Abstract
In an attempt to develop potent anticancer agents, a series of 4-arylideneamino/cycloalkylidineamino-1, 2-naphthoquinone thiosemicarbazones were synthesized and characterized using FT-IR, 1H NMR, 13C NMR spectroscopy and elemental analysis. The compounds were screened for antiproliferative activity against three human cancer cell lines (Hep-G2, MG-63 and MCF-7) using the MTT assay. Significant anticancer activity was observed for several members of the series. The compounds 4-(3, 4, 5-trimethoxybenzylidene amino) 1, 2-naphthoquinone-2-thiosemicarbazone (TS10) and 4-(4-hydroxy-3-methoxy benzylideneamino) 1, 2-naphthoquinone-2-thiosemicarbazone (TS13) were active cytotoxic agents in all three cancer cell lines, with IC50 values in the range of 3.5–6.4 µM. Further evaluation of some of these potent cytotoxic compounds demonstrated their good safety profile in a normal cell line (MCF-12A). Docking experiments showed a good correlation between the predicted glide scores and the IC50 values of these compounds. In silico ADME studies revealed that these compounds can be used for second generation development.
Introduction
Cancer is a severe human health problem worldwide and the second most common cause of death after cardiovascular diseasesCitation1. Despite of the significant advancement in chemotherapy over recent decades in the research and development of various anticancer drugs, current anticancer chemotherapy still suffers from major drawbacks, such as lack of selectivity of conventional chemotherapeutic agents for cancer tissues, unwanted side effects and the acquisition of multiple-drug resistance by cancer cellsCitation2. So, medicinal chemists are currently focusing on novel biological targets with the aim of providing new specific and potent chemotherapeutic agents that selectively destroy cancer cells while sparing their healthy neighbours.
In 1974, the National Cancer Institute (NCI; Bethesda, MD, USA) identified the quinone group as an important pharmacophoric moiety which can exert an antiproliferative effect in cancer cellsCitation3. Quinonoidal compounds from natural or synthetic origins represent the second largest class of clinically approved anticancer agentsCitation4. Numerous studies have demonstrated the anticancer effect of naturally occurring 1, 2-naphthoquinones, such as rhinacanthoneCitation5, beta-lapachoneCitation6 and mansononesCitation7 (–) as well as synthetic 1, 2-naphthoquinones including 4-hydroxy-3-methyl-1, 2-naphthoquinone thiosemicarbazone (HM-NQTS)Citation8, 1, 2-furano naphthoquinones, 1, 2-pyranonaphthoquinonesCitation9, 3-methyl phenylazolawsone derivatives and their copper complexes (HMPAL)Citation10 (–).
Figure 1. Structures of some potent natural and synthetic anticancer 1, 2-naphthoquinone derivatives.
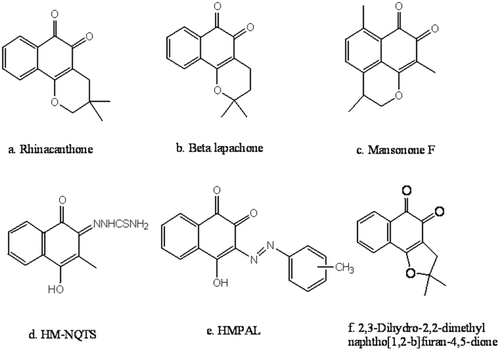
An extensive literature survey revealed that the presence of a 1, 2-dione group embedded in a six membered ring scaffold was essential for the anticancer activity of this class of compounds. Changes in the oxygen-oxygen donor atom system of quinonoidal compounds can have pronounced effects on their coordination propensities and biological activitiesCitation11. The anticancer activity of thiosemicarbazones has also received a great deal of attention and the combination of 1, 2-naphthoquinone with the potent cytotoxic thiosemicarbazone pharmacophore has been shown to synergistically increase the anticancer effects of parent 1, 2-naphthoquinone derivativesCitation12. In our previous research paper, we reported the synthesis of 1, 2-naphthoquinone analogues which can exert antiproliferative activityCitation13.
