Abstract
The aim of these studies was to synthesize twelve ester derivatives of dehydroepiandrosterone with therapeutic potential. The effect of 1–12 was demonstrated in the flank organs of gonadectomized hamsters treated with testosterone and the synthesized steroids. In vitro studies were carried out determining the IC50 values for the inhibition of the activity of 5α-reductase type 1 and 2, which are present in rat liver and human prostate respectively. The binding of 1–12 to the androgen receptors (AR) was determined using rat’s prostate cytosol. Steroids 1–12 containing different substituents in the phenyl group of the ester moiety in C-3 reduced the flank organs and inhibited the activity of 5α-R type 1; however only steroids 1 and 2 inhibited 5α-R type 2. 1–12 did not bind to the AR. The modification of one atom of the substituents in the phenyl group of the ester moiety in C-3 changed their biological potency (IC50).
Introduction
The 5α-reductase enzyme (EC 1.3.99.5) converts ▵4-3-ketosteroids to 5α-3-ketosteroids in androgen dependent tissues. Two types of 5α-reductase enzymes (5α-R) had been identified, 1 and 2, each encoded by a different gene, which have been characterized in several speciesCitation1. 5α-R type 2 plays a major role in prostate cancer and benign prostatic hyperplasia as it is predominantly expressed in this tissue. However, some evidence indicates that type 1 is expressed in the prostate epithelial cells while the type 2 is mainly located in the stromal compartmentCitation2. 5α-R type 1 is also located in the liver and skin and acts in a neutral or basic medium, whereas type 2 is active in acidic pH (1).
Hamster flank organs are two pigmented spots formed by a pilosebaceous complex, located in the dorsal skin surface of hamsters. In female hamsters the diameter of the pigmented spot measures 2 mm, whereas in males it is 8 mm. These nodules shrink after castration until they resemble the nodules of the female animals. However, it had been previously reported that daily injections or topical applications of testosterone 1 (T) or 2 (DHT) restore their original sizeCitation3. These glands can metabolize T to DHT in both intact and gonadectomized hamster since the 5α-R is present in this tissueCitation4. Hamster flank organs also have an androgen receptorCitation5. These glands had been used as a model to test antiandrogenic drugs as well as 5α-inhibitory compounds as finasteride and othersCitation6.
Some dehydroepiandrosterone derivatives had previously been reported as inhibitors of 5α-R enzyme and antagonists of the ARCitation7 which produced a decrease of prostate cancer tumorsCitation8 and antagonized the effects of DHT as well as androst-5-ene-3β-17α−diol (Adiol) on the growth of culture LNCaP cellsCitation9.
Previously, our group had reportedCitation7 that the ester moiety in C-3 of the dehydroepiandrosterone esters increases the 5α-R inhibitor activity as well as the antiandrogenic effect. These compounds contain an aliphatic ester moiety (propionyloxy, butyroyloxi and valeroyloxy) at C-3 position of the steroidal skeleton. In view of the fact that the aliphatic ester derivatives showed a high pharmacological activity, in this study we decided to synthesized similar compounds having an aromatic ester (benzoic and p-substituted benzoic acids) and compare the activity of the aromatic ester derivatives with that of the previously publicized aliphatic esters. In this paper we report the synthesis and the biological evaluation of the following compounds: 17-oxaandrost-5-ene-3β-yl-benzoate 1; 17-oxaandrost-5-ene-3β-yl-p-fluoroben-zoate 2; 17-oxaandrost-5-ene-3β-yl-p-chloroben-zoate 3; 17-oxaandrost-5-ene-3β-yl-p-bromoben-zoate 4; 17-oxaandrost-5-ene-3β-yl-p-iodobenzoate 5; 17-oxaandrost-5-ene-3β−yl-p-methylbenzoate 6; 17-chloro-16-formylandrost-5,16-diene-3β-yl-ben-zoate 7; 17-chloro-16-formylandrost-5,16-diene-3β-yl-p-fluorobenzoate 8; 17-chloro-16-formylandrost-5,16-diene-3β-yl-p-chlorobenzoate 9; 17-chloro-16-formylandros-5,16-diene-3β-yl-p-bromobenzoate 10; 17-chloro-16-formylandrost-5,16-diene-3β-yl-p-iodobenzoate 11; 17-chloro-16-formylandrost-5,16-diene-3β-yl-p-methylbenzoate 12 with therapeutic potential as antiandrogens.
