Abstract
The 90 kDa ribosomal S6 kinases (RSKs), especially RSK2, have attracted attention for the development of new anticancer agents. Through structural optimization of the hit compound 1 from our previous study, a series of barbituric acid aryl hydrazone analogues were designed and synthesized as potential RSK2 inhibitors. The most potent one, compound 9, showed a higher activity against RSK2 with an IC50 value of 1.95 μM. To analyze and elucidate their structure-activity relationship, the homology model of RSK2 N-terminal kinase domain was built and molecular docking simulations were performed, which provide helpful clues to design new inhibitors with desired activities.
Introduction
The 90 kDa ribosomal S6 kinases (RSKs) are serine/threonine kinases and lie downstream of Ras/mitogen-activated protein kinase (MAPK) pathway, which are responsible for activating some important cellular components including transcription factors and histonesCitation1–8. This family consists of four human isoforms (RSK1-4) with each being a product of a separate geneCitation9, among which RSK2 has attracted more attention, for its over expression and aberrant activation have been linked to many human diseases, including breast cancer, prostate cancerCitation10. These studies suggest that RSK2 is a potential therapeutic target in the treatment of metastatic cancers, which is of both research and clinical interest to develop potent RSK2 inhibitorsCitation11. In particular, RSK2 possesses a C-terminal kinase domain (CTKD) and an N-terminal kinase domain (NTKD) connected by a linker region. CTKD is believed to be activated by ERK and serves to regulate the phosphorylation of the linker regionCitation12, and the activated NTKD is believed to be responsible for phosphorylation of other substratesCitation13. However, it has been found that the linker region could be activated in the absence of CTKD, and CTKD-independent activation mechanisms of physiological RSK2 remain unknownCitation2. Therefore, present inhibitor studies against RSK2 mainly focus on NTKD.
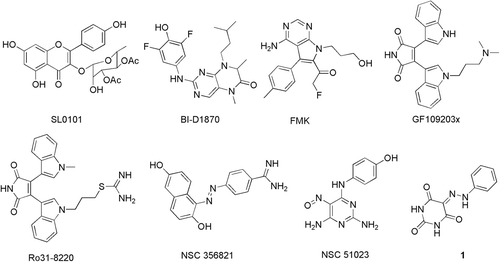
To date, a number of potent RSK2 inhibitors have been reported ()Citation14. There are mainly three RSK specific inhibitors identified and widely applied in RSK2 mediated biologic process as chemical probes. SL0101 is the first reported RSK2 specific inhibitor which was extracted from the tropical plant Forsteronia refracta and exhibits excellent inhibitory activity (IC50 = 89 nM)Citation15,Citation16. BI-D1870 is another specific inhibitor of the RSK family (RSK1-4) with an in vitro enzyme IC50 of 10–30 nMCitation17. Unlike SL0101 and BI-D1870, Fmk is an irreversible inhibitor of RSK2 with an IC50 of 15 nM by binding to the CTKDCitation18. Moreover, two bisindoylmaleimide compounds GF109203X and Ro31–8220, which were both potent but nonspecific RSK2 inhibitors, were obtained from kinase screening assays of PKC inhibitorsCitation19. Although RSK2 is proved to be a promising cancer therapeutic target, the crystal structure of NTKD of human RSK2 has not yet been determined. Therefore, Nguyen et al. identified two novel weakly binding inhibitors (NSC356821 and NSC51023) of RSK2 through homology modeling and virtual screeningCitation20. These inhibitors were widely applied for elucidating the context of the RSK2 signaling pathway as chemical probes, but none of them are applicable in clinical use. Therefore, there are ever-increasing needs to discover novel RSK2 inhibitors for further development into therapeutic candidates for cancer treatment.
Scheme 1. General synthesis route of the barbituric acid aryl hydrazone derivatives.
Figure 1. Selected structures of reported RSK2 inhibitors.
