
Abstract
Context: Bacterial sphingomyelinase (SMase) is thought to play a crucial role in bacterial evasion of the immune response during the early stages of infections.
Objective: The objective of this study was to predict the chemical structure required for competitive SMase inhibition, then synthesize and test the effect of potential inhibitors on the hydrolysis of sphingomyelin (SM) and protection against infection by Bacillus cereus.
Materials and methods: We synthesized 10 potential SMase inhibitors, derivatives of RY221B-a analogues, based on predictions from three-dimensional structural analysis. We then tested the effect of these compounds on the inhibition of SM hydrolysis and protection of mice inoculated with B. cereus.
Results: One compound, SMY-540, displayed a strong inhibitory effect (IC50 = 0.8 μM) upon SMase and prevented mortality in mice.
Conclusion: SMY-540 is an effective inhibitor of Bc-SMase and has potential for use in the development of drugs to treat infectious diseases caused by bacteria that produce SMase.
Introduction
Bacillus cereus sphingomyelinase (Bc-SMase) engages in phospholipase C activity, hydrolysing sphingomyelin (SM) to phosphocholine and ceramide. The hydrolysis of SM to form ceramide in the membrane of macrophages treated with Bc-SMase causes a decrease in membrane fluidity and interferes with phagocytosisCitation1. Bc-SMase also engages in hemolytic activityCitation2,Citation3. These Bc-SMase activities occur in a divalent metal ion-dependent mannerCitation4–7. In addition, Bc-SMase belongs to a family of Mg2+-dependent neutral SMase that includes SMase produced by Staphylococcus aureus, and Listeria ivanoviiCitation8.
Recently, we determined that Bc-SMase plays a crucial role in bacterial evasion of the immune response during the early stages of infectionsCitation1. In addition, Tajima et al.Citation9 reported that sphingomyelinase from Staphylococcus aureus suppresses TNF-α-induced IL-8 synthesis in endothelial cells and the transmigration of neutrophils. Therefore, an inhibitor of SMase might be useful for the prevention or treatment of infectious diseases caused by bacteria that produce sphingomyelinase.
To date, a number of naturally occurring inhibitors of SMase have been describedCitation10–28. Several synthetic inhibitors, such as a carbamate analog, carbon-analog or fluorinated carbon-analog, that mimic the phosphate ester moiety of phosphocholine by substituting it with stable functional groups have also been developedCitation29–34. We previously developed an inhibitor (RY221B-a) that is based on the crystal structure of Bc-SMaseCitation6,Citation34.
In this study, we investigated the inhibitory effect of various synthetic RY221B-a analogues on Bc-SMase-induced hydrolysis of SM. We then tested the effect of the most potent inhibitor, SMY-540, on the lethality of B. cereus inoculation using a mouse model.
Methods
Characterisation of synthetic compounds
All synthesized compounds were characterized using nuclear magnetic resonance (NMR), infrared spectroscopy (IR) and high-resolution mass spectrometry (HRMS) (see Appendix for details on syntheses).
Purification of Bc-SMase
Bc-SMase was over-expressed in B. subtilis (ISW1214) that were transformed with a plasmid vector, pHY300PLK, carrying the Bc-SMase gene. The expression and purification of the recombinant Bc-SMase were performed as described previouslyCitation6.
SMase activity
Bc-SMase activity was measured using a turbidometric assayCitation35. Briefly, Bc-SMase (50 ng/ml) was treated at 37 °C for 60 min with various compounds that were solubilized in dimethylacetoamide. The reaction mixture was incubated at 37 °C for 5 h with 1.0 mM SM from bovine brain (Nacalai, Kyoto, Japan) that was solubilized in a solution containing 20 mM Tris-HCl (pH 7.5), 1.0% sodium cholate, and 1 mM MgCl2. The turbidity of the reaction mixture was analysed by absorption spectrometer at 620 nm.
In silico docking simulation of SMY-540 with the catalytic domain of Bc-SMase
Molecular modeling studies were performed using Molecular Operating Environment software (MOE, Chemical Computing Group, Quebec, Canada). The X-ray crystallographic structure of the catalytic domain of Bc-SMase (PDB ID:2DDT) was used in the models. The enzyme was prepared for docking studies. The ligand was removed from the enzyme active site, hydrogen atoms were added to the structure with a standard geometry, and the structure was minimized using an MMFF94s force-field. Finally, MOE Alpha Site Finder was used to identify active sites within the enzyme structure, and dummy atoms were created from the obtained alpha spheres. The model was then evaluated using the ASE Dock program (Ryoka System Inc., Tokyo, Japan).
Mice
Six- to 8-week-old male wild-type mice (ICR and BALB/c strains; Nihon SLC, Shizuoka, Japan) were used in infection assays. Experimental protocols were approved by the Institute Animal Care and Use Committee at Tokushima Bunri University. The mice were housed in plastic cages under controlled environmental conditions (22 °C, 55% humidity). Food and water were available ad libitum.
Preparation of oil-in-water (o/w) emulsion
SMY-540 was solubilized in mineral oil (Nacalai). Saline containing 1.1% Tween 80 (Nacalai) and 5.6% D-mannitol (Nacalai) was added to the mineral oil-treated SMY-540, and the mixture was sonicated for 30 min using a bath-type sonicator. After sterilization, by heating at 60 °C for 30 min in a water bath, 3 or 15 μg of the o/w SMY-540 emulsion were each intravenously administered to 10 mice. The final concentrations of emulsion components were as follows: SMY-540 (1.0 or 5.0 mg/ml), mineral oil (3%), Tween 80 (1%) and D-mannitol (5.5%).
Infection and monitoring of mice
Bacillus cereus (1.0 × 107 CFU/mouse; JMU-06B-35, isolated from a patient with septicemia; Jichi Medical University) was intraperitoneally administered to each mouse 3 h after injection of the o/w emulsion of SMY-540. Mice were then monitored every 2 h for 100 h. All mice were euthanized 100 h following administration of B. cereus.