The cytotoxicities of quinonoidal compounds can be explained by a number of mechanisms including intercalation, DNA strand breakage, free radical mediated alkylation and inhibition of DNA repair enzymesCitation14. Frydman et alCitation15. reported that several 1, 2-naphthoquinone derivatives can induce DNA-TOPO II-mediated DNA cleavage, which is crucial for the cytotoxicity of these compounds.
These findings pave a way for research to be carried out in this area and attracted our interest towards the synthesis of 4-arylideneamino/cycloalkylidineamino 1, 2-naphthoquinone thiosemicarbazones as new anticancer compounds. A hypothetical binding model also proposed by docking studies to understand the binding preference of synthesized compounds with the active site of target enzyme (ATPase domain of Topoisomerase-II). Further, to get an insight about the drug likeliness of synthesized compounds, they were subjected to in silico ADME prediction studies.
Materials and methods
Reagents, instruments and cell lines
All the chemicals and solvents of analytical grade used in the present studies were procured from Merck, Genei (Bangalore), Himedia (Mumbai) and Aldrich. The synthesized compounds were characterized by MP, IR, 1H and 13C NMR spectral analysis. Melting points were determined by Electrothermal Stuart-SMP10, open capillary melting point apparatus and are were uncorrected. The IR spectra of the compounds were recorded on FT-IR spectrometer (SHIMADZU Infrared-spectrophotometer, FTIR-8400S) using KBr pellet technique. The 1H-NMR and 13C NMR spectra were recorded on JEOL AL300, FT-NMR spectrophotometer with Operating frequency, 300 MHz at temperature 25°C using DMSO-d6 as solvent and TMS as internal standard. Chemical shifts (δ) were reported in ppm. Elemental analysis was performed with CE-440 elemental analyzer (Exeter Analytical Inc., USA).
The cell lines MCF-7 (human breast cancer), Hep-G2 (liver sarcoma), MG-63 (Osteosarcoma) and MCF-12A (normal breast epithelial cells) were procured from National Centre for Cell Sciences Repository, Pune, India. These cells were maintained in Dulbecco’s minimum essential medium (DMEM) supplemented with 10% heat-inactivated foetal calf serum and 0.1% antibiotic solution (complete medium-CM) at 37°C in humidified air containing 5% CO2.
General procedure for synthesis of 4-arylideneamino/cycloalkylidineamino 1, 2-naphthoquinone
An equimolar mixture of substituted aryl aldehydes/cyclic ketones (0.01 M) and 4-amino-1, 2-naphthoquinone (0.01 M) in 20 mL ethanol/methanol was refluxed on a water bath for 6–10 h in the presence of few drops of glacial acetic acid/concentrated H2SO4 as catalyst. The progress of the reaction was monitored using thin layer chromatography (TLC) at appropriate time points. After completion of the reaction, the solution was cooled and the separated solid was filtered, washed with ice-cold water and dried. Finally, the product thus obtained was recrystallized from ethyl acetate and ethanol in different proportions.
General procedure for synthesis of 4-arylideneamino 1, 2-naphthoquinone-2-thiosemicarbazones
Thiosemicarbazide hydrochloride (1.0 mM) dissolved in 5 mL hot distilled water was added to a suspension of 4-arylideneamino-1, 2-naphthoquinone (1.0 mM) in 50.0 mL ethanol. The reaction mixture was refluxed on a water bath for 4–8 h. The progress of the reaction was monitored using TLC at appropriate time intervals. After completion of the reaction, the precipitate formed was filtered, washed several times with warm ethanol and dried. Compounds were purified by recrystallization or column chromatography whichever was desired. Spectral data of compound TS01 and TS02 are given below whereas spectroscopic data of remaining compounds are documented in Supplementary file.
4-benzylideneamino 1, 2-naphthoquinone-2-thiosemicarbazone (TS01)
Yield: 61.4%. M.P. 184–185°C; IR (KBr, υmax·cm−1): 3458.84 (–NH2 str.), 3053.19 (Ar.–C–H str.), 1683.49 (C=O str. of quinone), 1626.57 (–CH=N str.), 1576.59 (C=N str.), 1239.75 (C=S str.); 1H NMR (DMSO-d6, 300 MHz) δ (ppm): 13.89 (s, 1H, NH), 9.26, 8.86 (2s, 2H, NH2), 8.31 (s,1H, –CH=N), 6.89–7.79 (m, 9H, Ar–H); 13C NMR (DMSO–d6) δ (ppm): 118.29–140.06 (Aromatic–C, C5-C10, C1′-C6′), 154.29 (–C=N), 157.42 (–CH=N), 177.78 (C=O of quinone), 180.17 (C=S); anal. calcd. For C18H14N4OS (%): C, 64.65; H, 4.22; N, 16.75; S, 9.59. Found (%): C, 64.69; H, 4.26; N, 16.73; S, 9.63.
