Abstract
In some human cancer cases, the activity of p53 is inhibited by over-expressed Mdm2. The Mdm2 acts as an ubiquitin ligase, resulting in p53 ubiquitination and subsequent p53 proteasomal degradation. The disruption of the Mdm2-p53 interaction using small-molecule inhibitors is recognized as a promising strategy for anti-cancer drug design. Mushrooms are an important source of powerful compounds with anti-tumour properties. In this study, the first virtual screening of low molecular weight compounds present in mushroom is presented as potential Mdm2 inhibitors. A re-docking and cross-docking method was used to validate the virtual screening protocol. The steroids: ganoderic acids X (Ki = 16nM), Y (Ki = 22nM) and F (Ki = 69nM); 5,6-epoxy-24(R)-methylcholesta-7,22-dien-3β-ol (Ki = 74nM) and polyporenic acid C (Ki = 59nM) stand out as the top ranked potential inhibitors of Mdm2. The docking pose of the most promising compounds were carefully analysed and the information provided shows several interesting starting points for further development of Mdm2 inhibitors.
Keywords::
Introduction
The p53 tumour suppressor protein is a short-lived protein, which is stabilized in response to cellular stressCitation1. The post-transcriptional modification by ubiquitination is an important mechanism regulating protein activities in cellsCitation2. Ubiquitination of p53 leads to the protein degradation through the ubiquitin proteolysis pathway and can be accomplished by several proteins, including Mdm2 (Human E3 ubiquitin-protein ligase), COP1 and Pirh2 among othersCitation3,Citation4. The Mdm2-mediated ubiquitination is considered to be the most important mechanism of regulation of p53 abundanceCitation1–5. Stress signals lead to p53 stabilization either by induction of covalent modifications in Mdm2 and p53, or through altered protein–protein interactions. Mdm2 also harbours a post-ubiquitination function, probably enabling efficient targeting of ubiquitinated p53 to the proteasomeCitation1. In fact, Mdm2-mediated ubiquitination is responsible for maintaining low levels of p53 in a normal physiological state of a cell as well as for the rapid increase of p53 after genotoxic stressCitation5–7.
In some human cancer cases, the activity of p53 is inhibited by over-expressed Mdm2 that acts as an ubiquitin ligase, resulting in p53 ubiquitination and subsequent p53 proteasomal degradationCitation7. The disruption of the Mdm2-p53 interaction using small-molecule inhibitors (SMIs) is recognized as a promising strategy for anti-cancer drug designCitation8. Nutlins and spirooxindoles are examples of SMIs targeting Mdm2, which bind to its hydrophobic pocket with high affinity and selectivityCitation9.
Mushrooms are considered an important source of powerful compounds with anti-tumour properties. The anti-tumour properties of wild mushrooms have been extensively studied by this research group and by othersCitation10–14. Several low molecular weight (LMW) compounds, such as quinones, cerebrosides, isoflavones, catechols, amines, triacylglycerols, sesquiterpenes and steroids, have been isolated from mushrooms and proved to have anti-tumour propertiesCitation15–17. Mushrooms LMW compounds target processes, such as apoptosis, angiogenesis, metastasis, cell cycle regulation and other signal transduction cascades. Moreover, different molecular targets have been described for these compounds, including NF-κB transcription factors, protein kinases, aromatase, sulfatase, matrix metallo-proteinases (MMPs), cyclooxygenase and DNA polymeraseCitation16. Nevertheless, LMW compounds might have multiple molecular targets.
To understand the anti-tumour activities of mushrooms, it is important to understand how LMW present in mushrooms acts at a molecular level. In this study, a dataset of 40 LMW compounds isolated from mushrooms, representing different families of chemical compounds and described as having anti-tumour properties, were virtually screened to investigate its potential as Mdm2-p53 interaction inhibitors. Great care was taken in the validation process to insure the most reliable docking results. The docking tool AutoDock4 was used and the validation of the crystal structures was carefully performed using a re-docking and cross-docking approach.