Material and methods
Chemical and radioactive materials
Solvents were laboratory grade or better. Melting points were determined on a Fisher-Johns melting point apparatus and are uncorrected. 1H-NMR and 13C-NMR were taken on Varian Gemini 200 and VRX-300, respectively. Chemical shifts are given in ppm relative to that of Me4Si (δ = 0) in CDCl3 (the abbreviations of signal patterns are as follows: s, singlet; d, doublet, t, triplet, m, multiplet). Mass spectra were obtained with a HP5985-B spectrometer. IR spectra were recorded on a Perkin-Elmer 200s spectrometer.
(1, 2, 6, 7-3H) Testosterone [3H] T specific activity: 95 Ci/mmol and Mibolerone (17α-methyl-3H) [3H] MIB specific activity 70–87 Ci/mmol were provided by Perkin Elmer Life and Analytical Sciences. (Boston, MA). Radioinert T, 5α-dihydrotestosterone and MIB were supplied by Steraloids (Wilton, NH, USA). Sigma Chemical Co. (St. Louis, Mo) provided NADPH and Lubrol PX. Finasteride was obtained by extraction from Proscar (Merck, Sharp & Dohme). The tablets were crushed, extracted with chloroform and the solvent was eliminated in vacuum; the crude product was purified by silica gel column chromatography. The melting point of the isolated finasteride (252–254°C) was identical to that reported in the literature. Bio-gel Hydroxyapatite (HAP) was provided by Bio-Rad Laboratories, (CA, USA).
Synthesis of the steroidal derivatives
General procedure for the preparation of esters
To a solution of 3β-hydroxyandrost-5-ene-17-one (1000 mg, 3.5 mmol), DCC (2147 mg, 10.5 mmol) and DMAP (848 mg, 7.0 mmol) in chloroform (20 mL) was added the corresponding acid (14 mmol). The resulting solution was stirred at room temperature for 2 h. Hexane (15 mL) was added and the precipitated dicyclohexylurea was filtered. The organic phase was washed with 10% aqueous hydrochloric acid, 5% aqueous sodium bicarbonate and water. It was dried over anhydrous sodium sulfate, and the solvent was removed in vacuum. The crude ester was recrystallized from ethyl acetate.
17-oxaandrost-5-ene-3β-yl- benzoate (1). Yield 82% of pure product, mp 261–262°C. IR (KBr) cm–1: 2960, 1735, 1711, 1253, 1272. 1H-NMR (CDCl3) δ: 0.9 (3H, s, H-19), 1.1 (3H, s, H-18), 4.8 (1H, m, H-3), 5.4 (1H, d, J = 4 Hz, H-6), 7.4 (2H, t, J = 8 Hz, H-3′ and H-5′), 7.5 (1H, t, J = 8 Hz, H-4′), 8.0 (2H, d, J = 8 Hz, H-2′ and H-6′). 13C-NMR (CDCl3) δ: 13.5 (C-18), 19.4 (C-19), 74.3 (C-3), 121.9 (C-6), 128.6 (C-3′ and C-5′), 129.5 (C-2′ and C-6′), 130.7 (C-1′), 132.7 (C-4′), 139.9 (C-5), 168.1 (ester carbonyl), 221 (C-17). HRMS calc for C26H32O3 392.3287 found 392.5436. The spectral data for compounds 2–6 are given in the Info File.
General procedure for the preparation of 17-chloro-16-formylandrost-5,16-dien-3β-yl benzoates
A solution of the corresponding ester (500 mg, 1–1.3 mmol) in chloroform (5 mL) was added dropwise to a cold and stirred solution of phosphorus oxychloride (4.2 mL, 45.3 mmol) dissolved in dimethylformamide (4.2 mL, 55 mmol). The mixture was allowed to reflux under N2 for 5 h at 40°C. It was combined with 100 mL of a saturated cold solution of sodium bicarbonate and extracted with chloroform (3 × 125 mL); it was dried over anhydrous sodium sulfate and the solvent was removed to in vacuum. The yellow crude product was purified by flash column chromatography [FCC, silica gel 60 hexane:ethyl acetate 98:2].