Results
The effect of RY221B-a analogues on Bc-SMase-induced hydrolysis of SM
We evaluated the inhibitory effect of the RY221B-a synthetic derivatives () on Bc-SMase-induced hydrolysis of SM using a turbidometric assay. The IC50 of RY221B-a, SMY-471, SMY-579, SMY-540 and SMY-610 were 1.2, 0.9, 18.3, 0.8 and 3.3 μM, respectively (). However, other tested compounds (RY221B-b, SMY-584, SMY-632, SMY-469a, TOM-2-7, TOM-2-18, TOM-2-37) had little inhibitory effect. Lineweaver-Burk plot analysis, using various concentrations (0.1, 0.5, 1.0, 5.0 and 10) of inhibitors (RY221B-a, SMY-471 and SMY-540), revealed that the compounds competed with SM for access to the catalytic site. In addition, Ki values were calculated to be 5.2, 2.8 and 1.3 μM for RY221B-a, SMY-471 and SMY-540 based on the data of Lineweaver-Burk plot analysis, respectively ().
Figure 1. The structure of RY221B-a and its synthetic analogues.
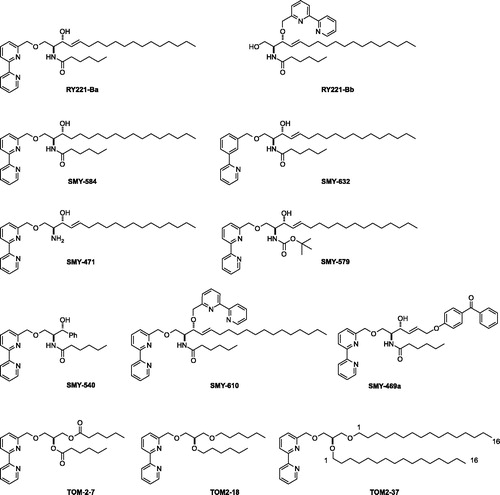
Table 1. Summary of the inhibitory effects of RY221B-a analogues on hydrolysis of SMase*.
Docking simulation analysis of Bc-SMase and SMY-540
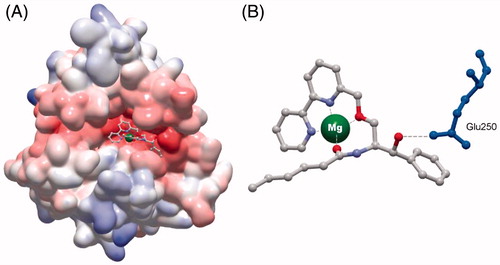
To investigate the mode of SMY-540 binding to the catalytic site of Bc-SMase, we conducted a tertiary structure analysis of this interaction using an in silico docking simulation, performed with MOE software. Our results showed that SMY-540 fit almost perfectly into the catalytic site of Bc-SMase. Moreover, the two nitrogen atoms of the bipyridyl moiety, the carbonyl oxygen of the amide and the ethereal oxygen, in SMY-540 coordinated with the Mg2+ in Bc-SMase (). The docking energy of SMY-540, as calculated from simulations (−204 kCal/mol), was lower than that of RY221B-a (−117 kCal/mol).
Figure 2. Docking simulation analysis of Bc-SMase and SMY-540. (A) Simulation analysis of the docking of SMY-540 to Bc-SMase. The most stable structure of SMY-540 on the molecular electrostatic potential surface is shown. The red and blue surfaces indicate the negative and positive electrostatic potential surfaces, respectively. (B) Interactions between Bc-SMase and SMY-540 at the catalytic site. The gray, white, red and purple in the compound indicate carbon, hydrogen, oxygen and nitrogen, respectively. Mg2+ is represented by green. Please view colour online.
The effect of SMY-540 on the lethality of Bc-SMase and B. cereus in mice
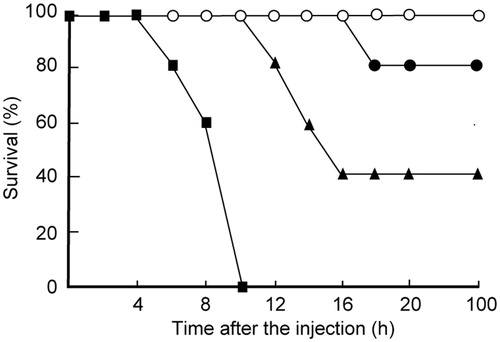
We reported that administration of anti-Bc-SMase antibody and immunization against Bc-SMase prevented death caused by the clinical isolate (JMU-06B-35) which is the Bc-SMase high expression strainCitation1. The administration of B. cereus (JMU-06B-35) results in the death of mice within 10 h. To investigate the therapeutic potential of SMY-540 against B. cereus infection, we tested the effect of SMY-540 on the mortality rate of mice that had been administered a B. cereus (JMU-06B-35). SMY-540 decreased the lethality of the B. cereus (JMU-06B-35) treatment in a dose-dependent manner (). Treatment with 0.5 mg/kg SMY-540 reduced lethality by 80% ().
Figure 3. Effect of SMY-540 on the lethality of B. cereus in mice. Mice were intravenously injected with 0.1 or 0.5 mg/kg SMY-540 emulsion. B. cereus (JMU-06B-35, 1.0 × 107 CFU/mouse) was intraperitoneally administered to each mouse 3 h after injection. Mice were monitored every 2 h, for 100 h after administration of B. cereus. The duration of the experiment was set at 100 h. ○, Saline; ▪, B. cereus; ▴, 0.1 mg/kg SMY-540 + B. cereus; •, 0.5 mg/kg SMY-540 + B. cereus.
Discussion
Here, we synthesized multiple compounds, derivatives of RY221B-a, that function as competitive inhibitors of Bc-SMase, by competing with SM for the catalytic site of the enzyme. Of these, one compound, SMY-540 was an especially effective inhibitor. In fact, SMY-540 was a more effective than RY221B-a and is comparable to the most potent SMase inhibitors that have been describedCitation34.
