
Abstract
Entamoeba histolytica, the parasite which causes amebiasis is responsible for 110 000 deaths a year. Entamoeba histolytica depends on glycolysis to obtain ATP for cellular work. According to metabolic flux studies, hexokinase exerts the highest flux control of this metabolic pathway; therefore, it is an excellent target in the search of new antiamebic drugs. To this end, a tridimensional model of E. histolytica hexokinase 1 (EhHK1) was constructed and validated by homology modeling. After virtual screening of 14 400 small molecules, the 100 with the best docking scores were selected, purchased and assessed in their inhibitory capacity. The results showed that three molecules (compounds 2921, 11275 and 2755) inhibited EhHK1 with an I50 of 48, 91 and 96 µM, respectively. Thus, we found the first inhibitors of EhHK1 that can be used in the search of new chemotherapeutic agents against amebiasis.
Introduction
Amebiasis caused by parasite Entamoeba histolytica, affects more than 10% of the world’s population, the untreated infection may lead to severe complications including hepatic amebiasis and intestinal tissue destruction. More than 50 million people worldwide are infected and up to 110 000 of these die every yearCitation1. Morphologically, E. histolytica exists as cyst and as trophozoite, which are the infective and the invasive forms of the disease, respectivelyCitation2. In general, amebiasis can be classified as intestinal, that ranges from asymptomatic to fulminating colitisCitation3,Citation4, it can also be extraintestinal, where the most common is amebic liver abscessCitation3,Citation5. Metronidazole and 5-nitroimidazoles are used as the first line drugs for the treatment of amebiasisCitation6; paromomycin, chloroquine, diiodohydroxyquin, diloxanide furoate and emetine have also been used as alternative drugsCitation6,Citation7. However, these drugs have important side effects such as neutrocytopenia, dizziness, anorexia, disulfiram-like alcohol intolerance, diarrhea, cardiotoxicity, tenderness, vomiting, nausea, local pain and occasionally encephalopathy and convulsionsCitation6,Citation8,Citation9. Moreover, they are ineffective against luminal cystsCitation6. Additionally, metronidazole has been described as a carcinogenic and mutagenic agent in rodents and bacteria; it is classified as a class B risk factor for pregnancy by the FDACitation10–12. Therefore, there is an urgent need of new drugs for the treatment of amebiasis.
Because E. histolytica trophozoites do not have mitochondria, the ameba is totally dependent on glycolysis for ATP supplyCitation6. Moreover, glycolysis regulation in ameba differs from that in humans. One of the principal differences is the pyrophosphate-dependent enzymes phosphofructokinase (PPi-PFK) and pyruvate phosphate dikinase (PPDK)Citation13. Another difference is hexokinase (HK) which is not inhibited by its product glucose-6-phosphate (G6P) like some vertebrate HKsCitation14, instead, it is inhibited by physiological concentrations of AMP and ADPCitation13,Citation15. Furthermore, metabolic flux studies showed that HK controls 73% of the flux in E. histolytica glycolysisCitation16. Therefore, EhHK is an excellent target in the search of specific inhibitors that can be developed into new drugs for the treatment of amebiasis. HK exists in two isoforms in E. histolytica, namely hexokinase 1 (EhHK1) and hexokinase 2 (EhHK2)Citation15,Citation17; these two enzymes have very similar molecular weights, 49.8 and 49.4 kDa for EhHK1 and EhHK2, respectivelyCitation17; both enzymes are formed by 445 residues with 89% sequence identity. In this work, EhHK1 was cloned, over-expressed and purified. A tridimensional model of the enzyme was obtained and a virtual screening strategy was applied to discover the first set of EhHK1 inhibitors.
Materials and methods
Homology modeling
EhHK1 model construction was made using the homology model tool in the computational package MOE (Molecular Operating Environment, www.chemcomp.com). First, a search in the Protein Data Bank for a suitable homology model template was made. After a multiple alignment of EhHK1 amino acid sequence obtained from the NCBI (www.ncbi.nlm.nih.gov) reference sequence: XP_653620.1, with HK sequences with known three-dimensional structures, the crystal structure of human HK1Citation18 (PDB ID: 1dgk), which includes an ADP molecule, was selected as starting model. Human HK1 has a molecular weight of 100 kDA, it is structurally formed by two halves of 50 kDa each, the N-terminal half (regulatory) and the C-terminal half (catalytic)Citation18. Here, the C-terminal half was used as a template, with a sequence identity of 30.2%Citation19 ().