17-chloro-16-formylandrost-5,16-dien-3β-yl benzoate (7). Yield 43% of pure product, mp 188–190°C. IR (KBr) cm–1: 2860, 1708, 1711, 1664, 1272, 1253. 1H-NMR (CDCl3) δ: 1.0 (3H, s, H-18), 1.1 (3H, s, H-19), 4.9 (1H, m, H-3), 5.4 (1H, d, J = 5.2 Hz, H-6), 7.4 (2H, t, J = 7.6 Hz, H-3′ and H-5′), 7.5 (1H, t, J = 7.6 Hz, H-4′), 8.0 (2H, d, J = 7.2 Hz, H-2′ and H-6′), 9.9 (1H, s, formyl group). 13C-NMR (CDCl3) δ: 15.3 (C-19), 19.4 (C-18), 74.4 (C-3), 122.1 (C-6), 128.4 (C-3′ and C-5′), 129.7 (C-2′ and C-6′), 130.8 (C-1′), 132.9 (C-4′), 136.6 (C-16), 140.1 (C-5), 162.5 (C-17), 166.1 (ester carbonyl),188.3 (formyl group). HRMS calc for C27H31ClO3 438.2452 found 438.7835. The spectral data for compounds 8–12 are given in the Info file.
Biological activity of the steroidal compounds
The human prostate of a man 53 years old, who died from diabetes and renal insufficiency was introduced to a solution of NaCl 150 mM and stored at –70ºC. This gland was obtained from the Department of Pathology, The General Hospital (SS) in Mexico City. Frozen human prostate was thawed on ice and minced with scissors. Unless specified, the following procedures were carried out at 4ºC.
Human prostate tissue was homogenized in two volumes of buffer A (20 mM sodium phosphate, pH 6.5 containing 0.32 M sucrose, 0.1 mM dithiothreitol Sigma-Aldrich Inc.) with a tissue homogenizer Ultra-Turrax IKA, T18 basic. (Wilmington, NC). Homogenates were centrifuged at 1500g for 60 min in a SW 60 Ti rotor (Beckman instruments, Palo Alto, CA). The pellets were separated, suspended in medium A and kept at –70ºC. The suspension, 5 mg of protein/mL for human prostates, determined by the Bradford methodCitation10 was used as source of 5α-R type 2 isozyme.
Animals and tissues
For the in vivo experiments, adult male golden hamsters 2.5 month old (150–200 g) were obtained from the Metropolitan University Xochimilco, Mexico. Hamster’s gonadectomies were performed under pentobarbital anesthesia and the castrated hamsters were kept in a room with controlled temperature (22ºC) and light–dark periods of 12 h. Food and water were provided ad libitum. After 36 days maintaining these conditions, the hamsters were sacrificed with CO2. This protocol was approved by the Institutional Care and Use Committee of the Metropolitan University of Mexico (UAM). The experiment with the gonadectomized animals was carried out on 8 groups of 4 animals/experiment.
With the aim to prepare microsomes as source of 5α-R type 1 isozyme, two livers from male adult rats, 8 months old were extirpated. The rats were fasted overnight to decrease glycogen levels before the removal of the livers.
Rat livers were minced (15–30 g) in one volume of buffer A using a tissue mill IKA A11 basic. Unless specified, the following procedures were carried out at 4ºC. The tissue was homogenized and the suspension centrifuged at 10,000g for 30 min (Beckman L70K ultracentrifuge); the pellet was discarded. The supernatant liquid was filtered through a strainer and centrifuged at 100,000g for 60 min. The microsomal pellet was washed by resuspension in five volumes of buffer A with a homogenaizer and the proteins were determined by the Bradford methodCitation10. The suspension was centrifuged at 100,000g and the pellet suspended in buffer A to a concentration of 10–20 mg of potein/mL; the microsomal solution was stored at –80°C prior to the preparation of the solubilized steroid 5α-R type 1. The solubilization of 5α-R type 1 isozyme from rat liver microomes was carried out according to Levy et al.Citation11.
In order to determine the binding of steroids 1–12 to the AR, the prostate of adult rats 8 months old weighing 500 g was extirpatedCitation12. In this study we used also rats because the prostate gland is bigger and there is no difference in the binding activity between rats and hamsters cytosol.
The prostate of the rats was blotted, weighed and soaked in cold TEMD (40 mM tris-HCl, 3 mM EDTA and 20 mM sodium molybdate, dithiotreitol 0.5 mM, 10% glycerol at pH 8) prior to their use. Unless specified, all procedures were carried out in an ice bath. Tissues were homogenized with a tissue homogenizer.
Tissues were homogenized in one volume of buffer TEMD plus protease inhibitors in an ice bath with a tissue homogenizer. Homogenates were centrifugedCitation13 in a SW 60 Ti rotor (Beckman Instruments, Palo Alto, CA).
The cytosolic fraction obtained from the supernatant liquid of the rat prostate homogenate described above, was stored at –70°C. Prostatic cytosol proteins (6 mg of protein in 200 µL) were determined by the Bradford methodCitation10.