Methods
LMW compounds dataset
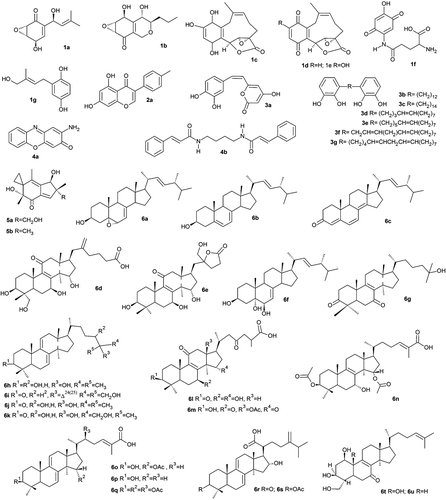
The LMW compounds dataset used is composed of 40 compounds isolated from mushrooms ()Citation15. The 2D structure of the compounds was constructed using the ACD/ChemSketch Freeware 12.0 softwareCitation18. The software VegaZZ 2.3.119 was then used to: convert all compounds from 2D to 3D, perform energy minimization and record files in PDB format. Next, AutoDockTools1.5.2 (ADT)Citation20 was used to: merge non-polar hydrogen, add Gasteiger charges and set up rotatable bonds through AutoTors. Finally, all compounds were recorded in PDBQT file format, a format required for AutoDock4 use. The octanol/water partition coefficient (LogP) was calculated using Open Babel softwareCitation21.
Figure 1. Chemical structure of the LMW mushroom compounds with anti-cancer potential isolated from mushrooms.
Protein structure preparation
The crystal structures of Mdm2 were obtained from the Protein Data Bank (PDB): 1T4ECitation22, 3LBKCitation23 and 3JZKCitation24 (PDB entries). The software AutoDockTools was used to extract the co-crystallized ligands from the PDB file, assign polar hydrogens, add gasteiger charges and save the structures in PDBQT file format required to use AutoDock4. AutoGrid4Citation25,Citation26 was used to create affinity grid maps for all the atoms present on the crystal structures used. The ADT was used to choose the correct parameters before using AutoGrid4. All affinity grid maps were centred on the active site and coordinates were selected to encompass all the protein active sites.
Virtual screening using AutoDock4
AutoDock4 (version 4.2)Citation25,Citation26 with the Lamarckian genetic algorithm was used to simulate ligand-receptor molecular docking. Docking parameters selected for AutoDock4 runs were as follows: 50 docking runs, population size of 200, random starting position and conformation, translation step ranges of 2.0 Å, mutation rate of 0.02, crossover rate of 0.8, local search rate of 0.06 and 2.5 million energy evaluations. Docked conformations were clustered using a tolerance of 2.0 Å root mean square deviation (RMSD). The entire virtual screening experiment was performed on a cluster of eight Intel Dual-Core 2.8 GHz computers using MOLA softwareCitation27. Inhibition constants (Ki) for all ligands were calculated by AutoDock4 as follows: Ki = exp((ΔG*1000.)/(Rcal*TK)), where ΔG is the binding energy, Rcal is 1.98719 and TK is 298.15. All figures with structure representations were prepared using PyMOL softwareCitation28.
Results and discussion
Structure selection, re-docking and cross-docking validation
The performance of a docking experiment is usually evaluated by re-docking the co-crystallized ligands into the protein binding site and then analysing the docking score and pose obtained. If more than one crystal structure is available, the docking experiment can also be evaluated by cross-docking the ligands to different crystal structures. This study was started by selecting adequate Mdm2 crystal structures and then validating them by performing re-docking and cross-dockingCitation29.
A total of 16 human Mdm2 crystal structures are currently available at the Protein Data Dank. As this study evaluates the potential Mdm2 inhibition activity of LMW compounds, only structures with co-crystallized SMIs were considered. From the 16 structures, 11 presented co-crystallized peptide inhibitors and only 5 presented co-crystallized SMIs: 1T4E, 3JZK and 3LBK, 3LBL and 1RV1 (PDB entries). After careful structural analysis, three were selected for this study: 1T4E, 3JZK and 3LBK. Structures 1RV1 and 3LBL were discarded as their crystal structure presents an asymmetric unit that contains three separate inhibitor-Mdm2 complexes, with crystal contacts near the binding site. These crystal contacts may provide a distorted binding site environment that is probably not equivalent to isolated Mdm2. These structures were thus considered unsuitable for docking as they could probably render unreliable docking scores and poses.