The in silico docking analysis supports the observation that SMY-540 is a more effective inhibitor than RY221B-a. We previously reported that the docking energy of RY221B-a was −117 kCal/molCitation34. Here, we measured the docking energy of SMY-540 at −240 kCal/mol, suggesting a more stable bond between this SMY-540 and Bc-SMase. Although the exact effect of the phenyl functional group is unclear, its addition was found to enhance Bc-SMase inhibition.
The evaluation of SMase inhibitors has been performed in vitroCitation10,Citation36–38, however, the effect of SMase inhibitors on the prevention of infectious diseases has not been attempted previously. Here, we show that SMY-540 has great potential as a drug for the treatment of infectious diseases caused by B. cereus. These results suggest that SMY-540 is an effective inhibitor of Bc-SMase and has potential for use in the development of drugs to treat infectious diseases caused by bacteria that produce SMase.
Conclusions
We developed a novel and potent neutral SMase inhibitor, SMY-540, using a molecular design that was based upon the phosphate-ligand analog hypothesis. This inhibitor, with an IC50 of 0.8, strongly reduced the lethality of B. cereus inoculum in mice. With refinement, it may be possible to combine SMY-540 with antibiotic therapy because SMY-540 may block the toxic effects of SMase from bacteria.
Declaration of interest
The authors report no declarations of interest.
Related Research Data
References
- Oda M, Hashimoto M, Takahashi M, et al. Role of sphingomyelinase in infectious diseases caused by Bacillus cereus. PLoS One 2012;7:e38054
- Oda M, Takahashi M, Matsuno T, et al. Hemolysis induced by Bacillus cereus sphingomyelinase. Biochim Biophys Acta 2010;1798:1073–80
- Ikezawa H, Mori M, Taguchi R. Studies on sphingomyelinase of Bacillus cereus: hydrolytic and hemolytic actions on erythrocyte membranes. Arch Biochem Biophys 1980;199:572–8
- Ikezawa H, Matsushita M, Tomita M, Taguchi R. Effects of metal ions on sphingomyelinase activity of Bacillus cereus. Arch Biochem Biophys 1986;249:588–95
- Ikezawa H, Mori M, Ohyabu T, Taguchi R. Studies on sphingomyelinase of Bacillus cereus. I. Purification and properties. Biochim Biophys Acta 1978;528:247–56
- Ago H, Oda M, Takahashi M, et al. Structural basis of the sphingomyelin phosphodiesterase activity in neutral sphingomyelinase from Bacillus cereus. J Biol Chem 2006;281:16157–67
- Oda M, Takahashi M, Tsuge H, et al. Role of side-edge site of sphingomyelinase from Bacillus cereus. Biochem Biophys Res Commun 2012;422:128–32
- Openshaw AE, Race PR, Monzo HJ, et al. Crystal structure of SmcL, a bacterial neutral sphingomyelinase C from Listeria. J Biol Chem 2005;280:35011–17
- Tajima A, Iwase T, Shinji H, et al. Inhibition of endothelial interleukin-8 production and neutrophil transmigration by Staphylococcus aureus beta-hemolysin. Infect Immun 2009;77:327–34
- Nara F, Tanaka M, Hosoya T, et al. Scyphostatin, a neutral sphingomyelinase inhibitor from a discomycete, Trichopeziza mollissima: taxonomy of the producing organism, fermentation, isolation, and physico-chemical properties. J Antibiot (Tokyo) 1999;52:525–30
- Nara F, Tanaka M, Masuda-Inoue S, et al. Biological activities of scyphostatin, a neutral sphingomyelinase inhibitor from a discomycete, Trichopeziza mollissima. J Antibiot (Tokyo) 1999;52:531–5
- Uchida R, Tomoda H, Dong Y, Omura S. Alutenusin, a specific neutral sphingomyelinase inhibitor, produced by Penicillium sp. FO-7436. J Antibiot (Tokyo) 1999;52:572–4
- Uchida R, Tomoda H, Arai M, Omura S. Chlorogentisylquinone, a new neutral sphingomyelinase inhibitor, produced by a marine fungus. J Antibiot (Tokyo) 2001;54:882–9
- Inoue M, Yokota W, Murugesh MG, et al. Total synthesis of (+)-scyphostatin, a potent and specific inhibitor of neutral sphingomyelinase. Angew Chem Int Ed Engl 2004;43:4207–9
- Katoh T, Izuhara T, Yokota W, et al. Enantiocontrolled synthesis of the epoxycyclohexenone moieties of scyphostatin, a potent and specific inhibitor of neutral sphingomyelinase. Tetrahedron 2006;62:1590–608
- Inoue M, Yokota W, Katoh T. Enantioselective total synthesis of (+)-scyphostatin, a potent and specific inhibitor of neutral sphingomyelinase. Synthesis 2007;4:622–37
- Takagi R, Miyanaga W, Tojo K, et al. Stereoselective total synthesis of (+)-Scyphostatin via a pi-facially selective Diels-Alder reaction. J Org Chem 2007;72:4117–25
- Fujioka H, Sawama Y, Kotoku N, et al. Concise asymmetric total synthesis of scyphostatin, a potent inhibitor of neutral sphingomyelinase. Chem Eur J 2007;13:10225–38
- Pitsinos E, Athinaios N, Xu Z, et al. Total synthesis of (+)-scyphostatin featuring an enantioselective and highly efficient route to the side-chain via Zr-catalyzed asymmetric carboalumination of alkenes (ZACA). Chem Comm 2010;46:2200–2