Figure 1. Sequence alignment between EhHK1 and the human HK1 C-terminal half used as template. Alignment was made in SWISS-MODEL WorkspaceCitation19.

Ten different intermediate models were constructed. These were the results of permutational selection of side chain rotamers and different loop candidates. The intermediate model with the best packing index according to the chosen scoring function was selected as the final model, and subjected to an energy minimization using force field Amber 99Citation20. Finally, stereochemical quality evaluation of the model was made with Ramachandran Plot using Protein geometry monitor in MOE and the external program Q-MeanCitation21,Citation22.
Virtual screening
To this end, a rigid body docking procedure was applied using MOE package. Library Hit Finder™ from Maybrige (www.maybridge.com), which comprises 14 400 small molecules, was used to identify inhibitors of EhHK1. These small molecules meet with Lipinski’s rulesCitation23. Their three-dimensional low energy conformations were generated with MOE using a fragment approach and a systematic search strategy. Then, atomic partial charges and an energy minimization were made applying the Gasteiger-Marsilli algorithmCitation24 and MMFF94x force field in MOE. Multiple conformations were predicted for each molecule and those with energy higher than 3 kcal/mol were eliminated from the process to avoid internal strains.
For virtual screening, atomic partial charges were assigned to the EhHK1 model and an energy minimization using CHARM27 force field in MOE was made. Residues forming the docking site (Gly62, Thr63, Asn64, Gly203, Thr204, Gly272, Ile273, Ser319, Arg320, Glu323, Gly395, Thr396, Val397 and Lys400) were determined by alignment according to those for ADP in human HK1 C-terminal half. Around 80 000 random orientations with variations in position and molecular rotation were assessed per conformer of each ligand. The score for each one was calculated using London dG Scoring function in MOE considering spatial compatibility to the binding site, internal energy of the ligand and protein–ligand interactions. A data base was obtained and the 100 molecules with the highest scores were purchased from Maybridge for inhibition studies.
EhHK1 gene cloning
Entamoeba histolytica strain HM1:IMSS genomic DNA was used for EhHK1 gene cloning by PCR. The oligonucleotides were 5′-GCGGATCCCAAGAAATCATTGATCAA-3′ (forward) including a BamH1 restriction site (underlined) and 5′-AAGCTTTTAGTGTTTACATGCAACAGCAGCAGC-3′ (reverse) including a HindIII restriction site (underlined). The PCR product was cloned using PCR BLUNT II® TOPO® vector (Invitrogen, Carlsbad, CA) and sequenced. Then it was subcloned into pET28a(+), that produced a protein with a 6his-tag in its N-terminal, and introduced by transformation into BL21(DE3)pLysS cells (Novagen, Madison, WI) for gen over-expression.
Enzyme purification
Minimal media (500 mL) containing 50 µg/mL of kanamycin were inoculated with bacteria supporting pET28a(+) plus the EhHK1 gene and grown at 37 °C until absorbance was 0.5. At this time, over-expression was induced using IPTG 2.5 mM; after 4 h, cells were harvested by centrifugation and the pellet was washed twice with buffer A (Tris-HCl 50 mM, pH 8.0). Cells were suspended in buffer A plus the protease inhibitor PMSF 200 µM, and lyzed by sonication. After centrifugation, the supernatant was passed through Ni-NTA affinity column, EhHK1 was purified with a gradient of 50–300 mM imidazole. The Bradford method was used to determine protein concentrationCitation25.
Molecular weight determination
A Superdex 200 10/300 GL column was washed with 50 mL distilled water followed by 50 mL of elution buffer (50 mM phosphate, pH 8.0, containing 0.15 M KCl) at a rate of 0.5 mL/min. EhHK1 (70 µg) was injected into the column and eluted with 30 mL elution buffer. EhHK1 molecular weight was determined using a calibration curve with different proteins of various molecular weights and adjusting the data to the equation:
where Kav is the distribution coefficient, Vo is the column void volume, Ve is the protein elution volume and Vt is the total matrix volume.
Enzyme activity
Activity was measured spectrophometrically at 37 °C in a G6P dehydrogenase (G6PDH) coupling system, following NADPH generation (ε = 6.19 × 103 M−1 cm−1) at 340 nmCitation13. The reaction mix contained buffer A plus glucose 1 mM, ATP 0.3 mM, MgCl2 5 mM, NADP+ 1 mM, 1U G6PDH (Leuconostoc mesenteroides, Roche) and 62.5 ng of EhHK1. Kinetics parameters for glucose and ATP were determined by varying concentration from 0.01 to 1 mM in both cases. Data were adjusted to the Michaelis–Menten equation.