In vitro experiments
Characterization of 5α-R type 1
Determination of protein concentration-dependent 5α-R type 1 activities
In order to determine the optimum concentration of protein to obtain the maximum activity of 5α-R enzyme type 1, different concentrations of solubilized microsomes (30, 60 and 90 µg of protein) were used as a source of this enzyme. A mixture containing a final volume of 1 mL, 1 mM DTT sodium phosphate buffer 20 mM, at pH 6.5, 2 nM of [1,2,6,7 3H] T, and 2 mM NADPHCitation11 was prepared. The reaction in duplicate was started when the enzymatic fraction was added to the mixture and this was incubated at 37°C for 60 min. The reaction was stopped by mixing with 1 mL of dichloromethane. Incubation without tissue was used as a control.
Determination of pH dependence of 5α-R activity
To measure the pH dependent 5α-R type 1 activities, the assays were carried out in a similar way that we described above but using 20 mM potassium phosphate buffers ranging from pH 6–8 and different concentrations of unlabeled T.
Determination of 5α-R type 1 activity
The activity of the 5α-R type 1 isozyme was determined by following the conversion of T to DHT at pH 7.5. 2 nM of [1,2,6,7 3H] T, and different concentrations of unlabeled T (5 × 10–7–6.3 × 10–6 M) or DHT (5 × 10–7–2.7 × 10–5M) and 2 mM NADPHCitation11 was prepared. The reaction in duplicate was started when 5α-R type 1 isozyme fraction (60 µg protein in a volume of 7.5 µL) was added to the mixture and incubated at 37°C for 60 min.
Determination of 5α-R type 2 activity
The activity of the isozyme 5α-R type 2 was assayed as previously describedCitation14,Citation15.
The radioactive zone that had identical chromatographic behavior as the standard T (Rf value of 0.56) corresponds to 70% of the accounted radioactivity in the plate. The radioactivity contained in the zone corresponding to DHT standard (Rf value of 0.67) of the experimental chromatogram was identified as the transformed DHT and corresponds to 20–27% of the total radioactivity accounted in the plate. This result was considered to be 100% of the activity of 5α-R (types 1 or 2) for the development of inhibition plots. Unmodified [3H]T was identified (Rf value of 0.56) from control incubations which did not contain tissue and had identical chromatographic behavior as the non labeled standard (identified by UV lamp, 254 nm). The radioactivity contained in the zone corresponding to DHT standard (Rf value of 0.67) of the control chromatogram was 1% of the total radioactivity accounted in the plate and was considered as an error; it was subtracted from the experimental chromatograms.
The 5α-R type 1 activity was calculated from the percentage of the labeled DHT, formed, taking into consideration the recovery, blank values, specific activity of [3H] T and ratio of added [3H] T to unlabeled T or DHT. The 5α-R type 2 activity was calculated taking into consideration the recovery, blank values and the specific activity of [3H] T.
Km and Vmax values were derived from Linewear–Burk plots, in which the regression lines were computed by the least square method. The statistical significance of the means was determined by Student’s t test in all experiments.
The efficiency of T 5α-R type 1 was estimated according to Weisser and Krieg reportCitation16.
Determination of 50% of the inhibitory concentration of steroids 1–12 in 5α-R type 1 and 2 activities
In order to calculate the IC50 values (the concentration of steroids 1–12 or finasteride required to inhibit 50% of the activity 5α-R types 1 and 2 enzyme, six series of tubes containing increasing concentrations of these steroids (10–11–10–3 M solubilized in DMSO) were incubated in duplicate: For type 2 isozyme assay, pH of 6.5; 2 mM [1,2,6,7-3H]T, 2 mM NADPH and 500 µg of protein from the enzymatic fraction obtained from human prostate were usedCitation14,Citation15. For 5α-R type 1 assay, we used a pH of 7.5; 2 mM NADPH, 2 nM [1,2,6,7-3H]T and 6.31 µM of non labeled T. The reaction was started by adding 60 µg of soluble 5α-R type 1 isozyme.
The concentration of finasteride and compounds 1–12 required for inhibiting 5α-R type 1 and 2 activities by 50% (IC50) were determined from the inhibition plots using SigmaPlot Software.
Androgen receptor competitive binding assay
For AR competitive binding studies, tubes containing 1 nM of [3H] MIB plus a range of increasing concentrations (1 × 10–10–4 × 10–7 M) of unlabeled MIB or steroids 1–12 in ethanol or acetone, or in the absence of competitor were prepared. The [3H] MIB-AR complex was separated by the hydroxyapatite (HAP) methodCitation17.
