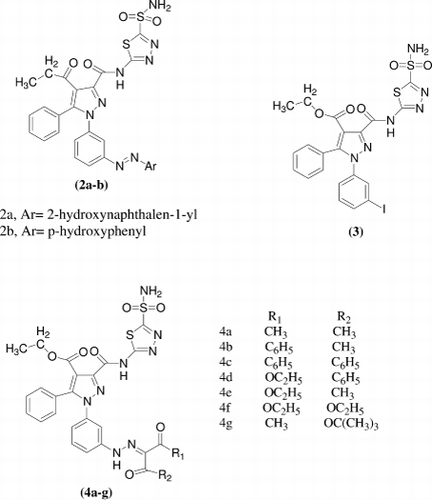
A series of benzimidazo[1,2-c][1,2,3]thiadiazole-7-sulphonamides [5a-g] were synthesized and there binding to two CAse isozymes measured by isothermal titration calorimetry (ITC). Human carbonic anhydrase I (hCAI) and bovine carbonic anhydrase II (bCAII) bound the inhibitors with observed association constants in the range from 1.1 × 106 to 2.6 × 107 M−1. The strongest binder to both isozymes of CAse was compound 5e with the observed Kd of about 0.04μM. The most specific binder was 5d. Compound 5d bound threefold tighter to bCAII that to hCAICitation23.
Quinoline
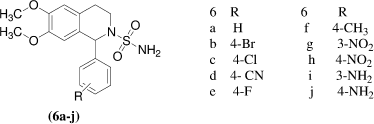
Quinoline derivatives having a substituted or unsubstituted sulfonamide moiety where their design complies with the general pharmacophore of the sulfonamide CA inhibitor, R. Gitto et al.Citation24 synthesized a series of 1-aryl-6,7-dimethoxy-3,4-dihydroisoquinoline-2(1H)-sulfonamides [6a-j]. These newly synthesized compounds incorporate the main features of the above mentioned anticonvulsants and a sulfonamide function capable to inhibit the enzyme carbonic anhydrase, which represents an attractive target in epilepsy. Compound 6h was found to be most interesting, demonstrating K1 value lower than that of topiramate against hCA XIV, more ever 6h and topiramate inhibited hCA I with similar potency.
Mostafa M. Ghorab et al.Citation25 synthesized some novel 4-(quinoline-1-yl) benzenesulphonamide derivatives [7a-u]. All the newly synthesized compounds were evaluated for their in vitro anticancer activity. Some compounds showed interesting in vitro anticancer activities when compared with doxorubicin as a reference drug. In addition, they also performed docking of the synthesized compounds into carbonic anhydrase isozyme II (CA II) active site in order to give a suggestion about the proposed mechanism of action. They found that quinoline derivative 7h carrying benzo[d][1,3]dioxol at 4-position with IC50 value <22.2 μM, quinoline derivative 7g having 4-methoxyphenyl at 4-position with IC50 = 22.9 μM and quinoline derivative 7e having styryl group at 4-position with IC50 = 23.1 μM showed higher significant cytotoxic activity which was even higher activity than that of the reference drug doxorubicin with IC50 = 69.9 μM.
Triazine
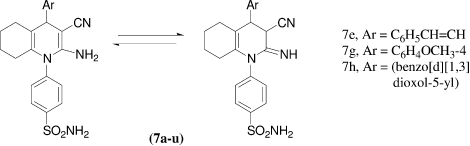
Sulfonamides incorporating triazinyl moieties previously reported were among the most potent and selective hCA IX inhibitors. In continuation of the investigation of this class of derivatives, reporting the synthesis and hCA I, II and IX inhibitory properties of a large series of such novel derivatives, V. Garaj et al.Citation26 reported synthesis of new series of aromatic benzenesulfonamides incorporating 1,3,5-triazine moieties in their molecules by reaction of cyanuric chloride with sulfanilamide, homosulfanilamide or 4-aminoethylbenzenesulfonamide. All the synthesized sulfonamides incorporating triazinyl moieties were screened for the inhibition of three physiologically relevant carbonic anhydrase isozymes, the cytosolic hCA I and II, and the transmembrane, tumor-associated hCA IX. The new compounds inhibited hCA I with inhibition constants in the range of 31–8500 nM, hCA II with inhibition constants in the range of 14–765 nM and hCA IX with inhibition constants in the range of 1.0–640 nM. The best hCA I inhibitors in this series were 8a-g, 9a-c and 10a-d, which showed KIs in the range of 31–95 nM, in the same range as the potent, clinically used inhibitors ethoxzolamide, methazolamide and indisulam. The most effective hCA II inhibitors (with potencies comparable to those of the clinically used sulfonamides were found to be 8a-g, with KIs in the range of 14–27 nM; The best hCA IX inhibitors were the amino, hydrazine, ethylamino, dimethylamino and amino acid derivatives, which showed inhibition constants in the range of 1–3 nM, being among the most potent hCA IX inhibitors reported up to now.