The three PDB structures used present co-crystallized inhibitors from three well-known families of mdm2 inhibitors: a benzodiazepine derivative in 1T4E, a imidazo-indole derivative for 3LBK and a chromenotriazolopyridine derivative for 3JZK (). To validate the docking approach for the PDB structures selected, the inhibitors were re-docked to the respective structure and then cross-docked to the other two selected PDB structuresCitation29. The docking scores are present as estimated average ΔG and Ki values and compared to experimental ΔG and Ki values (). Also the docking pose is analysed and compared to the experimental binding pose for each inhibitor (), and this alignment is quantified as RMSD values ().
Table 1. Re-docking and cross-docking results using the selected Mdm2 crystal structures.
Figure 2. Docking conformation of three known Mdm2 inhibitors. (a) Representation of the Mdm2 structure with the co-crystallized benzodiazepine inhibitor (PDB:1T4E). Superimposition of crystal (sticks and balls representation, green) and docked conformations (wire representation, yellow) for: (a) the benzodiazepine derivative, (b) the imidazo-indole derivative and (c) the chromenotriazolopyridine derivative. Superimpositions obtained by aligning the three Mdm2 structures using Pymol.
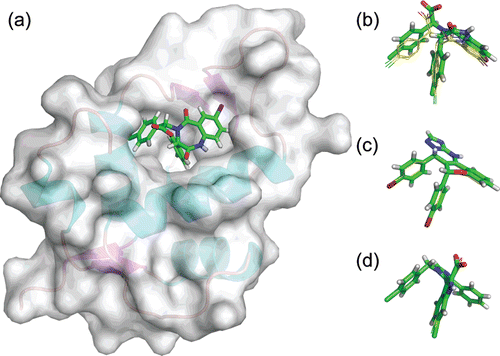
When comparing estimated and experimental ΔG values, differences of 0.51, 1.79 and 1.18 Kcal/mol were observed for the benzodiazepine, imidazo-indole and chromenotriazolopyridine derivatives, respectively (). These variations fall well within the residual standard error of 2.18 kcal/mol observed for AutoDock4Citation26. This is a strong indication that AutoDock4 is performing well with the selected Mdm2 crystal structures, thus validating them for docking with other LMW compounds. When calculating Ki values from ΔG values, the 2.18 kcal/mol residual standard error translates into an expected difference for Ki of 1.6 orders of magnitude (pKi difference). In this study, the estimated average Ki calculated by AutoDock4 was about 2 times lower than experimental Ki for benzodiazepine derivative (difference of 0.37 orders of magnitude), about 20 times lower for the imidazo-indole derivative (difference of 1.31 orders of magnitude) and about 10 times lower for the chromenotriazolopyridine derivative (difference of 0.86 orders of magnitude) (). These values are well within the 1.6 orders of magnitude difference considered acceptable for AutoDock4. Also, although the estimated Ki values obtained were consistently lower than the experimental Ki values, it is important to note that AutoDock 4 ranked correctly the inhibitors with the benzodiazepine being the most potent and the chromenotriazolopyridine derivative the less potent. This trend in ranking is observed when analysing the results for each individual PDB structure.
The average RMSD between the coordinates of the docked inhibitors on each PDB structure and the coordinates in their native crystal structure was calculated by aligning the structures using Pymol software. A near perfect fit was obtained between the binding mode of the docked inhibitors and the co-crystallized inhibitors, with average RMSD values of 1.16, 0.4 and 0.5 Å, respectively ( and ). These values shows that the difference between the crystal conformation and the predicted docked conformations of the compounds was very small thus further validating the PDB structures for molecular docking with the LMW compound dataset.
Virtual screening of LMW mushroom compound dataset
Using the selected Mdm2 structures, a virtual screening of the LMW compounds dataset () was then performed with AutoDock4, and the results are presented as estimated ΔG and estimated Ki ().
Table 2. Virtual screening of the LMW mushroom compound database using AutoDock4.
The LMW compounds screened represent different compound families discovered in mushrooms and presenting anti-tumoural activity, usually in tumoural cell lines (10). Some of those compounds are specific from each mushroom species (e.g. ganoderic acids, specific from Ganoderma lucidum), but other are common molecules, such as ergosterol derivatives.
The compounds with best docking scores were ganoderic acids (F, X, Y and W), 5,6-epoxy-24(R)-methylcholesta-7,22-dien-3β-ol (EMCD) and polyporenic acid C, all belonging to the steroid family. In fact all the steroids screened scored better than the other LMW compound families studied (). This is not surprising because, as can be observed in , Mdm2 inhibitors must present a large lipophilic skeleton to interact with the Mdm2 lipophilic pockets present in the Mdm2-p53 interaction site.