The IC50 value of each compound was calculated according to the plots of concentration versus percentage of binding using SigmaPlot Software.
In vivo experiments
For the daily subcutaneous injections, 2 mg/kg of the steroids 1–12 were dissolved in 200 µL of sesame oil and administered for 6 days to gonadectomized hamsters, together with 1 mg/kg of testosterone. Three groups of gonadectomized hamsters were kept as control; one was injected with 200 µL of sesame oil, the second with 1 mg/kg of testosterone and the third with T plus 1 mg/kg of finasteride for 6 days. After the treatment, the animals were sacrificed with CO2. The flank organs diameter size of each hamster was measured using a vernier. Two separate experiments were performed for each group of steroid treated animals. The results were analyzed using one-way analysis of variance and Dunnett’s Method to compare means, with JMP IN 5.1 software.
Results
Synthesis of the steroidal derivatives 1–12
The steroidal derivatives 1–12 were synthesized using dehydroepiandrosterone as the starting material. This compound was esterified with the corresponding acid in the presence of dicyclohexylcarbodiimide and dimethylaminopyridine. The resulting esters 1–6 were treated with phosphorus pentachloride in dimethylformamide (Vilsmeyer-Haack reaction) to form the final compounds, the esters of 17-chloro-16-formylandrost-5,16-diene moiety 7–12.
In vitro experiments
Protein concentration-dependent 5α-R type 1 activities
When different concentrations of protein obtained from solubilized microsomes of rat’s liver were used, 5α-R enzyme type 1 shows different activity. The optimum concentration of protein was of 60 µg.
pH Dependent 5α-R type 1 activities
5α-R type 1 present in the solubilized microsomes from rat’s liver, showed the conversion of T to DHT in the presence of NADPH. Furthermore, the pH profile of this enzyme was observed by using different concentrations of T. In the presence of 6.31 µM of T, 5α-R type 1 shows maximal conversion to DHT at pH 7.5. When smaller or higher concentrations of T were used, a lower activity of 5α-R type 1 at pH 7.5 was observed. The kinetic parameters were also calculated and these data are shown in . The higher Vmax value was determined at pH 7.5; however, this enzyme shows major affinity for T at pH 6.5 (lower value of Km = 2.97) see .
Table 1. Effect of the testosterone concentration on 5α-reductase type 1 activity at different pHs. Solubilized microsomes from rat liver were used as source of 5α-reductase type 1.
In addition, the efficiency of 5α-R type 1 for the conversion of T to DHT was calculated. This efficiency value (Vmax/Km)Citation16 was obtained from the rate between maximal velocities of production of DHT (Vmax), using different pHs and the Michaelis constants (Km).
Vmax/Km rate values showed that 5α-R type 1 was effective at pHs of 6.5, 7.5 and 8. However this efficiency was higher at pH 7.5 (40.16 value) as compared to that at pH 6.5 and 8 (37.03 and 18.78 values, respectively).
On the other hand, labeled T in the presence of increasing concentrations of unlabeled T showed a higher conversion to labeled DHT (higher 5α-R activity) Vmax = 111.1 pmol/mg of protein/h (Km = 27.6) as compared to that of the experiment where increasing concentrations of DHT and labeled T were used (Vmax = 0.597 pmol/mg of protein/h and Km 1.17); as a result of this, the efficiency of the enzyme 5α-R was higher in the absence of DHT.
However, the 5α-R type 1 showed a higher affinity (minor Km = 0.39 and Vmax = 169.49) when labeled T, unlabeled T [1 µM] and an increasing concentrations of DHT were present in the incubating medium. In this case, the efficiency value of 5α-R indicated by the rate of Vmax/Km was higher as compared to that of the experiment where T in minor concentration or T (in minor concentration) +DHT were present in the incubating medium. The affinity of the 5α-R type 1 was also evaluated in the presence of labeled T, unlabeled T [1.5 µM] and an increasing concentration of DHT; in this test, higher values of Km = 0.79 µM and Vmax = 188.67 were observed. This trial showed that the efficiency rate value was lower than that found for 1 µM of T.
The data from this experiment indicated very clearly that the affinity of 5α-R enzyme type 1 with DHT increases in the presence of 1 µM of T, (small Km value of 0.39) as compared to that using 1.5 µM of T giving a larger Km value (0.79) thus showing a reduced affinity. Nevertheless the presence of the product (DHT) did not inhibit the efficiency of the conversion of T to DHT at these concentrations (1 and 1.5 µM).