- Arenz C, Giannis A. Synthesis of the first selective irreversible inhibitor of neutral sphingomyelinase. Angew Chem Int Ed Engl 2000;39:1440–2
- Runcie KA, Taylor RJK. A short and efficient route to novel scyphostatin analogues. Org Lett 2001;3:3237–9
- Arenz C, Gartner M, Wascholowski V, Giannis A. Synthesis and biochemical investigation of scyphostatin analogues as inhibitors of neutral sphingomyelinase. Bioorg Med Chem 2001;9:2901–4
- Pitsinos EN, Wascholowski V, Karaliota S, et al. Synthesis and evaluation of three novel scyphostatin analogues as neutral sphingomyelinase inhibitors. Chembiochem 2003;4:1223–5
- Claus RA, Wustholz A, Muller S, et al. Synthesis and antiapoptotic activity of a novel analogue of the neutral sphingomyelinase inhibitor scyphostatin. Chembiochem 2005;6:726–37
- Wascholowski V, Giannis A. Sphingolactones: selective and irreversible inhibitors of neutral sphingomyelinase. Angew Chem Int Ed Engl 2006;45:827–30
- Wascholowski V, Giannis A, Pitsinos EN. Influence of the scyphostatin side chain on the mode of inhibition of neutral sphingomyelinase. ChemMedChem 2006;1:718–21
- Cha JY, Burnett GLT, Huang Y, et al. A strategy for the late-stage divergent syntheses of scyphostatin analogues. J Org Chem 2011;76:1361–71
- Fraser CJ, Howell GP, Harrity JP. A furan Diels-Alder cycloaddition approach to scyphostatin analogues. Org Biomol Chem 2012;10:9058–66
- Hakogi T, Monden Y, Iwama S, Katsumura S. Stereocontrolled synthesis of a sphingomyelin methylene analogue as a sphingomyelinase inhibitor. Org Lett 2000;2:2627–9
- Hakogi T, Monden Y, Taichi M, et al. Synthesis of sphingomyelin carbon analogues as sphingomyelinase inhibitors. J Org Chem 2002;67:4839–46
- Hakogi T, Taichi M, Katsumura S. Synthesis of a nitrogen analogue of sphingomyelin as a sphingomyelinase inhibitor. Org Lett 2003;5:2801–4
- Yokomatsu T, Takechi H, Akiyama T, et al. Synthesis and evaluation of a difluoromethylene analogue of sphingomyelin as an inhibitor of sphingomyelinase. Bioorg Med Chem Lett 2001;11:1277–80
- Taguchi M, Sugimoto K, Goda K, et al. Sphingomyelin analogues as inhibitors of sphingomyelinase. Bioorg Med Chem Lett 2003;13:1963–6
- Imagawa H, Oda M, Takemoto T, et al. Synthesis and evaluation of novel phosphate ester analogs as neutral sphingomyelinase inhibitors. Bioorg Med Chem Lett 2010;20:3868–71
- Torley L, Silverstrim C, Pickett W. A turbidometric assay for phospholipase C and sphingomyelinase. Anal Biochem 1994;222:461–4
- Fujii S, Nagata M, Morita M, et al. Novel inhibition mechanism of Bacillus cereus sphingomyelinase by beryllium fluoride. Arch Biochem Biophys 2004;424:201–9
- Fujii S, Itoh H, Yoshida A, et al. Activation of sphingomyelinase from Bacillus cereus by Zn2+ hitherto accepted as a strong inhibitor. Arch Biochem Biophys 2005;436:227–36
- Luberto C, Hassler DF, Signorelli P, et al. Inhibition of tumor necrosis factor-induced cell death in MCF7 by a novel inhibitor of neutral sphingomyelinase. J Biol Chem 2002;277:41128–39
- Suzuki H, Mori M, Shibakami M. Synthesis of ceramide mimics with a pseudo cyclic framework. Synlett 2003;14:2163–6
- Garner P, Park J, Malecki E. A stereodivergent synthesis of D-erythro-sphingosine and D-threo-sphingosine from L-serine. J Org Chem 1988;53:4395–8
- Garner P. Stereocontrolled addition to a penaldic acid equivalent: an asymmetric of threo-β-hydroxy-L-glutamic acid. Tetrahedron Lett 1984;25:5855–8
- Arena G, Contino A, Longo E, et al. Synthesis, characterization of a novel calixarene having dipyridyl pendants and study of its complexes with Cu(II) and Co(II). Tetrahedron Lett 2003;44:5415–8
- Thomas B, Lindemann C, Corcoran R, et al. Phosphonate lipid tubules II. J Am Chem Soc 2002;124:1227–33
Appendix: Syntheses of candidate SMase inhibitors
Appendix A1. Syntheses of RY-221-Ba, SMY-584, SMY-610 and SMY-632
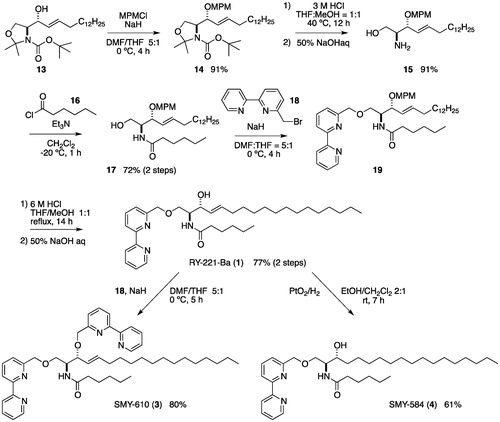
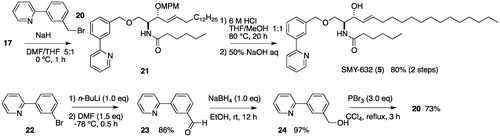
We slightly improved our previously reported methodCitation34 for the synthesis of RY-221-Ba [1, below]. The synthesis began with the known sphingosine derivative 13Citation39,Citation40 which was prepared from Garner’s aldehydeCitation41 in two steps. The alcohol 13 was protected as a p-methoxybenzyl ether (hereafter MPM ether). After hydrolysis of the oxazolidine ring and Boc protective groups of 14, the resulting amino group was treated with hexanoyl chloride (16) in the presence of Et3N to give a 72% yield of amide 17 in two steps. The hydroxyl group of 17 was converted to a bipyridylmethyl ether with NaH and 6-(bromomethyl)-2,2′-bipyridine (18)Citation42, followed by removal of the MPM ether under acidic conditions resulting in a 77% yield of 1 over two steps (). The conversion of 1 to SMY-610 (3) was accomplished by the etherification of 1 with 18 and 80% NaH. The synthesis of SMY-584 (4) was achieved by the selective hydrogenation of 1 with PtO2 under H2 in ethanol and dichloromethane ().