Using SHAFTS program, an in-house 3D similarity based computational virtual screening procedure combined with shape and chemical feature superposition, several RSK2 inhibitors with novel scaffolds and moderate inhibitory activities were identified in our previous studyCitation21. One of the hit compounds 1, a barbituric acid aryl hydrazine derivative, proved to have an inhibitory ratio of 47% at 10 μM against RSK2. In this study, we further report the synthesis and structural modification of a series of derivatives of compound 1 to improve the inhibitory potency against RSK2, of which compounds 9 and 14 showed improved inhibitory activities against RSK2 with IC50 values of 1.95 μM and 2.19 µM, respectively. Finally, molecular docking simulations were performed using a reliable RSK2 homology model to elucidate the relationship between SAR and binding mode.
Methods
Chemistry
Preparation of target compounds was carried out from the coupling of corresponding aniline diazonium salt and barbituric acid derivatives (). In detail, aniline (20.0 mmol) dissolved in 6.7 mL concentrated hydrochloride was mixed with 21 mL water, stirred and cooled to 0°C. Then a solution of sodium nitrite (25 mmol) in 6 mL water was added carefully to form a diazonium solution. Separately, a mixture of NaOAc (9.2 g, 0.1 mol) dissolved in 16 mL water and barbituric acid (20.0 mmol) dissolved in ethanol (62 mL). The diazonium solution was added dropwise when the mixture was cooled below 10°C. After stirring overnight, the resulting precipitation was filtered and washed by ethanol/water (1/1). The product was separated and purified by recrystallization or silica gel chromatography and the structure was identified by 1H-NMR and MS.
5-(2-(4-hydroxyphenyl)hydrazono)pyrimidine-2,4,6 (1H,3H,5H)-trione (9). Brown solid; mp > 300°C; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 14.34 (s, 1H), 11.39 (s, 1H), 11.17 (s, 1H), 9.79 (d, J = 3.2 Hz, 1H), 7.43 (d, J =8.8 Hz, 2H), 6.85 (d, J = 8.8 Hz, 2H); HRMS (ESI, m/z): 247.0474 (M-H+).
5-(2-(4-methoxyphenyl)hydrazono)pyrimidine-2,4,6 (1H,3H,5H)-trione (12). Yellow solid; mp 291–292°C; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 14.28 (s, 1H), 11.42 (s, 1H), 11.20 (s, 1H), 7.55 (d, J = 9.2 Hz, 2H), 7.04 (d, J = 9.2 Hz, 2H), 3.78 (s, 3H); HRMS (ESI, m/z): 261.0629 (M-H+).
5-(2-(4-morpholinophenyl)hydrazono)pyrimidine-2,4,6 (1H,3H,5H)-trione (13). Red solid; mp 279–280°C; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 14.39 (s, 1H), 11.38 (s, 1H), 11.17 (s, 1H), 7.48 (d, J =8.8 Hz, 2H), 7.04 (d, J = 8.8 Hz, 2H), 3.74 (t, J =4.8 Hz, 2H), 3.15 (t, J =4.8 Hz, 2H); MS (ESI, m/z): 318 (M-H+).
5-(2-(4-hydroxyphenyl)hydrazono)-dihydro-2-thioxopyrimidine-4,6 (1H,5H)-dione (14). Brown solid; mp > 300°C; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 14.49 (s, 1H), 12.50 (s, 1H), 12.33 (s, 1H), 9.88 (s, 1H), 7.48 (d, J =9.2 Hz, 2H), 6.86 (d, J = 9.2 Hz, 2H); HRMS (ESI, m/z): 263.0242 (M-H+).
5-(2-(4-morpholinophenyl)hydrazono-dihydro-2-thioxopyrimidine-4,6 (1H,5H)-dione (15). Brown solid; mp 242–243°C; 1H NMR(400 MHz, DMSO-d6) δ (ppm): 14.55 (s, 1H), 12.49 (s, 1H), 12.32 (s, 1H), 7.53 (d, J = 9.2 Hz, 2H), 7.05 (d, J = 9.2 Hz, 2H), 3.74 (t, J = 4.4 Hz, 2H), 3.18 (t, J = 4.4 Hz, 2H); HRMS (ESI, m/z): 332.0811 (M-H+).