Biological activity of the novel compounds
Determination of the 50% inhibitory concentration of the synthesized compounds in human prostate
The concentration of finasteride and compounds 1–12 required for inhibiting 5α-R type 1 and 2 activities by 50% (IC50) were determined from the inhibition plots, using different concentrations of the tested steroids; the results ± standard deviations are shown in . Finasteride inhibited 5α-R type 2 with a low value of IC50 (0.0085 µM), whereas this value was higher for type 1 isozyme (0.63 µM). Steroids 1 and 2 were the only one from the synthesized compounds that inhibited the enzyme 5α-R type 2, with IC50 values of 100 and 0.00358 µM, respectively ().
Figure 1. Steroidal structures and effect of different dehydroepiandrosterone derivatives on the activity of 5α-R enzyme type 1 and 2 (5α-R): 1–12. The IC50 values indicate the required concentration of the novel steroids for the inhibition of 50% of the activity of 5α-R1 and 5α-R2. This figure shows also the relative binding affinity (RBA) of these derivatives to the androgen receptor (AR). The following abbreviations were used: NA, non active compound.
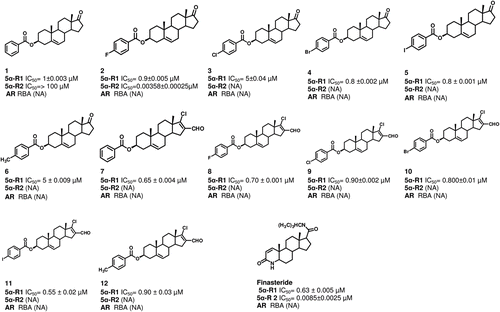
Steroids 1–12 inhibited the enzyme 5α-R type 1 activity; however, compounds 3 and 6 showed a higher IC50 value (0.9 and 5 µM, respectively; lower activity) as compared to that of the remaining steroids. On the other hand compound 11 and Finasteride showed a lower IC50 value (0.55 ± 0.02 and 0.63 ± 0.005 µM, respectively; a higher inhibitory activity).
Binding of the synthesized compounds to the androgen receptor
The binding of steroids 1–12 to the AR was determined from the IC50 and the relative binding affinity (RBA) values for each compound. The IC50 value was calculated according to the plots of concentration of the non labeled MIB or the synthesized compounds versus percentage of binding to MIB (). As can be seen in , none of the studied compounds bound to the AR.
In vivo experiments
Flank organ test
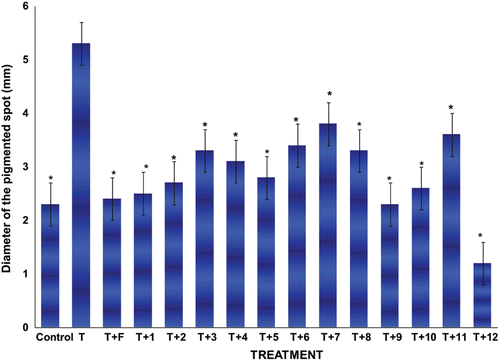
After castration, the diameter of the pigmented spot of the male hamster flank organs decreased (p < 0.005) as compared to that of the normal glands. Treatment with vehicle alone did not change this condition, whereas s.c. injections of 1mg/kg of T for 6 days significantly increased (p < 0.005) the diameter of the pigmented spot in castrated male hamsters flank organs (). The diameter of the pigmented spot significantly decreased (p < 0.005) when testosterone (T) and finasteride (1 mg/kg) or steroids 1–12 (2 mg/kg) were injected together, as compared to that of testosterone-treated animals (). Compound 12 showed the highest pharmacological activity (small diameter of the pigmented spot 1.0 mm).
Figure 2. Diameter size ± standard deviations of the pigmented spot from gonadectomized hamster’s flank organs receiving different treatments with T plus the novel steroids for 6 days. Control gonadectomized hamsters were treated with vehicle only. The following abbreviations were used: Testosterone (T), Finasteride (F). *Significant differences with the T-treatment were observed.
Discussion
Since the optimal concentration of the protein obtained from the solubilized microsomes of rat’s liver as 5α-R type 1 source was of 60 µg, thus at this protein concentration the active sites of the enzyme were saturated by the substrate and coenzyme.
The kinetic data for 5α-R type 1 enzyme indicated that Vmax was of 1.1 nmol/mL/h at pH 7.5. This result correlates well with the previously reported data for this enzymeCitation18 and is corroborated by the efficiency rate value of the reaction at this pH. On the other hand, it had been previously demonstratedCitation19 that a different activity of the enzymes is observed when a range of pHs are used. This fact had been explained on the ground that some electrostatic changes were effected in the molecules involved in the reactionCitation19. This could be in agreement with the variation in the activity of the 5α-R type 1 observed in this study.