Scheme A1. Syntheses of RY-221-Ba, SMY-584 and SMY-610.
SMY-584: SMY-584 was purified by silica-gel column chromatography (hexane/ethyl acetate 2:1) and separated as a white powder; − 3.3° (c 0.09, CHCl3); FTIR (neat) 3287, 2916, 2849, 1641, 1542, 1467, 1431 cm−1; lH NMR (300 MHz in CDCl3) δ 0.88 (6H, t, J = 6.6 Hz), 1.23–1.34 (30H, m), 1.43–1.58 (4H, m), 2.16 (2H, t, J = 4.2 Hz), 3.65 (1H, m), 3.80 (1H, dd, J = 2.7, 9.6 Hz), 3.97 (1H, dd, J = 3.3, 9.6 Hz), 4.01 (1H, m), 4.65 (1H, d, J = 13.1 Hz), 4.75 (1H, d, J = 13.1 Hz), 6.44 (1H, d, J = 7.8 Hz), 7.32 (2H, t, J = 7.1 Hz), 7.83 (2H, t, J = 7.1 Hz), 8.32 (1H, d, J = 7.8 Hz), 8.40 (1H, d, J = 7.8 Hz), 8.68 (1H, dd, J = 2.1, 4.0 Hz); Citation13C NMR (75 MHz in CDCl3) δ 13.98, 14.14, 22.41, 22.71, 25.45, 29.10, 29.13, 29.27, 29.47, 29.61, 29.62, 29.63 (2C), 29.67 (3C), 31.38, 31.94, 32.20, 36.70, 52.79, 70.70, 72.31, 74.39, 120.19, 121.31, 121.50, 123, 77, 136.88, 137.60, 149.19, 155.90, 155.98, 156.89, 173.35; MS (FAB) m/z 590 (M + Na)+; HRMS (FAB) Calcd for C35H57N3O3Na (M + Na)+ 590.4298, found 590.4314.
SMY-610: SMY-610 was purified by silica-gel column chromatography (hexane/ethyl acetate 2:1) and separated as a white solid; − 19.5° (c 0.3, CHCl3); FTIR (neat) 2950, 2921, 1635, 1542, 1436 cm−1; lH NMR (400 MHz in CDCl3) δ 0.85 (3H, t, J = 7.2 Hz), 0.87 (3H, t, J = 6.8 Hz), 1.20–1.36 (26H, m), 1.56 (2H, m), 2.03–2.12 (4H, m), 3.77 (1H, dd, J = 3.6, 9.6 Hz), 4.02 (1H, dd, J = 4.4, 9.6 Hz), 4.06 (1H, t, J = 8.0 Hz), 4.36 (1H, m), 4.56 (1H, d, J = 13.6 Hz), 4.75 (1H, d, J = 13.6 Hz), 4.69 (1H, d, J = 13.6 Hz). 4.80 (1H, d, J = 13.6 Hz), 5.46 (1H, dd, J = 8.4, 15.2 Hz), 5.78 (1H, dt, J = 6.8, 15.2 Hz), 5.98 (1H, d, J = 9.2 Hz), 7.29 (2H, ddd, J = 1.2, 4.8, 7.6 Hz), 7.36 (1H, d, J = 7.2 Hz), 7.39 (1H, d, J = 7.2 Hz), 7.70 (1H, d, J = 8.0 Hz), 7.74 (1H, d, J = 8.0 Hz), 7.78 (2H, dt, J = 1.6, 7.6 Hz), 8.23 (2H, d, J = 8.0 Hz), 8.37 (2H, dd, J = 1.2, 7.6 Hz), 8.67 (2H, m); 13C NMR (100 MHz in CDCl3) δ 13.94, 14.14, 22.41, 22.70, 25.45, 29.23, 29.29, 29.37, 29.53, 29.67 (2C), 29.70 (3C), 31.42, 31.93, 32.35, 36.94, 51.91, 69.34, 71.33, 74.26, 80.50, 119.60, 119.77, 121.15, 121.19, 121.30, 121.39, 123.67, 123.70, 127.01, 136.86, 136.87, 137.39 (2C), 137.45, 149.19 (2C), 155.35, 155.51, 156.07, 156.17, 157.88, 158.35, 172.61; MS (FAB) m/z 772 (M + K)+; HRMS (FAB) Calcd for C46H63N5O3K (M + K)+ 772.4568, found 772.4561.
SMY-632 (5) was synthesized by a similar procedure to that used for the synthesis of 1, using 2-(3-(bromomethyl)phenyl)pyridine (20, prepared from commercially available 2-(3-bromophenyl) pyridine in three steps) instead of 18 ().
Scheme A2. Synthesis of SMY-632.
SMY-632: SMY-632 was purified by silica-gel column chromatography (hexane/ethyl acetate 1:2) and separated as a white solid; + 1.79° (c 0.13, CHCl3); FTIR (neat) 3299, 2925, 2853, 1731, 1645, 1463 cm−1; lH NMR (300 MHz in CDCl3) δ 0.88 (6H, t, J = 6.3 Hz), 1.23-1.30 (26H, m), 1.61–1.64 (2H, m), 2.00 (2H, m), 2.18 (2H, t, J = 7.6 Hz), 3.61 (1H, dd, J = 3.0, 9.6 Hz), 3.83 (1H, J = 2.7, 9.6 Hz), 4.07 (1H, m), 4.21 (1H, m), 4.54 (1H, d, J = 12.0 Hz), 4.63 (1H, d, J = 12.0 Hz), 5.47 (1H, dd, J = 6.0, 15.3 Hz), 5.72 (1H, dt, J = 6.3, 15.3 Hz), 6.29 (1H, d, J = 8.1 Hz), 7.24-7.36 (2H, m), 7.47 (1H, t, J = 7.5 Hz), 7.72–7.80 (2H, m), 7.88 (1H, d, J = 7.5 Hz), 8.69 (1H, d, J = 5.1 Hz); 13C NMR (75 MHz in CDCl3) δ 14.00, 14.09, 22.46, 22.74, 25.39, 29.20, 29.23, 29.36, 29.66, 29.68 (2C), 29.71 (2C), 31.47, 31.90, 32.33, 36.80, 52.79, 70.66, 74.40, 74.48, 120.20, 121.32, 121.39, 123, 87, 128.9, 129.14, 133.49, 137.23, 137.54, 148.66, 155.80, 155.88, 156.48, 173.35; MS (FAB) m/z 565 (M + H)+; HRMS (FAB) Calcd for C36H56N2O3Na (M + Na)+ 587.4189, found 587.4171.