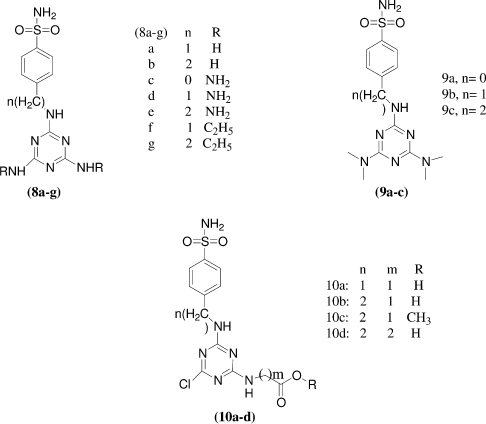
Vladimir Garaj et al.Citation27 synthesized a series of benzenesulfonamide derivatives incorporating triazine moieties in their molecules by reaction of cyanuric chloride with sulfanilamide, homosulfanilamide, or 4-aminoethylbenzenesulfonamide. All the synthesized sulfonamides incorporating triazinyl moieties were evaluated for the inhibition of three physiologically relevant carbonic anhydrase isozymes, the cytosolic hCA I and II, and the transmembrane, tumor associated hCA IX. The new compounds reported inhibited hCA I with KIs in the range of 75–136 nM. The best hCAI inhibitors were the amines 11b-d, the chloroderivative 11a and the ethoxyderivative 11e. Against isozyme hCA II the new derivatives showed a better inhibitory power, with KIs in the range of 13–278 nM. Against the tumor associated isozyme hCA IX, compound 11e was found to be a potent inhibitor (KI of 12 nM being more potent than the clinically used sulphonamide including indisulam).
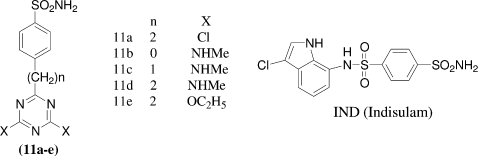
A series of bicyclic 1,3,4-thiadiazolo[3,2-a]triazine-7-sulfonamides [12] and 1,3,4-thiadiazolo[3,2-a]pyrimidine [13a-c] were synthesized by Alan R. Katritzky et al.Citation28 from 5-amino-l,3,4-thiadiazole-2-sulfonamide and evaluated for topical efficacy as ocular hypotensive agents. All compounds tested showed approximately the same activity against the enzyme with IC50 value ranging from 1.4 to 5 × 10−8 M.
Pyridine sulphonamide
Z. Brzozowski et al.Citation29 synthesized a series of novel 4-substituted-3-pyridinesulfonamides [14–22] and [23a-c] and investigated as inhibitors of five isoforms of zinc enzyme carbonic anhydrase that is, the cytosolic, ubiquitous isozymes CA I and II, and transmembrane isozymes CA IX, XII (cancer-associated) and XIV. The newly synthesized compounds showed inhibition constants in the range of 0.078–11.7 μM. (i) Against the slow cytosolic isoform hCA I, compounds 22e, 23a and 23b showed hCA I inhibitory activity (KIs = 0.078e1.52 μM) in the same range as the clinically used compounds. (ii) Against the ubiquitous and dominant rapid cytosolic isozyme hCA II, compounds 21d acting as a stronger CA II inhibitors, with KIs = 9.9nM, of the some order magnitude as most of the clinically used compounds.(iii) A quite larger variation of inhibitory activity was observed for the inhibition of the tumor-associated isoform, hCA IX, compound 15a, 16a, 16b, 16d, 17a-d, 21b, 21c, 21e, 22a-e, 23a and 23b were very effective hCA IX inhibitors (KIs in range of 4.6–12.0 nM). (iv) A quite good inhibition profile of the second tumor-associated isoform, hCA XII has also been observed, most of the compounds were very effective hCA XII inhibitors (KIs = 3.4–8.9 nM). (v) All of the investigated sulfonamides showed moderate to weak inhibition properties towards hCA XIV isozyme with KIs in the range of 50.9–160.0 nM.