The kinetic results obtained in this experiment indicated also that when DHT was added to the incubated medium, the conversion of T to DHT was not inhibited at concentrations of 1 and 1.5 µM of T. On the contrary, the presence of 1 µM of T induced an increase of the affinity of 5α-R type 1 for its product, DHT (lower Km value) and an increase in Vmax was observed. On the other hand, the efficiency rate value for this enzyme was higher when 1 µM of T plus increasing concentrations of DHT were used, as compared to the experiment containing 1.5 µM of T plus increasing concentrations of DHT. The result of these experiments is similar to that previously reported by our group for 5α-R type 2Citation20.
The overall data suggested that the decrease of T observed in elderly man could produce higher affinity of 5α-R for its product DHT. As a consequence of this, an increase of the affinity for both isozymesCitation21 could produce a higher synthesis of DHT in the target organs. This metabolic imbalance could induce a pathological illness such as prostate cancer, where an increase of 5α-R type 1 activity had been noticedCitation21.
On the other hand, it is important to consider that high levels of intracellular DHT result in cellular proliferation and delay cellular differentiationCitation22. In this context previously it had been reportedCitation23 that in advanced prostatic cancer the 5α-R type 1 enzyme is expressed to a higher degree than the type 2. Furthermore, these authors demonstrated also that in the tissue located around cancer cells, the gene expression of 5α-R type 1 is increased as compared with that found in benign prostatic hypertrophyCitation23.
On the ground of the findings given above, the effect of steroids 1–12 described in this study as inhibitors of 5α-R type 1 could have a therapeutic potential for prostate cancer, since they are specific for the inhibition of 5α-R type 1 enzyme, with the exception of 1 and 2 that also inhibited type 2 isozyme. These compounds have also the advantage that they did not bind to the AR and this phenomenon decreases the adverse effects and improves also their therapeutic potential. However one of the important goals is to decrease the IC50 value of the5 α-R types 1 and 2 inhibitors; these studies demonstrated that the change of the nature of the substituent in phenyl group of the ester moiety in C-3 of dehydroepiandrosterone skeleton is sufficient to change the inhibitory potency of synthesized compounds for the5α-R enzymes.
In this respect, the results of these experiments showed also that steroid 2 having a fluorine atom attached to the phenyl ring in the ester moiety, exhibited higher inhibitory activity for 5α-R type 2 enzyme than steroid 1 which lacks the halogen atom. It appears that the higher electronegativity of compound 2 increases its binding affinity for this enzyme.
In vivo experiments showed also that finasteride and the synthesized steroids decreased the hamster’s flank organs diameter size, thus indicating the presence of 5α-R type 1 in this tissue. The result obtained for finasteride of the flank organ test in this study is similar to the previously reported by Chen et al.Citation6 On the other hand the effect of steroids 1–12 in the flank organs correlated very well with the results obtained in vitro, since a decrease in the diameter of the flank organ spot produced by 1–12, corresponds to an inhibition of 5α-R type 1 in the in vitro experiments.
It is interesting to note that the aliphatic ester derivatives of dehydroepiandrosterone exhibited a higher 5α-R type 2 enzyme inhibitory activityCitation7 as compared to their aromatic counterparts. On the other hand, the aromatic derivatives described in this paper showed an opposite phenomenon (the aromatic steroids exhibited a higher 5α-R type 1 enzyme inhibitory activity as compared to their aliphatic homologues).
Furthermore, these studies demonstrated also that the treatment with steroids 1–12 applied to the hamsters for 6 days in the dose used did not produce any toxicological effect.
Future studies are planned in order to test the effect of these compounds in PC-3 line cells that exhibit some 5α-R activity.
Declaration of interest
We thank CONACYT for its support for the project No 165049. This research did not have any real or perceived conflicts of interest and disclosure arising from intellectual, personal or financial circumstances of the research.
References
- Russell DW, Wilson JD. Steroid 5 alpha-reductase: two genes/two enzymes. Annu Rev Biochem 1994;63:25–61.
- Bonkhoff H, Stein U, Aumüller G, Remberger K. Differential expression of 5 alpha-reductase isoenzymes in the human prostate and prostatic carcinomas. Prostate 1996;29:261–267.
- Cabeza M, Heuze Y, Quintana H, Bratoeff E. Comparison between two different hamster models used for the determination of testosterone and finasteride activity. Asian Journal Animal and Veterinary Advances 2010;5:202–209.