From the steroids analysed, ganoderic acids X (Ki = 44 nM), Y (Ki = 47 nM) and F (Ki = 59 nM) stand out as potential Mdm2 inhibitors. These ganoderic acids were isolated from Ganoderma lucidum and have been shown to exert a cytotoxic effect in some tumour cell lineCitation30. Furthermore, ganoderic acid X induced apoptosis in human hepatoma cells, suggesting that the basic lanostane structure is necessary for the biological activity of purified triterpeneCitation31. Treatment of human hepatoma HuH-7 cells with ganoderic acid X caused immediate inhibition of DNA synthesis as well as activation of nitrogen-activated protein kinases and cell apoptosis. The apoptotic molecular events were elucidated and included degradation of chromosomal DNA, a decrease in the level of Bc1-xL, disruption of mitochondrial membrane, release of cytochrome c into the cytosol and activation of caspase-3. Calviño et al. used extracts of Ganoderma lucidum against interleukin 3-dependent lymphoma cells (DA-1) and described an increase of p53 and Mdm2 after 19 h and a reduction of these two proteins after 24 h32.
Another promising Mdm2 inhibitor was EMCD (Ki = 106 nM), isolated from Cordyceps sinensis and that inhibited the proliferation of K562, Jurkat, WM-1341, HL-60 and RPMI-8226 tumour cell linesCitation33.
Polyporenic acid C also stands out as a top ranked potential Mdm2 inhibitors (Ki = 158 nM). It is known as MMPs inhibitor and it was isolated from the mushroom Piptoropus betulinus, traditionally used in Czech Republic as a functional food for the treatment of rectal cancerCitation13–16. It was also isolated from Daedalea dickinsii.
It was hypothesized that the anti-tumoural activities observed for the top ranked compounds may result from the disruption of the Mdm2-p53 interaction with the blockage of Mdm2 mediated targeting of p53 for proteosomal degradation and consequent increase in p53 levels. Also, the potential Mdm-2 inhibition may be synergistically amplified by the presence of several of the studied compounds, specially the different ganoderic acids, as they all presented low Ki values. Although an experimental demonstration of Mdm2 inhibition ability is required, the studied steroids may prove to be a new class of Mdm-2 inhibitors.
Structural analysis of the top ranked steroids as Mdm2 inhibitors
The docking poses of the compounds with best docking scores were structurally analysed. To better understand the key interactions with Mdm2, the detailed binding mode of three compounds representing the steroids with lowest ΔG: ganoderic acid X, EMCD and polyporenic acid C are presented in .
Figure 3. Docking conformation of the top ranked LMW mushroom compounds in the Mdm2 interaction site: (a) ganoderic acid X, (b) 5,6-epoxy-24(R)-methylcholesta-7,22-dien-3β-ol and (c) polyporenic acid C. All the compounds are represented in green sticks and balls representation. The Mdm2 residues from the interaction site interacting with the compounds are represented in white wire representation. Hydrogen bonds are represented in traced red (bond distances between 2.8 and 3.2 Å), hydrophobic interactions in traced yellow (bond distances between 3.5 and 4 Å).
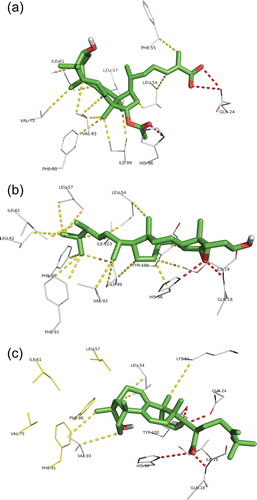
The docking pose of ganoderic acid X shows that the hydrophobic steroid skeleton is stabilized by hydrophobic interactions with several residues (LEU54, PHE55, LEU57, ILE61, VAL75, PHE91, VAL93 and ILE99) present at the hydrophobic Mdm2 cleft that is responsible for interacting with p53. The ganoderic acid X is further stabilized by the formation of H-bonds between the carbonyl group and GLN24 and between the acetyl group and HIS96 (). An important docking conformation feature is the positioning of all ganoderic acid X polar groups (hydroxyl and acetyl) outwards the Mdm2 hydrophobic cleft allowing interactions with the solvent. The ganoderic acid X docking conformation respects the typical interaction pattern observed for the best known Mdm2 inhibitors, with a larger hydrophobic section of the compounds stabilized by a network of hydrophobic interactions in the Mdm2-p53 interaction site, and a number of solvent exposed polar groups (see ). It is important to note that, although ganoderic acids X and Y presented the lowest estimated Ki, they present a high calculated LogP due to the predominant hydrophobic steroid skeleton (). This may render these compounds too insoluble for any potential Mdm2 inhibition. However ganoderic acid F presents a lower LogP with just a slightly higher estimated Ki. This profile, with high potential Mdm2 inhibition activity and adequate hydrophobicity, may render ganoderic acid F more effective as an Mdm2 inhibitor, when present in edible mushrooms.