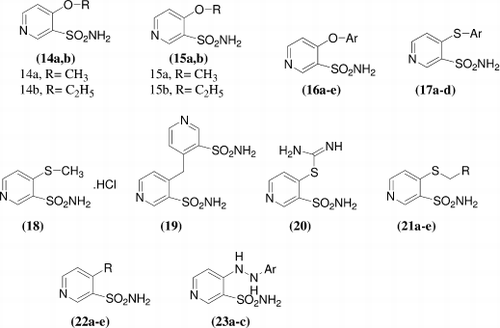
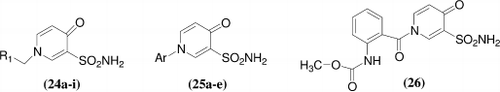
A series of 1-substituted 1,4-dihydro-4-oxo-3-pyridinesulfonamide [24–26] had been synthesized and investigated as inhibitors of four isoforms of zinc enzyme carbonic anhydrase, that is the cytosolic ubiquitous CA I and II, and cancer-associated isozymes CA IX and XII. Against the human isozymes hCA I the new compounds showed inhibition constants in the range of 1.09–12.1 μM, against hCA II in the range of 50.5–172 nM, against hCA IX in the range of 5.2–118 nM, and against hCA XII in the range of 8.7-381 nM, respectively. Compounds 24a, 24b, 24d-j, 25a and 25c-d showed excellent hCA IX inhibitory efficacy, with KIs = 5.2–11.0 nM, being much more effective as compared to the clinically used sulfonamides AAZ, MZA, EZA, DCP and IND (KIs = 24–50 nM). Compounds 24a, 24b, 24d-j, 25a and 25c were also very effective hCA XII inhibitors (KIs = 8.7–45.2 nM) which were comparable or more effective than those clinically used EZA and DCP (KIs = 22 and 50 nM), respectivelyCitation30.
Indole
Graham et al.Citation31 synthesized indole-2-sulfonamide [27a-d, 28b-c, 29b-c and 30] and all the newly synthesized compounds were evaluated as topically active ocular hypotensive agents. These compounds were found to be excellent inhibitors of carbonic anhydrase and to lower intraocular pressure in a rabbit model of ocular hypertension. Compound 27b and 29b were found to be better intraocular pressure lowering agent with IC50 value 16 nM and 11 nM, respectively.

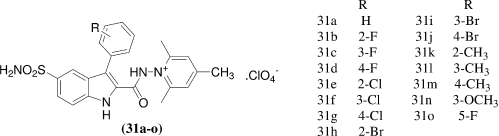
Özlen Güzel et al.Citation32 observed the effect of indole-sulphonamide derivatives on mammalian carbonic anhydrase isozymes. In their research work they synthesized a series of 2-(hydrazinocarbonyl)-3-aryl-1H-indole-5-sulfonamides [31a-o] possessing various 2-, 3- or 4- substituted phenyl groups with methyl-, halogeno- and methoxy-functionalities, or a perfluorophenyl moiety, by reaction with 2,4,6-trimethylpyrylium perchlorate. All the newly sulfonamides were evaluated as inhibitors of four mammalian carbonic anhydrase isoforms, that is, CA I, II (cytosolic), CA IX and XII (transmembrane, tumor-associated forms). Most of these sulfonamides were found to be potent inhibitors of hCA IX, and against hCA XII with some of the new compounds. These compounds were generally less effective inhibitors of hCA II. Being membrane impermeant, these positively-charged sulfonamides were interesting candidates for targeting the tumor-associated CA IX and XII, as possible diagnostic tools or therapeutic agents. Against the cytosolic isoform hCA I compound 31g and 31m were found to be very active with KIs of 3.2 and 5.1 nM, respectively. Against the ubiquitous and dominant rapid cytosolic isozyme hCA II, compound 31o was found to be most potent with KIs in subnanomolar range (0.93nM). The tumor associated isoform hCA IX, one of the important targets for obtaining novel antitumor therapies which ultimately emerged, was generally excellently inhibited by the new compounds reported here, many of these derivatives [31c-e, 31g-k and 31m] had KIs < 10 nM, being thus more than 10 times better hCA IX inhibitors compared to lead. Against the second tumor-associated isoform, hCA XII (less widespread as compared to hCA IX) the new compounds reported here showed a mixed behavior, with three of them acting as very effective inhibitors (5, 6 and 9, KIs in the range of 9.5–12.6 nM), whereas 31a, 31b, 31e and 31f being moderate inhibitors (KIs in the range of 38–97 nM).