Biological assay
The inhibition assays against RSK2 in vitro were determined by ADP Quest® (DiscoveRx) in 96-well flat-bottom plates according to the manufacturer’s instructionsCitation21. The kinase (20 ng millipore) in 40 μL assay buffer (15 mM HEPES, pH 7.4, 20 mM NaCl, 1 mM EGTA, 0.02% Tween 20, 10 mM MgCl2, and 0.1 mg/mL BGG) containing 25 μM S6 peptide (AKRRRLSSLRA, Anaspec) was incubated for 20 min at room temperature with indicated concentrations of the compounds to be tested. Reactions were initiated by adding 10 μL of ATP to a final ATP concentration of 10 μM and terminated after 60 min at room temperature by adding 20 μL ADP Reagent A and 40 μL ADP Reagent B. Ro31–8220 was used as a positive control. Compounds dilutions were prepared from stock in DMSO and diluted with assay buffer for inhibition assay. The fluorescence signal detecting the amount of ADP produced as a result of enzyme activity was recorded on the Synergy™ 2 Multi-Mode Microplate Reader (BioTek) at an excitation wavelength of 530 nm and an emission wavelength of 590 nm at 30 min after the addition of the ADP Reagent B. The inhibition rate (%) was calculated using the following equation:
IC50 values were determined from the results of at least three independent tests and calculated from the inhibition curves. The accuracy of the in vitro screening assays was confirmed by the IC50 value of control inhibitor Ro31–8220 at 10 nM, which is in compliance with the reported valueCitation19.
Molecular modeling
To investigate the binding poses of these compounds and then elucidate corresponding relationship with SAR, the homology model of RSK2 NTKD built in our previous study was used for molecular dockingCitation22. Compounds 1, 9 and 13 were picked out for the docking studies and prepared by the Ligprep module in Maestro (Schrödinger Inc, version 7.5). Then Glide program in extra-precision (XP) mode was used to perform the molecular docking simulations with default parameter sets. The grid-enclosing box was centered on the centroid of aligned ligand (Staurosporine) and defined so as to enclose residues located within 20 Å around the ATP binding site, and a scaling factor of 1.0 was set to van der Waals (VDW) radii of those receptor atoms with the partial atomic charge less than 0.25. The top 10 docked poses of each compound ranked by GlideScore were remained for further analysis.
Results and discussion
SAR analysis
The structures of synthesized compounds and corresponding potencies against RSK2 are listed in . Compound 1 was obtained through the molecular 3D similarity (shape-densities and chemical feature overlaps) method SHAFTS in our previous study using NSC 51023 as the query templateCitation19, suggesting NSC 51023 and compound 1 may present similar binding modes in the RSK2 NTKD ATP site. The docking results revealed that the amide moieties of barbituric acid ring can form pivotal contacts with the hinge region of RSK2 NTKD by H-bonds interaction. Comparing with compound 1 as shown in , the enzyme inhibitory activity of di-methylated derivate (compound 2) decreases significantly with both 1, 3-positions of the barbituric acid ring methylated, highlighting the pivotal role of the polar hydrogen atoms on the barbituric acid scaffold in forming putative H-bonds. Therefore, in the subsequent investigations, the polar hydrogen atoms in 1, 3-positions were retained, and various substituted groups were introduced to different positions on the benzene ring to further optimize the inhibitory activity. In general, the inhibitory potencies were more sensitive to structural optimizations at the para-position of the benzene ring. Introduction of halogen atoms including F, Cl, Br and I on the benzene ring in any position (compound 3–8) did not contribute to the improvement of inhibitory activities, which was even deteriorated with the meta-position substitute (compound 6). Meanwhile, introduction of electron-withdrawing groups such as nitro, cyano, carboxyl and sulfonamide greatly decreases the inhibitory activities (compounds 16, 19, 22 and 23), in which the para-nitro substitute led to a complete ablation of the inhibitory activity. Only electron-donating groups like hydroxyl or morpholinyl at the para-position such as compounds 9, 13–15, can make a positive contribution to the inhibitory activity remarkably, suggesting that electron factor on the conjugation structure may affect the strength of the hydrogen bonds formed with the active site. Similarly, electron-donating groups (e.g. OH, compound 10) are more favored at the meta-position over electron-withdrawing groups (e.g. Cl, compound 6). At the ortho-position, moderate electron-withdrawing groups like CONH2 of compound 21 or Cl of compound 5 were tolerated while more electron-negative groups such as NO2 and CN or electron-donating groups like OH would further reduce the activities as in compounds 11, 17 and 18. Furthermore, it also may be that the ortho CONH2 forms a planar conformation through internal H-bonding to the hydrazone which may form an additional H-bonding with the hinge, whereas the ortho NO2 or CN substitute disrupts the planarity and undermines the extra H-bond with the hinge. Replacement of the carbonyl at 2-position to a thiocarbonyl was also tolerated, which only led to a slight reduction in activity by comparisons of 9 with 14 and 13 with 15.