- Takayasu S, Adachi K. The in vivo and in vitro conversion of testosterone to 17 -hydroxy-5 -adrosten-3-one (dihydrotestosterone) by the sebaceous gland of hamsters. Endocrinology 1972;90:73–80.
- Shiba K, Hamaguchi T, Kataoka K, Yamaguchi Y, Handa H, Adachi K. Cloning of the hamster androgen receptor gene. J Dermatol Sci 2001;26:163–168.
- Chen C, Puy LA, Simard J, Li X, Singh SM, Labrie F. Local and systemic reduction by topical finasteride or flutamide of hamster flank organ size and enzyme activity. J Invest Dermatol 1995;105:678–682.
- Arellano Y, Bratoeff E, Garrido M, Soriano J, Heuze Y, Cabeza M. New ester derivatives of dehydroepiandrosterone as 5a-reductase inhibitors. Steroids 2011;76:1241–1246.
- Long BJ, Grigoryev DN, Nnane IP, Liu Y, Ling YZ, Brodie AM. Antiandrogenic effects of novel androgen synthesis inhibitors on hormone-dependent prostate cancer. Cancer Res 2000;60:6630–6640.
- Marwah P, Marwah A, Lardy HA, Miyamoto H, Chang C. C19-steroids as androgen receptor modulators: design, discovery, and structure-activity relationship of new steroidal androgen receptor antagonists. Bioorg Med Chem 2006;14:5933–5947.
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–254.
- Levy MA, Brandt M, Greway AT. Mechanistic studies with solubilized rat liver steroid 5 alpha-reductase: elucidation of the kinetic mechanism. Biochemistry 1990;29:2808–2815.
- Davies P, Thomas P, Griffiths K. Measurement of free and occupied cytoplasmic and nuclear androgen receptor sites in rat ventral prostate gland. J Endocrinol 1977;74:393–404.
- Cabeza M, Vilchis F, Lemus AE, Díaz de León L, Pérez-Palacios G. Molecular interactions of levonorgestrel and its 5 alpha-reduced derivative with androgen receptors in hamster flanking organs. Steroids 1995;60:630–635.
- Hirosumi J, Nakayama O, Fagan T, Sawada K, Chida N, Inami M et al. FK143, a novel nonsteroidal inhibitor of steroid 5 alpha-reductase: (1) In vitro effects on human and animal prostatic enzymes. J Steroid Biochem Mol Biol 1995;52:357–363.
- Bratoeff E, Sainz T, Cabeza M, Heuze I, Recillas S, Pérez V et al. Steroids with a carbamate function at C-17, a novel class of inhibitors for human and hamster steroid 5alpha-reductase. J Steroid Biochem Mol Biol 2007;107:48–56.
- Weisser H, Krieg M. Kinetic analysis of androstenedione 5 alpha-reductase in epithelium and stroma of human prostate. Steroids 1997;62:589–594.
- Hechter O, Mechaber D, Zwick A, Campfield LA, Eychenne B, Baulieu EE et al. Optimal radioligand exchange conditions for measurement of occupied androgen receptor sites in rat ventral prostate. Arch Biochem Biophys 1983;224:49–68.
- Normington K, Russell DW. Tissue distribution and kinetic characteristics of rat steroid 5 alpha-reductase isozymes. Evidence for distinct physiological functions. J Biol Chem 1992;267:19548–19554.
- Segel IH. Enzyme kinetics. Behavior and analysis of rapid equilibrium and steady state enzyme systems. New York: John Wiley & Sons, Inc., 1993.
- Cabeza M, Trejo KV, González C, García P, Soriano J, Heuze Y et al. Steroidal 5a-reductase inhibitors using 4-androstenedione as substrate. J Enzyme Inhib Med Chem 2011;26:712–719.
- Thomas LN, Douglas RC, Rittmaster RS, Too CK. Overexpression of 5 alpha-reductase type 1 increases sensitivity of prostate cancer cells to low concentrations of testosterone. Prostate 2009;69:595–602.
- Dadras SS, Cai X, Abasolo I, Wang Z. Inhibition of 5alpha-reductase in rat prostate reveals differential regulation of androgen-response gene expression by testosterone and dihydrotestosterone. Gene Expr 2001;9:183–194.
- Goldenberg L, So A, Fleshner N, Rendon R, Drachenberg D, Elhilali M. The role of 5-alpha reductase inhibitors in prostate pathophysiology: Is there an additional advantage to inhibition of type 1 isoenzyme? Can Urol Assoc J 2009;3:S109–S114.