Thiophene
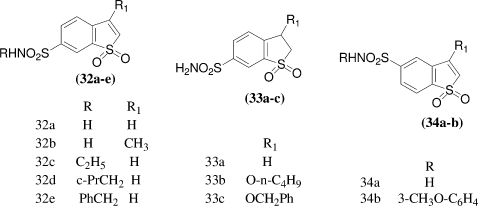
A. Innocenti et al.Citation33 reported synthesis and screening of some selected benzo[b]thiophene-5- and 6-sulfonamide derivatives for cytotoxic activity and the interactions with several CA isozymes, some of which are known to be involved in tumorigenesis (hCA IX), whereas others are ubiquitously found in many normal tissues (the cytosolic isoforms hCA I and II). They concluded that hCA I was inhibited by unsubstituted sulfonamides with inhibition constants in the range of 63–138 nM, hCA II with inhibition constants in the range of 6.3–8.8 nM, and hCA IX with inhibition constants in the range of 2.8–15 nM, being thus more active than clinically used inhibitors such as acetazolamide, methazolamide, ethoxzolamide, dichlorophenamide or indisulam. Some of these derivatives also showed some selectivity for the inhibition of the tumor-associated (hCA IX) over the cytosolic isozyme hCA II. Against isozyme hCA I the primary sulfonamides 32a, 32b, 33a-c, and 34a showed moderate inhibitory activity, with inhibition constants in the range of 63–138 nM, Against the ubiquitous, physiologically relevant isozyme hCA II, again the unsubstituted sulfonamides 32a, 32b, 33a-c, and 34a showed very good inhibition, with KIs in the range of 6.3-8.8 nM, The tumor associated isozyme hCA IX was also inhibited well by the unsubstituted sulfonamides 32a, 32b, 33a-c, and 34a, which showed KIs in the range of 2.8–15 nM. They concluded that compound 32a, 32c, 32e, 34a, and 34b were cytotoxic.
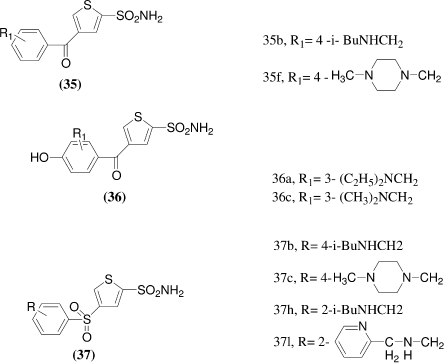
Hartman et al.Citation34 synthesized a series of 4-substituted thiophene and was found to possess nanomolar-level potency for inhibition of hCA II in vitro. Selected compounds from this group were further evaluated for their potential to act as topically effective ocular hypotensive agents in the ocular normotensive albino rabbit and the ocular α-chymotrypsinized rabbit. A sequence of in vitro, ex vivo, and in vivo studies were used to evaluate the ocular hypotensive profile of this series of thiophene sulfonamides. Several functionalized 4-substituted thiophene- and furan-2-sulfonamides showed sub-10 nM potencies. Specifically, 4-acyl analogs, such as 35b and 35f, bearing monosubstitution in the 3- or 4-positions of the phenyl ring, displayed equivalent or somewhat enhanced activity as compared to their corresponding sulfonyl counterparts, 37b and 37c. Similarly, the thiophene-based Mannich adducts 36a and 36c were of comparable potency to furan adducts. In ex vivo assay for their ability to penetrate the albino rabbit eye and to inhibit the target enzyme in a homogenate of the iris-ciliary body at 1h postdosing. All compounds were screened at 0.5 % (1 drop, 50 pL), while the more potent compounds were screened at lower concentrations. The most potent compounds proved to be 35b, 35d, 37c, and 37e, all of which showed appreciable ex vivo activity at a concentration of 0.1 %. Compounds 36a, 37h, and 37l were found to be most potent for their ability to lower IOP in the α-chymotrypsinized (α -CT) rabbit model of ocular hypertension.