4-(4-nitrobenzylideneamino) 1, 2-naphthoquinone-2-thiosemicarbazone (TS02)
Yield: 53.2%. M.P. 198–199°C; IR (KBr, υmax·cm−1): 3443.31 (–NH2 str.), 3219.02 (–NH str.) 3078.14 (Ar.–C–H str.), 1678.12 (C=O str. of quinone), 1623.49 (–CH=N str.), 1571.19 (C=N str.), 1367.26, 1520.28 (–N=O str.), 1219.10 (C=S str.); 1H NMR (DMSO–d6, 300 MHz) δ (ppm): 14.08 (s, 1H, NH), 9.42, 8.97 (2s, 2H, NH2), 8.57 (s,1H,–CH=N), 7.48–8.27 (m, 8H, Ar–H); 13C NMR (DMSO–d6) δ (ppm): 127.29–142.53 (Aromatic–C, C5–C10 C1′–C6′), 155.71 (–C=N), 159.33 (–CH=N), 178.67 (C=O of quinone),181.31 (C=S); anal. calcd. For C18H13N5O3S (%): C, 56.98; H, 3.45; N, 18.46; S, 8.45. Found (%): C, 57.01; H, 3.42; N, 18.50; S, 8.41.
General procedure for synthesis of 4-cycloalkylidineamino 1, 2-naphthoquinone-2-thiosemicarbazones
To the boiling solution of 4-cycloalkylidineamino 1, 2-naphthoquinone (0.01 M) in 50 mL of ethanol, few drops of glacial acetic acid were added. Thereafter, thiosemicarbazide hydrochloride (0.01 M) dissolved in 15 mL of water was added drop wise with stirring. The reaction mixture was refluxed for 10–12 h. On cooling the precipitate formed was filtered and recrystallized with methanol.
In vitro antiproliferative activity
The cytotoxicity of compounds TS01-TS18 was evaluated against MCF-7 (human breast cancer), Hep-G2 (liver sarcoma) and MG-63 (Osteosarcoma) cell lines using the standard MTT assayCitation16. Doxorubicin, a quinone derivative and a clinical antitumor drug, was used as a marker to evaluate the relative cytotoxic efficiency of synthesized compounds. Compounds TS09, TS10, TS11 and TS13 were further evaluated against MCF-12A, a normal epithelial cell line.
The cells were seeded in a 96-well plate at the density of approximately 20,000 cells per well and allowed to attach for 24 h. Various concentrations (1–25 µM) of the test compounds were prepared by dissolving them in DMSO and then by serially diluting with culture medium. Each concentration of compounds was added in triplicate to every well having monolayer of cells and incubated at 37°C in a CO2 incubator for 48 h. After incubation, 30 µL of 5 mg/mL solution of 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) was added to each well, and the plate was incubated for another 4 h. Incubation at 37°C for 4 h allowed the reduction of MTT by viable cells to an insoluble formazan product, which was dissolved by addition of 0.04 N HCl in isopropanol, and the absorbance of each well was measured by using ELISA plate reader at 540 nm. Statistical analysis was performed using Graph Pad prism software, version 5 (Graph Pad software) by nonlinear regression analysis. IC50 values were calculated for each set of triplicate wells.
Molecular docking
Molecular docking studies were conducted in order to validate the obtained pharmacological data and to provide understandable evidence for the observed anticancer activity of the compounds. The computation was carried out using the Schrödinger 2011 molecular modelling software package. Docking was performed by using the Glide 5.7 integrated with Maestro 9.2 (Schrödinger, LLC, 2011) interface on the Linux operating system.