The EMCD revealed a similar docking pose to ganoderic acid X, with the hydrophobic steroid skeleton stabilized by hydrophobic interactions with residues present at the MDM2-p53 interaction site (LEU54, LEU57, ILE61, LEU82, PHE86, PHE91, VAL93 and ILE99 and TYR100). The epoxy group is stabilized by H-bonds with HIS96 and the amine group of the peptide bond between GLN18 and ILE19. The calculated LogP was even higher compared to ganoderic acids, making EMCD even more insoluble.
Finally with polyporenic acid C, the docking conformation shows a steroid skeleton not as deeply buried in the Mdm2 cleft as the ganoderic acids and EMCD. Because of this, it promotes fewer hydrophobic interactions and is not able to interact to a number of hydrophobic residues previously observed to interact with ganoderic acids and EMCD (, yellow residues). One point of notice is that the hydroxyl group of polyporenic acid C is directed inward to the Mdm2 cleft, as opposed to ganoderic acid X were the hydroxyl group is positioned outward towards the solvent. This probably makes the polyporenic acid C conformation less stable and is probably the reason for the higher estimated Ki value. On the other hand, polyporenic acid C presents a lower calculated LogP of 6.3, making it more water soluble and thus easier to reach Mdm2 in the cell when the mushrooms are ingested.
The docking conformations analysed portrait the main difficulties in developing Mdm2 inhibitors where the delicate balance between a predominant hydrophobic character, to allow Mdm2 interaction with p53, and hydrophilic traits to promote solvent interaction is clearly observed.
Conclusions
In conclusion, the first virtual screening of LMW mushroom compounds as potential Mdm2 inhibitors was presented. The Mdm2 structures used were carefully analysed and a method using re-docking and cross-docking was used to validate the docking protocol used. From the compounds studied, a number of steroids stand out as the most promising potential Mdm2 inhibitors, with estimated Ki in the tenths of nanomolar range: ganoderic acids, EMCD and polyporenic acid C. The docking pose was structurally analysed in detail and the most promising steroid compounds presented may be regarded as good starting points in the development of new Mdm2 inhibitors.
Declaration of interest
The authors report no declarations of interest. The authors are grateful to Fundação para a Ciência e a Tecnologia (FCT, Portugal) and COMPETE/QREN/EU for financial support to this work (research project PTDC/AGR-ALI/110062/2009) and to CIMO (strategic project PEst-OE/AGR/UI0690/2011).
References
- Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol 2003;13:49–58.
- Hershko A. The Ubiquitin System for Protein Degradation and Some of Its Roles in the Control of the Cell-Division Cycle Nobel Lecture. Israel J Chem 2006;46:113–120.
- Brooks CL, Gu W. Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr Opin Cell Biol 2003;15:164–171.
- Hochstrasser M. Ubiquitin-dependent protein degradation. Annu Rev Genet 1996;30:405–439.
- Brazhnik P, Kohn KW. HAUSP-regulated switch from auto- to p53 ubiquitination by Mdm2 (in silico discovery). Math Biosci 2007;210:60–77.
- Iwakuma T, Lozano G. MDM2, an introduction. Mol Cancer Res 2003;1:993–1000.
- Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature 1997;387:299–303.
- Chène P. Inhibiting the p53-MDM2 interaction: an important target for cancer therapy. Nat Rev Cancer 2003;3:102–109.
- Riedinger C, Noble ME, Wright DJ, Mulks F, Hardcastle IR, Endicott JA et al. Understanding small-molecule binding to MDM2: insights into structural effects of isoindolinone inhibitors from NMR spectroscopy. Chem Biol Drug Des 2011;77:301–308.