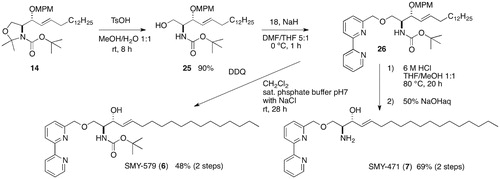
Appendix A2. Syntheses of SMY-579 and SMY-471
SMY-579 (6) and SMY-471 (7) were prepared from the common intermediate 14 in the manner described below. The selective hydrolysis of oxazolidine 14 was achieved by using a catalytic amount of TsOH in MeOH and H2O. The resulting alcohol 25 was converted to the bipyridylmethyl ether 26, followed by deprotection of the MPM ether with DDQ to provide a 48% yield of 6 in two steps. Acid treatment of 26 produced a 69% yield of 7 over two steps ().
Scheme A3. Syntheses of SMY-579 and SMY-471.
SMY-579: SMY-579 was purified by silica–gel column chromatography (hexane/ethyl acetate 2:1) and separated as a white solid; − 3.3° (c 1.6, CHCl3); FTIR (neat) 3400, 2930, 2854, 1714, 1582, 1566, 1504, 1462, 1432, 1366 cm−1; lH NMR (300 MHz in CDCl3) δ 0.88 (3H, t, J = 6.9 Hz), 1.23–1.31 (22H, m), 1.44 (9H, s), 2.00 (2H, dt, J = 6.6, 6.6 Hz), 3.35 (1H, brs), 3.75 (1H, dd, J = 3.0, 9.0 Hz), 3.78 (1H, m), 3.92 (1H, dd, J = 2.7, 9.0 Hz), 4.25 (1H, m), 4.67 (1H, d, J = 13.2 Hz), 4.75 (1H, d, J = 13.2 Hz), 5.38 (1H, brd, J = 7.6 Hz), 5.50 (1H, dd, J = 6.5, 16.0 Hz), 5.74 (1H, dt, J = 6.8, 16.0 Hz), 7.32 (1H, ddd, J = 1.4, 4.9, 7.7 Hz), 7.36 (1H, d, J = 7.7 Hz), 7.83 (2H, t, J = 7.8 Hz), 8.30 (1H, d, J = 7.8 Hz), 8.42 (1H, d, J = 7.8 Hz), 8.68 (1H, ddd, J = 0.8, 2.0, 4.7 Hz); 13C NMR (125 MHz in CDCl3) δ 14.12, 22.67, 28.36, 29.14, 29.20, 29.35, 29.48, 29.61, 29.64, 29.67, 31.91, 32.28, 54.04, 70.81, 74.30, 74.32, 79.51, 120.05, 121.25, 121.36, 123.76, 129.28, 133.36, 136.95, 137.56, 149.15, 155.82, 155.90, 156.09, 157.09; MS (CI) m/z 568 (M + H)+; HRMS (Cl) Calcd for C34H54N3O4 (M + H)+ 568.4115, found 568.4130.
SMY-471: SMY-471 was purified by recrystallization from pentane and separated as a white solid; + 11.7° (c 0.06, CHCl3); FTIR (neat) 3363, 2919, 2849, 1681, 1582, 1564, 1467, 1432 cm−1; lH NMR (200 MHz in CDCl3) δ 0.88 (3H, t, J = 6.6 Hz), 1.22–1.28 (22H, m), 1.97 (2H, dt, J = 7.6, 11.4 Hz), 3.36 (lH, dt, J = 5.7, 11.4 Hz), 3.80 (2H, d, J = 5.0 Hz), 4.35–4.40 (3H, m), 4.75 (1H, d, J = 16.5 Hz), 4.80 (1H, d, J = 16.5 Hz), 5.41 (1H, dd, J = 6.1, 15.2 Hz), 5.77 (1H, dt, J = 8.6, 15.2 Hz), 7.30–7.36 (2H, m), 7.81 (2H, t, J = 7.6 Hz), 8.15 (1H, d, J = 7.6 Hz), 8.21 (1H, d, J = 7.6 Hz), 8.74 (1H, d, J = 4.5 Hz); 13C NMR (75 MHz in CDCl3) δ 14.16, 22, 72, 29.05, 29.26, 29.39, 29.49, 29.69 (2C), 29.72 (3C), 31.95, 32.30, 55.38, 69.99, 71.80, 73.50, 120.21, 121.37 (2C), 123.97, 127.42, 134.49 (2C), 137.19, 137.78, 149.52, 155.60, 157.41; MS (CI) m/z 468 (M + H)+; HRMS (CI) Calcd for C29H46N3O2 (M + H)+ 468.3590, found 468.3595.
Appendix A3. Syntheses of SMY-540 and SMY-469a
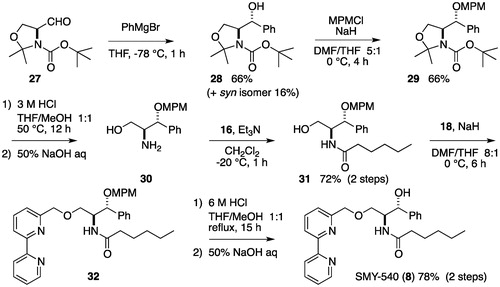
The synthesis of SMY-540 (8) began with the alkylation of Garner’s aldehyde (27) using phenylmagnesium bromide. The resulting alcohol 28 was protected as the MPM ether 29, which, after hydrolysis of the oxazolidine ring and Boc protective groups, gave amino alcohol 30. After the selective acylation of the amino group with hexanoyl chloride, the hydroxyl group of 31 was etherified with NaH and 18 to produce 32. Finally, the MPM ether of 32 was deprotected with 6 M HCl. Basic treatment of the resulting salt resulted in a 78% yield of 8 over two steps ().