Oxadiazole and triazole
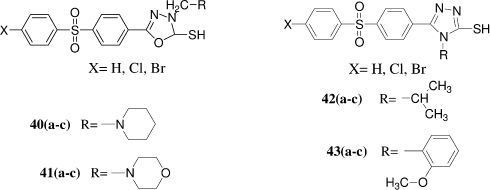
Novel series of mercapto-1,3,4-oxadiazole and -1,2,4-triazole derivatives were synthesized by G. L. Almajan et al.Citation35 by various pathways starting from 4-(4-halogeno-phenylsulfonyl)benzoic acid hydrazides which were reacted with carbon disulfide or isothiocyanates. All the newly synthesized heterocyclic mercaptans were assayed as inhibitors of three physiologically relevant isoforms of the zinc enzyme carbonic anhydrase i.e. the cytosolic CA I and II, and the tumor-associated, transmembrane isozyme CA IX. Interesting biological activity was detected for some of the new mercaptans, with inhibition constants in the low micromolar range. Against the slow cytosolic isozyme hCA I, the heterocycles 38–41 showed good inhibitory activity, with inhibition constants in the range 5.1–9.3 μM, being on the other hand weaker inhibitors as compared to the classical sulfonamide inhibitor acetazolamide (5-acetamido-1,3,4-thiadiazole-2-sulfonamide), which had a KI of 0.25 μM. Against the physiologically most important cytosolic isoform, hCA II, the new compounds investigated here showed good inhibitory activity, with KIs in the range 0.63–31 μM, being again less efficient inhibitors as compared to the sulfonamide acetazolamide (KI of 12 nM). Against the tumor associated isoform hCA IX, compounds 38–41 showed a moderate - weak inhibitory activity, with KIs in the range of 1.25–59 μM, whereas the bulky triazoles 42 and 43 were quite weak inhibitors, with KIs in the range 129–476 μM.
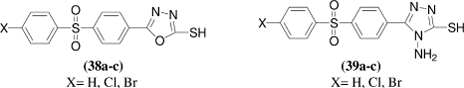
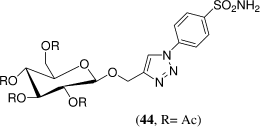
Wilkinson et al.Citation18 reported synthesis of a series of benzene sulfonamides containing triazole-o-glycoside tails by the 1,3-dipolar cycloaddition reaction of 4-azidobenzenesulfonamide with o-propynyl glycosides. These newly synthesized glycoconjugates were assessed for their ability to inhibit the enzymatic activity of the physiologically dominant isozymes hCA I and II and the tumor-associated isozyme hCA IX. Against hCA I these compounds were either micromolar or low-nanomolar inhibitors, while against hCA II and IX inhibition in the range of 6.8–53 and 9.7–107 nM, respectively, was observed. The most potent inhibitor against hCA IX was the galactose derivative 44 (KI = 9.7 nM); this is so far the most potent glycoconjugate inhibitor reported for the tumor-associated hCA IX. These carbohydrate-tethered sulfonamides may prove interesting lead candidates to target tumor associated CA isozymes, wherein the CA domain is located extracellularly.
Pyrrole-3-carboxamide
S. Gluszok et al.Citation36 reported the synthesis and the pharmacological evaluation of a new class of human carbonic anhydrase (hCA) inhibitors, 1,5-diarylpyrrole-3-carboxamides prepared by a solid-phase strategy involving a PS(HOBt) resin. The inhibitory potency (KI, nM) of all the derivatives was evaluated against the tumor-associated hCA IX and against the ubiquitous and physiologically relevant hCA I and hCA II. All the compounds exhibited a weak inhibitory activity against hCA I. The KI values of the R1-phenyl derivatives 45a-e were found between 781 and 2151 nM as observed for the R1-naphthyl molecules 46a-i (427–10,000 nM). The 4-biphenylyl derivatives 47a-i were also weak hCA I inhibitors with KI values higher than 1736 nM. Against hCA II All the synthesized molecules were found much more potent (KI: 9-620 nM) than against hCA I. The phenyl 45a-e and the 4-biphenylyl 47a-i derivatives were moderate hCA II inhibitors with Ki values ranging from 127 to 620 nM. The best hCA II inhibitors (KI≤ 70 nM) belonged to the R1-naphthyl series (46a-e and 46h). The most potent hCA IX inhibitors were the 2-naphthyl compounds 46a, 46b, 46d, and 46h (KI: 10–22 nM) whereas 46c, 46e-g, and 45i were less active on hCA IX (KI: 125–147 nM).