Table 1. The structures of babituric acid aryl hydrone derivatives and their inhibitory activities against RSK2.
Homology model and binding modes
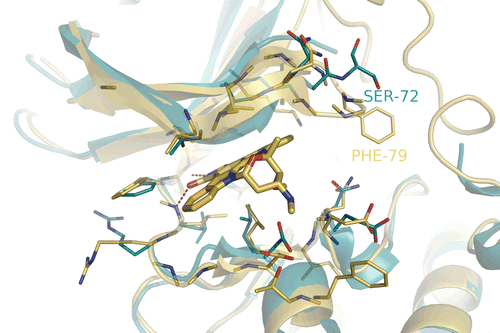
The RSK2 homology model used in this study proved reliable for elucidating the binding modes and inhibitory activity relationship for known RSK2 inhibitors in our previous study. As shown in , the domains of these two kinases are generally similar, since the residue Ser 72 in RSK1 has changed to a larger Phe 79 in the same position of RSK2, the ATP binding pocket of RSK2 appears more closed at the bottom compared with RSK1.
Figure 2. Superimpose RSK1 NTD complexed with Staurosporine (blue) onto the modeled RSK2 NTD (golden). The hydrogen bond interactions are shown with red dashed lines.
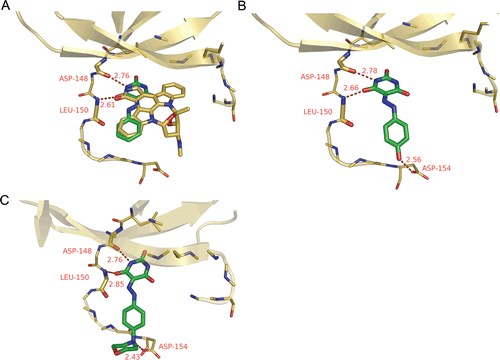
The potential binding modes of docking result for compounds 1, 9 and 13 are shown in . Similar to the other well-known kinase inhibitorsCitation23, barbituric acid inhibitors reported here bind to active conformation of ATP-binding site of RSK2; Like Staurosporine in the homology model, the barbituric acid core of compound 1 () occupies the adenine binding pocket of the enzyme and interacts with the kinase hinge region by two H-bond interactions including amide moiety of barbituric acid with the backbone oxygen atom of Asp 148 (2.76 Å) and the backbone nitrogen atom of Leu 150 (2.61 Å), whilst the extended hydrazone stretches along the hinge with the terminal phenyl ring to a solvent exposed region. However, comparing with compound 1, compounds 9 and 13 show alternative conformations of the hydrazone (), which coincide with the report of similar compounds as GSK3β inhibitors by Arnost et al.Citation24. For compounds 9 and 13 the H-bonds interactions with the hinge region are conserved but additional H-bond contacts are formed with Asp154 by the para hydroxyl or morpholinyl, which presumably, contribute to the improved the inhibitory potency. As for the ortho- and meta-substituent analogues such as 10 and 11, the deterioration of activities may also result from the introduction to inappropriate positions as well except for the electron effects, which hinders the hinge H-bonds formation by steric clash and abolishes the H-bond with Asp154 by an inappropriate distance and dihedral angle.