- Vaz JA, Heleno SA, Martins A, Almeida GM, Vasconcelos MH, Ferreira IC. Wild mushrooms Clitocybe alexandri and Lepista inversa: in vitro antioxidant activity and growth inhibition of human tumour cell lines. Food Chem Toxicol 2010;48:2881–2884.
- Borchers AT, Keen CL, Gershwin ME. Mushrooms, tumors, and immunity: an update. Exp Biol Med (Maywood) 2004;229:393–406.
- Poucheret P, Fons F, Rapior S. Biological and pharmacological activity of higher fungi: 20-year retrospective analysis. Cryptogamie Mycol 2006;27:311–333.
- Lindequist U, Niedermeyer TH, Jülich WD. The pharmacological potential of mushrooms. Evid Based Complement Alternat Med 2005;2:285–299.
- Russell R, Paterson M. Ganoderma – a therapeutic fungal biofactory. Phytochemistry 2006;67:1985–2001.
- Ferreira IC, Vaz JA, Vasconcelos MH, Martins A. Compounds from wild mushrooms with antitumor potential. Anticancer Agents Med Chem 2010;10:424–436.
- Zaidman BZ, Yassin M, Mahajna J, Wasser SP. Medicinal mushroom modulators of molecular targets as cancer therapeutics. Appl Microbiol Biotechnol 2005;67:453–468.
- Moradali MF, Mostafavi H, Ghods S, Hedjaroude GA. Immunomodulating and anticancer agents in the realm of macromycetes fungi (macrofungi). Int Immunopharmacol 2007;7:701–724.
- ACD/ChemSketch Freeware 12.0 software. Available at: (http://www.acdlabs.com/resources/freeware/). Accessed on 03, December 2012.
- Pedretti A, Villa L, Vistoli G. VEGA–an open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J Comput Aided Mol Des 2004;18:167–173.
- Sanner MF. A component-based software environment for visualizing large macromolecular assemblies. Structure 2005;13:447–462.
- O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An open chemical toolbox. J Cheminform 2011;3:33.
- Grasberger BL, Lu T, Schubert C, Parks DJ, Carver TE, Koblish HK et al. Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells. J Med Chem 2005;48:909–912.
- Popowicz GM, Czarna A, Wolf S, Wang K, Wang W, Dömling A et al. Structures of low molecular weight inhibitors bound to MDMX and MDM2 reveal new approaches for p53-MDMX/MDM2 antagonist drug discovery. Cell Cycle 2010;9:1104–1111.
- Allen JG, Bourbeau MP, Wohlhieter GE, Bartberger MD, Michelsen K, Hungate R et al. Discovery and optimization of chromenotriazolopyrimidines as potent inhibitors of the mouse double minute 2-tumor protein 53 protein-protein interaction. J Med Chem 2009;52:7044–7053.
- Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem 1998;19:1639–1662.
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem 2009;30:2785–2791.
- Abreu RM, Froufe HJ, Queiroz MJ, Ferreira IC. MOLA: a bootable, self-configuring system for virtual screening using AutoDock4/Vina on computer clusters. J Cheminform 2010;2:10.
- The PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC. Available at: (http://www.pymol.org/). Accessed on 03 December, 2012.
- Phosrithong E, Ungwitayatorn J. Molecular docking study on anticancer activity of plant-derived natural products. Med Chem Res 2010;19:817–835.
- Jiang J, Grieb B, Thyagarajan A, Sliva D. Ganoderic acids suppress growth and invasive behavior of breast cancer cells by modulating AP-1 and NF-kappaB signaling. Int J Mol Med 2008;21:577–584.
- Li CH, Chen PY, Chang UM, Kan LS, Fang WH, Tsai KS et al. Ganoderic acid X, a lanostanoid triterpene, inhibits topoisomerases and induces apoptosis of cancer cells. Life Sci 2005;77:252–265.
- Calviño E, Pajuelo L, Casas JA, Manjón JL, Tejedor MC, Herráez A et al. Cytotoxic action of Ganoderma lucidum on interleukin-3 dependent lymphoma DA-1 cells: involvement of apoptosis proteins. Phytother Res 2011;25:25–32.
- Bok JW, Lermer L, Chilton J, Klingeman HG, Towers GH. Antitumor sterols from the mycelia of Cordyceps sinensis. Phytochemistry 1999;51:891–898.