Scheme A4. Synthesis of SMY-540.
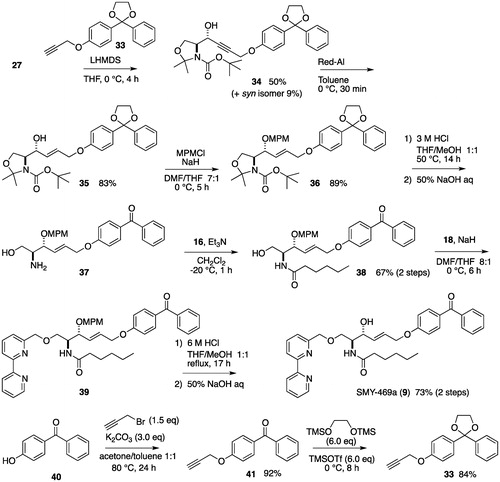
The synthesis of SMY-469a (9) started with the alkylation of 27 using lithium acetylide, which was prepared from LHMDS and 2-phenyl-2-(4-(prop-2-yn-1-yloxy)phenyl)-1,3-dioxolane (33). The resulting propargyl alcohol 34 was reduced to the allylic alcohol 35 using Red-Al, followed by protection of the alcohol as an MPM ether to produce an 89% yield of 36. The hydrolysis of one ketal and an oxazolidine ring of 36 using 3M HCl yielded amino alcohol 37. Following a similar method as used for the synthesis of 8, the synthesis of SMY-615 (9) was the achieved from the crude amino alcohol 37 in three steps ().
Scheme A5. Synthesis of SMY-469a.
SMY-540: SMY-540 was purified by silica–gel column chromatography (CH2Cl2/CH3OH 5:1) and separated as a colorless oil; − 10.7° (c 0.10, CHCl3); FTIR (neat) 3363, 2918, 2867, 1581, 1565, 1430 cm−1; lH NMR (400 MHz in CDCl3) δ 0.89 (3H, t, J = 7.2 Hz), 1.23–1.34 (4H, m), 1.56 (2H, m), 2.09 (2H, t, J = 7.2 Hz), 3.87 (1H, brd, J = 4.8 Hz), 4.01 (1H, m), 4.09 (1H, dd, J = 2.8, 12.0 Hz), 4.68 (1H, d, J = 16.5 Hz), 4.73 (1H, d, J = 16.5 Hz), 5.02 (1H, d, J = 3.2 Hz), 6.26 (1H, d, J = 8.0 Hz), 7.30–7.43 (7H, m), 7.82 (2H, t, J = 8.0 Hz), 8.33 (1H, d, J = 8.0 Hz), 8.43 (1H, d, J = 8.0 Hz), 8.71 (1H, d, J = 5.0 Hz); Citation13C NMR (100 MHz in CDCl3) δ 13.88, 22.31, 25.1 6, 31.26, 36.54, 59.23, 70.03, 73.08, 73.32, 120.27, 121.35, 121.68, 126.20, 126.66 (2C), 126.74, 128.58 (2C), 137.00, 137.36, 138.23, 149.40, 155.86, 156.13, 157.51, 173.11; MS (CI) m/z 433 (M + H)+; HRMS (CI) Calcd for C26H3lN3O3 (M + H)+ 433.2365, found 433.2356.
SMY-469a: SMY-469a was purified by silica–gel column chromatography (hexane/ethyl acetate 2:1) and separated as a colorless oil; − 3.0° (c 0.03, CHCl3); FTIR (neat) 3233, 2943, 2935, 2887, 1700, 1653, 1607, 1581, 1560, 1430, 1366 cm−1; lH NMR (500 MHz in CDCl3) δ 0.88 (3H, t, J = 7.2 Hz), 1.24–1.37 (4H, m), 1.59 (2H, m), 2.10 (2H, t, J = 7.2 Hz), 3.77 (1H, d, J = 7.0 Hz), 3.95 (1H, d, J = 7.0 Hz), 4.13 (1H, m), 4.23 (1H, m), 4.77 (1H, d, J = 12.5 Hz), 4.91 (1H, d, J = 5.0 Hz), 4.98 (1H, d, J = 12.5 Hz), 5.86 (1H, brd, J = 15.0 Hz), 5.99 (1H, t, J = 15.0 Hz), 7.04 (2H, d, J = 8.4 Hz), 7.33 (2H, t, J = 8.0 Hz), 7.48 (2H, t, J = 7.2 Hz), 7.57 (1H, t, J = 7.2 Hz), 7.76 (2H, d, J = 8.4 Hz), 7.80–7.87 (4H, m), 8.33 (1H, d, J = 8.0 Hz), 8.43 (1H, d, J = 8.0 Hz), 8.69 (1H, d, J = 5.0 Hz); Citation13C NMR (125 MHz in CDCl3) δ 13.80, 22.21, 25.19, 31.30, 36.54, 54.28, 62.66, 69.89, 69.90, 74.35, 114.42 (2C), 120.25, 121.27, 121.49, 123.87, 128.11, 128.23 (2C), 129.75 (2C), 130.92, 132.00, 132.44 (2C), 133.33, 136.99, 137.64, 138.08, 149.24, 155.87, 156.03, 156.89, 160.98, 173.94, 195.46; MS (CI) m/z 594 (M + H)+; HRMS (CI) Calcd for C36H40N3O5 (M + H)+ 594.2878, found 594.2871.