Figure 3. Docking results of compounds (A) 1, (B) 9, and (C) 13 against RSK2 NTD. The protein model and Staurosporine are colored in golden, and the docked inhibitors are colored in green. The hydrogen bonds interactions between RSK2 NTD and inhibitors are shown with red dashed lines.
Conclusion
In summary, through structural optimization of the hit compound 1 from our previous study, a series of barbituric acid aryl hydrazone analogues were designed and synthesized as RSK2 inhibitors. The SAR was analyzed to highlight the pivotal role of the barbituric acid scaffold, thus the assumed interaction mode was verified. Among them, four compounds exhibited inhibitory rates more than 50% at 10 μM concentration, and the most potent of which, compound 9 inhibited RSK2 with an IC50 value of 1.95. The potential binding modes were explored to further elucidate the SAR, which was also consistent with the previously proposed interaction mode by a ligand-similarity method. To our knowledge, although hydrazone-containing compounds with different scaffolds were reported as kinase inhibitors, especially as moderate RSK2 inhibitorsCitation25, this is the first report that barbituric acid aryl hydrazone derivatives can act as RSK2 inhibitors. These inhibitors might serve as new lead compounds for further development of therapeutics to treat cancer, especially to suppress cell migration.
Declaration of interest
This work was supported by the Fundamental Research Funds for the Central Universities, the National Natural Science Foundation of China (grants 21173076, 81102375 and 81102420), the Special Fund for Major State Basic Research Project (grant 2009CB918501), the Shanghai Committee of Science and Technology (grants 11DZ2260600, 09dZ1975700 and 10431902600), the Innovation Program of Shanghai Municipal Education Commission (grant 10ZZ41), and the 863 Hi-Tech Program of China (Grant 2012AA020308). H. L. is also sponsored by Shanghai Rising-Star Program (grant 10QA1401800) and Program for New Century Excellent Talents in University (grant NCET-10-0378). X. L. is also sponsored by Research Fund for the Doctoral Program of Higher Education (20110074120009).
Related Research Data
References
- Doehn U, Hauge C, Frank SR, Jensen CJ, Duda K, Nielsen JV et al. RSK is a principal effector of the RAS-ERK pathway for eliciting a coordinate promotile/invasive gene program and phenotype in epithelial cells. Mol Cell 2009;35:511–522.
- Anjum R, Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nat Rev Mol Cell Biol 2008;9:747–758.
- Carriere A, Ray H, Blenis J, Roux PP. The RSK factors of activating the Ras/MAPK signaling cascade. Front Biosci 2008;13:4258–4275.
- Cho YY, Yao K, Bode AM, Bergen HR 3rd, Madden BJ, Oh SM et al. RSK2 mediates muscle cell differentiation through regulation of NFAT3. J Biol Chem 2007;282:8380–8392.
- Xing J, Ginty DD, Greenberg ME. Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science 1996;273:959–963.
- Yang X, Matsuda K, Bialek P, Jacquot S, Masuoka HC, Schinke T et al. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin-Lowry Syndrome. Cell 2004;117:387–398.
- Peng C, Cho YY, Zhu F, Xu YM, Wen W, Ma WY et al. RSK2 mediates NF-{kappa}B activity through the phosphorylation of IkappaBalpha in the TNF-R1 pathway. FASEB J 2010;24:3490–3499.
- Cho YY, Yao K, Kim HG, Kang BS, Zheng D, Bode AM et al. Ribosomal S6 kinase 2 is a key regulator in tumor promoter induced cell transformation. Cancer Res 2007;67:8104–8112.