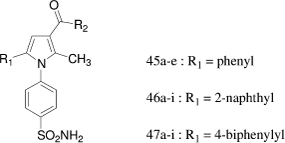
Assay of CA activity
C. T. Supuran et al.Citation37 had shown that the pH of the tumor environment can be changed by modulating CA activity, which may favorably influence the anticancer effect of drugs, or other agents, used concomitantly with a CA inhibitor/activator.
CA activity was determined by following the hydration of CO2 according to the method of Wilbur and AndersonCitation38. In this method, the rate of pH reduction is measured using an electrode as soluble CO2, in a standardised solution, is converted to HCO3− by the action of CA, when compared to the uncatalysed reaction. Enzyme units were calculated according to the formula:
where to and tc are the times (seconds) needed for the pH change, without and with enzyme reactions, respectively. Assays were performed at least twice on each lysate and the mean value determinedCitation39.
Conclusion
Carbonic anhydrase isozymes are associated with several biological disorders i.e. glaucoma, acid-base disequilibria, epilepsy, and other neuromuscular diseases, altitude sickness, edema, obesity and hypoxic tumor. The present review demonstrated that heterocyclic sulphonamide derivatives have potent carbonic anhydrase inhibition activity, so these heterocyclic compounds can be utilized for managing above mentioned disorders. Furthermore, the substitutions to these heterocyclic moieties can be utilized for the development of more potentially active inhibitors with better therapeutic value.
Declaration of interest
The authors declared no conflict of interest.
Related Research Data
References
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–181.
- Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2010;20:3467–3474.
- Supuran CT. Carbonic anhydrase inhibition/activation: trip of a scientist around the world in the search of novel chemotypes and drug targets. Curr Pharm Des 2010;16:3233–3245.
- Güzel O, Innocenti A, Scozzafava A, Salman A, Supuran CT. Carbonic anhydrase inhibitors. Aromatic/heterocyclic sulfonamides incorporating phenacetyl, pyridylacetyl and thienylacetyl tails act as potent inhibitors of human mitochondrial isoforms VA and VB. Bioorg Med Chem 2009;17:4894–4899.
- Nishimori I, Vullo D, Innocenti A, Scozzafava A, Mastrolorenzo A, Supuran CT. Carbonic anhydrase inhibitors. The mitochondrial isozyme VB as a new target for sulfonamide and sulfamate inhibitors. J Med Chem 2005;48:7860–7866.
- Hilvo M, Innocenti A, Monti SM, De Simone G, Supuran CT, Parkkila S. Recent advances in research on the most novel carbonic anhydrases, CA XIII and XV. Curr Pharm Des 2008;14:672–678.
- Pastorekova S, Parkkila S, Pastorek J, Supuran CT. Review article. J Enz Inhib Med Chem 2004;19:199–229.
- Supuran CT. Carbonic anhydrase inhibitors in the treatment and prophylaxis of obesity. Expert Opin Ther Pat 2003;13(10):1545–1550.
- Svastová E, Hulíková A, Rafajová M, Zat’ovicová M, Gibadulinová A, Casini A et al. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett 2004;577:439–445.
- Supuran CT. Indisulam: an anticancer sulfonamide in clinical development. Expert Opin Investig Drugs 2003;12:283–287.
- Scozzafava A, Owa T, Mastrolorenzo A, Supuran CT. Anticancer and antiviral sulfonamides. Curr Med Chem 2003;10:925–953.
- Vullo D, Franchi M, Gallori E, Pastorek J, Scozzafava A, Pastorekova S et al. Carbonic anhydrase inhibitors. inhibition of cytosolic isozymes I and II and transmembrane, cancer-associated isozyme IX with anions. J Enzyme Inhib Med Chem 2003;18:403–406.