Appendix A4. Syntheses of TOM-2-7, TOM-2-18 and TOM-2-37
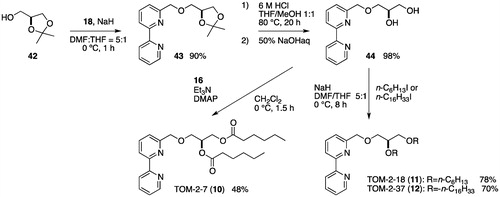
The syntheses of TOM-2-7 (10), TOM-2-18 (11) and TOM-2-37 (12) began with a glycerol derivative 42 prepared from D-Mannitol in four stepsCitation43. The [2,2′-bipyridin]-6-ylmethyl ether moiety was constructed by Williamson ether synthesis using NaH and 18, and the hydrolysis of the resulting ketal 43 gave a 98% yield of diol 44. Acylation of 44 with hexanoyl chloride resulted in a 48% yield of 10. In contrast, the treatment of 44 with NaH and alkyl iodides resulted in a 78% and 70% yield of 11 and 12, respectively ().
Scheme A6. Syntheses of TOM-2-7, TOM-2-18 and TOM-2-37.
TOM-2-7: TOM-2-7 was purified by silica-gel column chromatography (hexane/ethyl acetate 2:1) and separated as a colorless oil; + 3.3° (c 0.4 CHCl3); FTIR (neat) 3068, 3025, 2957, 2932, 2860, 1741, 1698, 1603, 1558, cm−1; lH NMR (200 MHz in CDCl3) δ 0.88 (6H, t, J = 6.2 Hz), 1.23–1.33 (8H, m), 1.53–1.68 (4H, m), 2.30 (2H, t, J = 7.4 Hz), 2.34 (2H, t, J = 7.4 Hz), 3.75 (2H, d, J = 5.0 Hz), 4.24 (1H, dd, J = 6.2, 12.2 Hz), 4.42 (1H, dd, J = 4.0, 12.2 Hz), 4.72 (H, d, J = 14.0 Hz), 4.79 (1H, d, J = 14.0 Hz), 5.30 (1H, m), 7.30 (1H, ddd, J = 1.2, 4.8, 7.6 Hz), 7.44 (1H, dd, J = 0.8, 7.6 Hz), 7.80 (1H, dt, J = 1.8, 7.6 Hz), 7.83 (1H, t, J = 7.6 Hz), 8.28 (1H, d, J = 8.0 Hz), 8.38 (1H, dt, J = 1.2, 8.0 Hz), 8.66 (1H, m); Citation13C NMR (100 MHz in CDCl3) δ 13.92, 22.31, 24.57, 24.64, 31.23, 31.25, 34.09, 34.32, 62.63, 69.17, 70.05, 74.44, 77.23, 119.81, 121.14, 121.20, 123.72, 136.91, 137.58, 149.21, 155.49, 156.10, 157.65, 173.16, 173.48; MS (CI) m/z 457 (M+); HRMS (CI) calcd for C26H37N2O5 (M+) 457.2703, found 457.2724.
TOM-2-18: TOM-2-18 was purified by silica-gel column chromatography (hexane/ethyl acetate 4:1) and separated as a colorless oil; − 3.8° (c 1.1 CHCl3); FTIR (neat) 2954, 2930, 2858, 1581, 1430 cm−1; lH NMR (300 MHz in CDCl3) δ 0.86 (6H, t, J = 6.2 Hz), 1.23–1.40 (12H, m), 1.53–1.68 (4H, m), 3.45 (2H, t, J = 6.6 Hz), 3.54 (2H, m), 3.61 (2H, t, J = 6.6 Hz), 3.68-3.75 (3H, m), 4.77 (1H, s), 7.30 (1H, m), 7.50 (1H, d, J = 7.8 Hz), 7.80 (1H, dt, J = 1.4, 7.8 Hz), 7.82 (1H, t, J = 7.8 Hz), 8.26 (1H, d, J = 7.8 Hz), 8.40 (1H, d, J = 8.2 Hz), 8.68 (1H, d, J = 5.8 Hz); Citation13C NMR (75 MHz in CDCl3) δ 14.07 (2C), 22.63, 22.65, 25.79 (2C), 29.63, 30.08, 31.68, 31.71, 70.61, 70.70, 71.08, 71.71, 74.40, 77.89, 119.55, 121.11, 121.17, 123.60, 136.82, 137.40, 149.13, 155.34, 156.21, 158.37; MS (CI) m/z 429 (M+); HRMS (CI) Calcd for C26H41N2O3 (M + H)+ 429.3117, found 429.3121.
TOM-2-37: TOM-2-37 was purified by silica-gel column chromatography (hexane/ethyl acetate 8:1) and separated as a colourless oil; − 1.2° (c 1.0 CHCl3); FTIR (neat) 2954, 2930, 2858, 1581, 1564 cm−1; lH NMR (300 MHz in CDCl3) δ 0.88 (6H, t, J = 6.9 Hz), 1.19–1.30 (52H, m), 1.57 (4H, m), 3.45 (2H, t, J = 6.9 Hz), 3.52–3.78 (5H, m), 3.61 (2H, t, J = 6.6 Hz), 4.77 (2H, s), 7.29 (1H, ddd, J = 0.9, 4.8, 7.5 Hz), 7.49 (1H, d, J = 7.5 Hz), 7.79 (1H, dt, J = 1.8, 5.7 Hz), 7.81 (1H, t, J = 7.5 Hz), 8.26 (1H, d, J = 7.0 Hz), 8.39 (1H, dt, J = 0.9, 7.0 Hz), 8.67 (1H, m); Citation13C NMR (100 MHz in CDCl3) δ 14.01, 14.03, 22.58, 22.60, 25.74, 25.75, 29.58, 30.03, 31.64, 31.67, 70.56, 70.65, 71.02, 71.65, 74.34, 77.84, 119.50, 121.06, 121.13, 123.57, 136.78, 137.36, 149.10, 155.29, 156.16, 158.34; MS (CI) m/z 710 (M + 2H)+; HRMS (CI) Calcd for C46H80N2O3 (M+) 708.6169, found 708.6187.