In silico ADME predictions of the best fit molecules
The ultimate goal of drug discovery is the identification of innovative small molecular scaffolds exhibiting high binding affinity and selectivity for the target enzyme together with a reasonable absorption, distribution, metabolism and excretion (ADME) profile. Such chemical entities are likely to carve a niche in the horizon of drug development process. The best fit ligands were neutralized and checked for their ADME properties using QikpropCitation17.
Since quinones are good electrophiles so they can easily be attacked by Glutathione (GSH) in the liver, resulting in the formation of GSH-quinone conjugates, which is the natural defence exerted by our body to reduce toxicity of electrophillic drugs like quinoneCitation18,Citation19. Hence possibility of most active compound (TS10) to form glutathione conjugate was predicted by using software Pallas v3.1.1.2 which generated a pool of possible metabolites of TS10Citation20.
Results and discussion
Synthesis of 4-arylideneamino/cycloalkylidineamino 1, 2-naphthoquinone thiosemicarba-zones
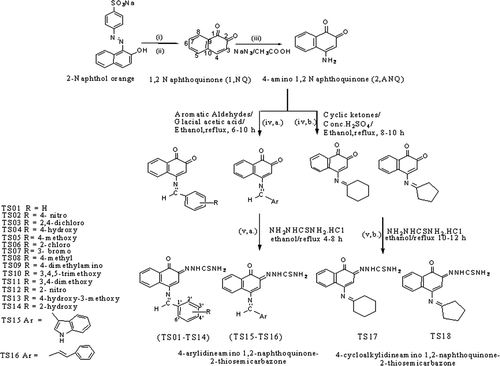
The synthetic route of the target compounds TS01–TS18 is illustrated in . According to previously reported method, synthesis of starting materials 1, 2-naphthoquinone was accomplished from water soluble dye β-naphthol orangeCitation13. 4-amino 1, 2-naphthoquinone (ANQ), the key intermediate for the synthesis of target compounds was obtained through nucleophilic substitution of 1, 2-naphthoquinone with sodium azide in acetic acid. Condensation of ANQ with appropriate aryl aldehydes/cyclic ketones in presence of glacial acetic acid/conc. H2SO4 as catalyst afforded the formation of 4-arylideneamino/cycloalkylidineamino 1, 2-naphthoquinones. Derivatization of one of the carbonyl group of 4-arylideneamino/cycloalkylidineamino 1, 2-naphthoquinones with thiosemicarbazide side chain led to synthesis of proposed compounds [TS01-TS18].
Scheme 1. General pathway for the synthesis of 4-arylideneamino/cycloalkylidineamino 1, 2-naphthoquinone-2-thiosemicarbazones, reagents & conditions (i) Na2S2O4, stirring at 40–50°C; (ii) FeCl3 in conc. HCl with crushed ice, add with stirring; (iii) sodium azide in water and glacial acetic acid, heated to 40°C; (iv) a. aromatic aldehydes, glacial acetic acid, ethanol/methanol, reflux 6–10 h (iv) b. cyclic ketones, concentrated H2SO4, ethanol/methanol, reflux 8–10 h and (v) a. thiosemicarbazide hydrochloride, ethanol, reflux 4–8 h, (v) b. thiosemicarbazide hydrochloride, ethanol, reflux 10–12 h.
Derivatizing one of the quinone carbonyl oxygen in 4-arylideneamino/cycloalkylidineamino 1, 2-naphthoquinone with thiosemicarbazide side chain gave rise to additional bands in the range of 3219–3381 and 3411–3491 cm−1 in the IR spectrum of synthesized compounds which revealed the asymmetric and symmetric stretches of the amino group. Strong imine absorption at 1562–1593 cm−1 due to C=N linkage also confirmed the successful derivatization.
1H NMR spectrum of thiosemicarbazone derivatives showed a singlet in the range of 13.78-14.11 ppm due to NH proton of thiosemicarbazone and 2 singlet were observed at 9.17–9.45 and 8.79–8.98 ppm which showed that 2 protons of NH2 group are magnetically inequivalent. In 13C NMR spectrum of thiosemicarbazone derivatives, peak was observed in the range of 182–180 ppm due to presence of C=S group of compounds. Presence of a characteristic singlet peak in 1H NMR in the range of 8.11–8.68 δ ppm confirmed the presence of azomethine proton (–CH=N–).