- Vullo D, Franchi M, Gallori E, Pastorek J, Scozzafava A, Pastorekova S et al. Carbonic anhydrase inhibitors: inhibition of the tumor-associated isozyme IX with aromatic and heterocyclic sulfonamides. Bioorg Med Chem Lett 2003;13:1005–1009.
- Winum JY, Vullo D, Casini A, Montero JL, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: inhibition of transmembrane, tumor-associated isozyme IX, and cytosolic isozymes I and II with aliphatic sulfamates. J Med Chem 2003;46:5471–5477.
- Winum JY, Vullo D, Casini A, Montero JL, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of cytosolic isozymes I and II and transmembrane, tumor-associated isozyme IX with sulfamates including EMATE also acting as steroid sulfatase inhibitors. J Med Chem 2003;46:2197–2204.
- Abdel-Hamid MK, Abdel-Hafez AA, El-Koussi NA, Mahfouz NM, Innocenti A, Supuran CT. Design, synthesis, and docking studies of new 1,3,4-thiadiazole-2-thione derivatives with carbonic anhydrase inhibitory activity. Bioorg Med Chem 2007;15:6975–6984.
- Santos MA, Marques S, Vullo D, Innocenti A, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: inhibition of cytosolic/tumor-associated isoforms I, II, and IX with iminodiacetic carboxylates/hydroxamates also incorporating benzenesulfonamide moieties. Bioorg Med Chem Lett 2007;17:1538–1543.
- Wilkinson BL, Bornaghi LF, Houston TA, Innocenti A, Vullo D, Supuran CT et al. Carbonic anhydrase inhibitors: inhibition of isozymes I, II, and IX with triazole-linked O-glycosides of benzene sulfonamides. J Med Chem 2007;50:1651–1657.
- Smaine FZ, Pacchiano F, Rami M, Barragan-Montero V, Vullo D, Scozzafava A et al. Carbonic anhydrase inhibitors: 2-substituted-1,3,4-thiadiazole-5-sulfamides act as powerful and selective inhibitors of the mitochondrial isozymes VA and VB over the cytosolic and membrane-associated carbonic anhydrases I, II and IV. Bioorg Med Chem Lett 2008;18:6332–6335.
- Temperini C, Cecchi A, Boyle NA, Scozzafava A, Cabeza JE, Wentworth P Jr et al. Carbonic anhydrase inhibitors. Interaction of 2-N,N-dimethylamino-1,3,4-thiadiazole-5-methanesulfonamide with 12 mammalian isoforms: kinetic and X-ray crystallographic studies. Bioorg Med Chem Lett 2008;18:999–1005.
- Kasimogullari R, Bülbül M, Günhan H, Güleryüz H. Effects of new 5-amino-1,3,4-thiadiazole-2-sulfonamide derivatives on human carbonic anhydrase isozymes. Bioorg Med Chem 2009;17:3295–3301.
- Kasimogullari R, Bülbül M, Arslan BS, Gökçe B. Synthesis, characterization and antiglaucoma activity of some novel pyrazole derivatives of 5-amino-1,3,4-thiadiazole-2-sulfonamide. Eur J Med Chem 2010;45:4769–4773.
- Baranauskiene L, Hilvo M, Matuliene J, Golovenko D, Manakova E, Dudutiene V et al. Inhibition and binding studies of carbonic anhydrase isozymes I, II and IX with benzimidazo[1,2-c][1,2,3]thiadiazole-7-sulphonamides. J Enzyme Inhib Med Chem 2010;25:863–870.
- Gitto R, Ferro S, Agnello S, De Luca L, De Sarro G, Russo E et al. Synthesis and evaluation of pharmacological profile of 1-aryl-6,7-dimethoxy-3,4-dihydroisoquinoline-2(1H)-sulfonamides. Bioorg Med Chem 2009;17:3659–3664.
- Al-Said MS, Ghorab MM, Al-Qasoumi SI, El-Hossary EM, Noaman E. Synthesis and in vitro anticancer screening of some novel 4-[2-amino-3-cyano-4-substituted-5,6,7,8-tetrahydroquinolin-1-(4H)-yl]benzenesulfonamides. Eur J Med Chem 2010;45:3011–3018.