- Jones SW, Erikson E, Blenis J, Maller JL, Erikson RL. A Xenopus ribosomal protein S6 kinase has two apparent kinase domains that are each similar to distinct protein kinases. Proc Natl Acad Sci USA 1988;85:3377–3381.
- Kang S, Elf S, Lythgoe K, Hitosugi T, Taunton J, Zhou W et al. p90 ribosomal S6 kinase 2 promotes invasion and metastasis of human head and neck squamous cell carcinoma cells. J Clin Invest 2010;120:1165–1177.
- Kang S, Chen J. Targeting RSK2 in human malignancies. Expert Opin Ther Targets 2011;15:11–20.
- Smith JA, Poteet-Smith CE, Malarkey K, Sturgill TW. Identification of an extracellular signal-regulated kinase (ERK) docking site in ribosomal S6 kinase, a sequence critical for activation by ERK in vivo. J Biol Chem 1999;274:2893–2898.
- Cho YY, Yao K, Pugliese A, Malakhova ML, Bode AM, Dong Z. A regulatory mechanism for RSK2 NH(2)-terminal kinase activity. Cancer Res 2009;69:4398–4406.
- Nguyen TL. Targeting RSK: an overview of small molecule inhibitors. Anticancer Agents Med Chem 2008;8:710–716.
- Smith JA, Maloney DJ, Clark DE, Xu Y, Hecht SM, Lannigan DA. Influence of rhamnose substituents on the potency of SL0101, an inhibitor of the Ser/Thr kinase, RSK. Bioorg Med Chem 2006;14:6034–6042.
- Smith JA, Poteet-Smith CE, Xu Y, Errington TM, Hecht SM, Lannigan DA. Identification of the first specific inhibitor of p90 ribosomal S6 kinase (RSK) reveals an unexpected role for RSK in cancer cell proliferation. Cancer Res 2005;65:1027–1034.
- Sapkota GP, Cummings L, Newell FS, Armstrong C, Bain J, Frodin M et al. BI-D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo. Biochem J 2007;401:29–38.
- Cohen MS, Zhang C, Shokat KM, Taunton J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science 2005;308:1318–1321.
- Alessi DR. The protein kinase C inhibitors Ro 318220 and GF 109203X are equally potent inhibitors of MAPKAP kinase-1beta (Rsk-2) and p70 S6 kinase. FEBS Lett 1997;402:121–123.
- Nguyen TL, Gussio R, Smith JA, Lannigan DA, Hecht SM, Scudiero DA et al. Homology model of RSK2 N-terminal kinase domain, structure-based identification of novel RSK2 inhibitors, and preliminary common pharmacophore. Bioorg Med Chem 2006;14:6097–6105.
- Lu W, Liu X, Cao X, Xue M, Liu K, Zhao Z et al. SHAFTS: a hybrid approach for 3D molecular similarity calculation. 2. Prospective case study in the discovery of diverse p90 ribosomal S6 protein kinase 2 inhibitors to suppress cell migration. J Med Chem 2011;54:3564–3574.
- Li S, Zhou Y, Lu W, Zhong Y, Song W, Liu K et al. Identification of inhibitors against p90 ribosomal S6 kinase 2 (RSK2) through structure-based virtual screening with the inhibitor-constrained refined homology model. J Chem Inf Model 2011;51:2939–2947.
- Noble ME, Endicott JA, Johnson LN. Protein kinase inhibitors: insights into drug design from structure. Science 2004;303:1800–1805.
- Arnost M, Pierce A, ter Haar E, Lauffer D, Madden J, Tanner K et al. 3-Aryl-4-(arylhydrazono)-1H-pyrazol-5-ones: Highly ligand efficient and potent inhibitors of GSK3beta. Bioorg Med Chem Lett 2010;20:1661–1664.
- Andreani A, Granaiola M, Leoni A, Locatelli A, Morigi R, Rambaldi M et al. Imidazo[2,1-b]thiazole guanylhydrazones as RSK2 inhibitors. Eur J Med Chem 2011;46:4311–4323.