Antiproliferative activity
The synthesized compounds were evaluated for antiproliferative activity against a panel of three cancer cell lines MCF-7, Hep-G2 and MG-63 using MTT assay with doxorubicin as the positive control and the results are expressed as IC50 in Supplementary Table S1.
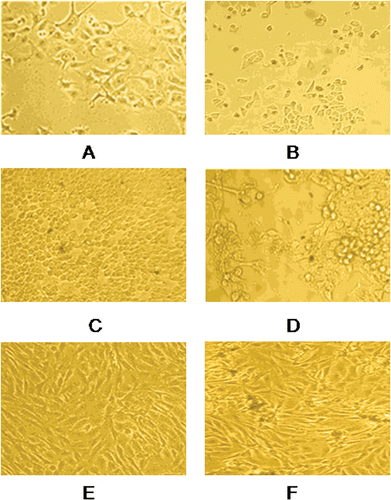
Majority of the compounds exhibited promising anticancer potency against MCF-7 and Hep-G2 cell lines but were found to be less active against MG-63. Compounds TS10 & TS13 displayed remarkable cytotoxicity against the MCF-7 cell line with IC50 values 3.5, 3.81 µM respectively which is very close to standard drug doxorubicin. Microscopic images of morphological changes in cells after treatment with compound TS10 are shown in . Compounds TS01, TS06, TS07, TS08, TS12, TS15 and TS16 were significantly active against MCF-7 and Hep-G2 but nonsignificant for MG-63. Compounds TS02 and TS03 were found to be least active in the series (IC50 12.17–20.07 µM).
Figure 2. Morphological changes in MCF-7 cells incubated for 48 h, (A) negative control (B) with 5 µM concentration of compound TS10, morphological changes in Hep-G2 cells incubated for 48 h, (C) negative control (D) with 5 µM concentration of compound TS10, morphological changes in MG-63 cells incubated for 48 h, (E) negative control (F) with 5 µM concentration of compound TS10. The cells were observed under phase contrast microscope 100×.
On the basis of cytotoxicity screening, the preliminary structure activity relationship study suggested that compounds with electron donating groups on phenyl ring were generally more potent than compounds having phenyl ring with electron withdrawing substituents. The pharmacological data indicated that 3, 4, 5-trimethoxy-phenyl analogue (TS10) showed approximate 3–4 fold improvement in cytotoxicity against all the cell lines (IC50 3.5–6.14 µM) compared with phenyl analogue having electron withdrawing nitro group at 4th position (TS02, IC50 13.98–20.07 µM).
Inspection of cytotoxicity data of compounds TS04 and TS14 showed that migration of hydroxyl group from 2nd position of phenyl ring (TS14, IC50 7.24–9.04 µM) to 4th position of phenyl ring (TS04, IC50 5.42–9.08 µM) enhanced the activity. In nitro-substituted analogues, presence of electron withdrawing group on the 4th position (TS02, IC50 13.98–20.07 µM) led to decreased activity as compared to nitro group at 2nd position (TS12, IC50 7.55–12.41 µM) in all tested cell lines. The cytotoxic activity gets enhanced by the replacement of the phenyl ring with the cyclopentyl or cyclohexyl ring.
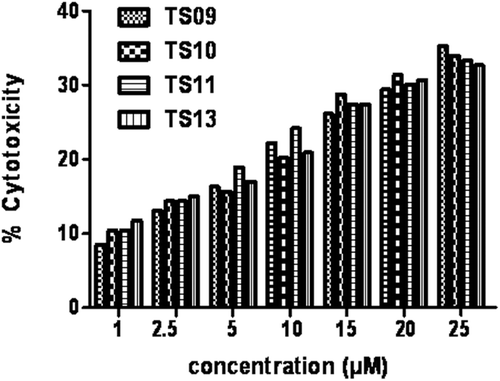
The major challenging aspect of synthesizing new chemotherapeutic agents is selectivity towards cancerous cells compared to the normal cells. Therefore, we have studied the cytotoxic efficacy of some of the potent compounds TS09, TS10, TS11 & TS13 on MCF-12A, a normal epithelial cell line. Compounds showed only 25–35% cell death in the MCF-12A cell line (); thus the compounds are selective toward cancerous cells with high margin of safety.