- Garaj V, Puccetti L, Fasolis G, Winum JY, Montero JL, Scozzafava A et al. Carbonic anhydrase inhibitors: synthesis and inhibition of cytosolic/tumor-associated carbonic anhydrase isozymes I, II, and IX with sulfonamides incorporating 1,2,4-triazine moieties. Bioorg Med Chem Lett 2004;14:5427–5433.
- Garaj V, Puccetti L, Fasolis G, Winum JY, Montero JL, Scozzafava A et al. Carbonic anhydrase inhibitors: novel sulfonamides incorporating 1,3,5-triazine moieties as inhibitors of the cytosolic and tumour-associated carbonic anhydrase isozymes I, II and IX. Bioorg Med Chem Lett 2005;15:3102–3108.
- Katritzky AR, Caster KC, Maren TH, Conroy CW, Bar-Ilan A. Synthesis and physicochemical properties of thiadiazolo [3,2-a] pyrimidinesulfonamides and thiadiazolo [3,2-a]triazinesulfonamides as candidates for topically effective carbonic anhydrase inhibitors. J Med Chem 1987;30:2058–2062.
- Brzozowski Z, Slawinski J, Saczewski F, Innocenti A, Supuran CT. Carbonic anhydrase inhibitors: synthesis and inhibition of the human cytosolic isozymes I and II and transmembrane isozymes IX, XII (cancer-associated) and XIV with 4-substituted 3-pyridinesulfonamides. Eur J Med Chem 2010;45:2396–2404.
- Brzozowski Z, Slawinski J, Innocenti A, Supuran CT. Carbonic anhydrase inhibitors. Regioselective synthesis of novel 1-substituted 1,4-dihydro-4-oxo-3-pyridinesulfonamides and their inhibition of the human cytosolic isozymes I and II and transmembrane cancer-associated isozymes IX and XII. Eur J Med Chem 2010;45:3656–3661.
- Graham SL, Hoffman JM, Gautheron P, Michelson SR, Scholz TH, Schwam H et al. Topically active carbonic anhydrase inhibitors. 3. Benzofuran- and indole-2-sulfonamides. J Med Chem 1990;33:749–754.
- Güzel O, Maresca A, Scozzafava A, Salman A, Balaban AT, Supuran CT. Carbonic anhydrase inhibitors. Synthesis of 2,4,6-trimethylpyridinium derivatives of 2-(hydrazinocarbonyl)-3-aryl-1H-indole-5-sulfonamides acting as potent inhibitors of the tumor-associated isoform IX and XII. Bioorg Med Chem Lett 2009;19:2931–2934.
- Innocenti A, Villar R, Martinez-Merino V, Gil MJ, Scozzafava A, Vullo D et al. Carbonic anhydrase inhibitors: inhibition of cytosolic/tumor-associated carbonic anhydrase isozymes I, II, and IX with benzo[b]thiophene 1,1-dioxide sulfonamides. Bioorg Med Chem Lett 2005;15:4872–4876.
- Hartman GD, Halczenko W, Smith RL, Sugrue MF, Mallorga PJ, Michelson SR et al. 4-substituted thiophene- and furan-2-sulfonamides as topical carbonic anhydrase inhibitors. J Med Chem 1992;35:3822–3831.
- Almajan GL, Barbuceanu SF, Innocenti A, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of the cytosolic and tumor-associated carbonic anhydrase isozymes I, II and IX with some 1,3,4-oxadiazole- and 1,2,4-triazole-thiols. J Enzyme Inhib Med Chem 2008;23:101–107.
- Gluszok S, Frédérick R, Foulon C, Klupsch F, Supuran CT, Vullo D et al. Design, solid-phase synthesis, and biological evaluation of novel 1,5-diarylpyrrole-3-carboxamides as carbonic anhydrase IX inhibitors. Bioorg Med Chem 2010;18:7392–7401.
- Supuran CT, Scozzafava A, Casini A. Carbonic anhydrase inhibitors. Med Res Rev 2003;23:146–189.
- Wilbur KM, Anderson NG. Electrometric and colorimetric determination of carbonic anhydrase. J Biol Chem 1948;176:147–154.
- Ozensoy O, Kockar F, Arslan O, Isik S, Supuran CT, Lyon M. An evaluation of cytosolic erythrocyte carbonic anhydrase and catalase in carcinoma patients: an elevation of carbonic anhydrase activity. Clin Biochem 2006;39:804–809.