Figure 3. Percentage cytotoxicity of MCF-12 A, a normal epithelial cell line at various concentration (1–25 µM) of compounds TS09, TS10, TS11 and TS13.
Molecular docking
The structure of the human Topoisomerase-II ATPase (TP-II) co-crystallized with AMP-PNP complex [PDB:1ZXM] was retrieved from the Protein Data Bank [www.rcsb.org (DOI: 10.2210/pdb1zxm/pdb)] and further modified for docking calculations by deleting the substrate cofactor as well as the crystallographically observed water molecules. The active site of the protein was defined for generating the grid. Glide calculations were carried out with Impact version v57111 and the Glide scores are shown in Supplementary Table S1.
It was observed that the most active compounds of the series, TS10 and TS13 were predicted to be most active in silico. Some other compounds like TS04, TS05, TS06, TS09, TS11, TS15 having significant biological activity are also found to have good glide scores. The afore findings confirms that the experimental results of wet lab are in good agreement with the predicted ones as observed from the glide score. Standard drug doxorubicin was used to probe the hydrophilic and hydrophobic interactions between synthesized ligands and topoisomerase-II. Glide predicted binding interactions of doxorubicin and compound TS13 were shown in Supplementary Figures S1 and S2, respectively.
However, some compounds like TS17 and TS18 having cycloalkylidene group did not exhibit good glide scores but were found to have significant activity. It is possible that these compounds may act through a different mechanism, and further studies are required to elucidate their mechanism of action.
In silico ADME predictions of the best fit molecules
Qikprop helps in analysing the pharmacokinetics and pharmacodynamics of the ligand by accessing the drug like properties. As revealed in Supplementary Tables S2 and S3, all the best-fit compounds were found to be non toxic for CNS, value of permeability less than 500 through MDCK Cells indicated the non permeability of compounds through blood-brain barrier, a desirable aspect. Apart from this, majority of compounds were predicted to possess significant oral absorption. None of the best fit compounds violated the Lipinski rule of five as well as log P, donor HB and acceptor HB were within the range indicated the drug like behaviour of compounds.
The possible metabolites prediction of most active compound with software Pallas v 3.1.1.2 indicated that the compound can form GSH conjugate so the toxic metabolites of electrophillic quinone compound can be scavenged by natural defence biomarker GSH (Supplementary Figure S3).
Conclusion
In summary, we have successfully synthesized and characterized a library of 4-arylideneamino/ cycloalkylidineamino 1, 2-naphthoquinone thiosemicarbazone derivatives of biological interest. These compounds exhibited promising anticancer activity against selected human cancer cell lines (MCF-7, Hep-G2 and MG-63). Some of the potent compounds were further evaluated for cytotoxicity against one normal cell line (MCF-12A) and exhibited selective killing of cancer cells as compared to normal cell line. The results of in vitro antiproliferative screening of synthesized compounds revealed that the compounds TS09, TS10, TS11, TS13, TS14, TS17 and TS18 showed significant cytotoxicity against all the cancer cell lines. The wet lab findings are also supported by molecular docking studies. Along with that, results of in silico pharmacokinetic predictions revealed the drug likeliness of synthesized compounds, so they are more likely to move on to higher phases of the drug development process and provide useful insights about the structural requirements necessary for carrying out rationally based pharmacophoric modifications in order to obtain promising compounds with anticancer effects.
Supplementary Material
Download PDF (466.2 KB)Acknowledgments
The authors express their sincere thanks to the Head, Department of Chemistry, Faculty of Science, Banaras Hindu University, Varanasi, India for providing the facilities of 1H NMR and 13C NMR spectroscopy. We gratefully acknowledge the financial assistance given by Jawahar Lal Nehru Memorial Fund, New Delhi for the grant of fellowship to Mrs. Shubhanjali Shukla.
Declaration of interest
The authors report no conflicts of interest.
References
- WHO Cancer. Available at: http://www.who.int/hpr/NPH/docs/gs_cancer.pdf, http://www.who.int/diet physical activity/publications/facts/cancer/en/
- Menta E, Palumbo M. Novel antineoplastic agents. Exp Opin Ther Patents 1997;7:1401–1426.
- Liu KK, Li J, Sakya S. Synthetic approaches to the 2003 new drugs. Mini Rev Med Chem 2004;4:1105–1125.
- Powis G, Hacker MP, eds. (1991). The toxicity of anticancer drugs. New York: Pergamon Press, 106–151.
- Siripong P, Hahnvajanawong C, Yahuafai J, Piyaviriyakul S, Kanokmedhakul K, Kongkathip N et al. Induction of apoptosis by rhinacanthone isolated from Rhinacanthus nasutus roots in human cervical carcinoma cells. Biol Pharm Bull 2009;32:1251–1260.
- Pardee AB, Li YZ, Li CJ. Cancer therapy with beta-lapachone. Curr Cancer Drug Targets 2002;2:227–242.
- Wang D, Xia M, Cui Z, Tashiro S, Onodera S, Ikejima T. Cytotoxic effects of mansonone E and F isolated from Ulmus pumila. Biol Pharm Bull 2004;27:1025–1030.
- Saha DK, Padhye S, Sinn E, Newton C. Synthesis, structure, spectroscopy and antitumor activity of hydroxynaphthoquinone thiosemicarbazone and its metal complexes against MCF-7 human breast cancer cell line. Ind J Chem 2002;41A:279–283.
- Kongkathip N, Kongkathip B, Siripong P, Sangma C, Luangkamin S, Niyomdecha M et al. Potent antitumor activity of synthetic 1,2-Naphthoquinones and 1,4-Naphthoquinones. Bioorg Med Chem 2003;11:3179–3191.
- Gokhale N, Padhye S, Newton C, Pritchard R. Hydroxynaphthoquinone metal complexes as antitumor agents x: synthesis, structure, spectroscopy and in vitro antitumor activity of 3-methyl-phenylazo lawsone derivatives and their metal complexes against human breast cancer cell line mcf-7. Met Based Drugs 2000;7:121–128.
- Afrasiabi Z, Sinn E, Chen J, Ma Y, Rheingold AL, Zakharov LN et al. Appended 1,2-naphthoquinones as anticancer agents 1: synthesis, structural, spectral and antitumor activities of ortho-naphthaquinone thiosemicarbazone and its transition metal complexes. Inorg Chim Acta 2004;357:271–278.
- Chen J, Huang YW, Liu G, Afrasiabi Z, Sinn E, Padhye S et al. The cytotoxicity and mechanisms of 1,2-naphthoquinone thiosemicarbazone and its metal derivatives against MCF-7 human breast cancer cells. Toxicol Appl Pharmacol 2004;197:40–48.
- Shukla S, Srivastava RS, Shrivastava SK, Sodhi A, Kumar P. Synthesis, characterization and antiproliferative activity of 1,2-naphthoquinone and its derivatives. Appl Biochem Biotechnol 2012 167:1430–1445.
- Halinska A, Belej T, O’Brien PJ. Cytotoxic mechanisms of anti-tumour quinones in parental and resistant lymphoblasts. Br J Cancer Suppl 1996;27:S23–S27.
- Frydman B, Marton LJ, Sun JS, Neder K, Witiak DT, Liu AA et al. Induction of DNA topoisomerase II-mediated DNA cleavage by beta-lapachone and related naphthoquinones. Cancer Res 1997;57:620–627.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63.
- Sengupta D, Verma D, Naik PK. Docking mode of delvardine and its analogues into the p66 domain of HIV-1 reverse transcriptase: screening using molecular mechanics-generalized born/surface area and absorption, distribution, metabolism and excretion properties. J Biosci 2007;32:1307–1316.
- Zheng XG, Kang JS, Kim Y, You YJ, Jin GZ, Ahn BZ. Glutathione conjugates of 2- or 6-substituted 5,8-dimethoxy-1,4-naphthoquinone derivatives: formation and structure. Arch Pharm Res 1999;22:384–390.
- Priebe W, Krawczyk M, Kuo MT, Yamane Y, Savaraj N, Ishikawa T. Doxorubicin- and daunorubicin-glutathione conjugates, but not unconjugated drugs, competitively inhibit leukotriene C4 transport mediated by MRP/GS-X pump. Biochem Biophys Res Commun 1998;247:859–863.
- Pallas 3.1.1.2, ADME-Tox software. CompuDrug International Inc., U.S.A., 2000.