Inhibition assays
Inhibition studies with small molecules were made as described above, adding the molecules at the indicated concentration. The I50 value was obtained from the fitting of concentrations curves to equation:
where Vi is the activity at a given inhibitor concentration, Vo indicates the initial activity, I50 is the concentration that inhibits 50% of enzyme activity, I the concentration of the tested inhibitor and n is a measure of cooperativity.
Results and discussion
In this work, we searched for inhibitors of recombinant E. histolytica HK1 (EhHK1) through virtual screening of a small molecules library. Because of the crystal structure of EhHK1 is not available; a tridimensional model of the enzyme was constructed using human HK1 as template. According to the Ramachandran map generated with the Protein geometry monitor tool in MOE, the model had 90.52% of the residues in the core region, 7.91% in allowed regions and only 1.57% in disallowed region (). The amino acids out of the permitted area were Glu13, Lys33, Pro34, Glu74, Gln241, Thr309 and Ser329, none of which belongs to the docking site used for virtual screening. Moreover, additional evaluation using Q-MEAN serverCitation21,Citation22 reported a QMEAN6 score of 0.639 and a z-score of −1.52, supporting the quality of the model ().
Figure 2. EhHK1 model Ramachandran map. Image shows residues in disallowed regions.
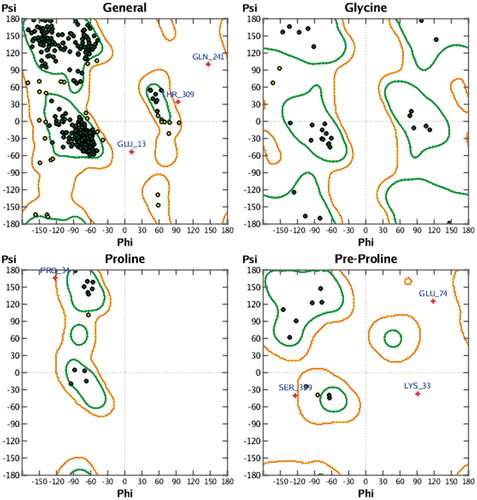
Figure 3. EhHK1 model QMEAN evaluation. (A) Normalised QMEAN6 score graphic showing the Z-score value, and the position (circle) of the model in the set of PDB structures used for evaluation. (B) EhHK1 model in ribbons, virtual screening site was located at the center in a region with less than 1 Å deviation, according to the estimated per-residue inaccuracy.

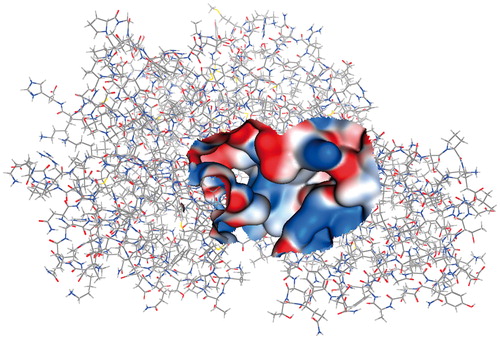
After obtaining the EhHK1 3D-model, we performed a virtual screening strategy to find small molecules with potential affinity for the enzyme. To this end, the dock tool in MOE was used to predict the binding mode of a 14 400 small molecule library into the predicted ATP binding site of the enzyme (). A data base with 319 195 poses was generated. One hundred compounds, with the best binding energies (from −17.95 to −11.05 kcal/mol), were selected to assess their capacity to inhibit EhHK1.
Figure 4. EhHK1 3D model showing the Gaussian surface of predicted ATP binding site used for virtual screening.
To this end, we cloned, over-expressed, purified and characterized EhHK1 (see “Materials and methods” section). Size exclusion chromatography in native conditions yielded one peak with a molecular weight of 53.8 kDa, indicating that the enzyme exists as a monomer (). This result differs from those of Saavedra et al.Citation13, who reported that EhHK1 is a dimer. The difference could be due to the type of size exclusion chromatography column that was employed, the concentration of the protein injected, and the elution buffer, in particular its ionic strength, which has been found to be an important factor in the stability of proteins in different oligomeric sates of HKCitation26,Citation27. Unfortunately, there are no other reports on the oligomeric state of EhHK1. In view of our sized exclusion chromatography data and earlier reportsCitation15,Citation17, we have considered that our EhHK1 is a monomer. It is noted that the kinetic parameters of our EhHK1 () are similar to those reported elsewhereCitation13,Citation15.
Figure 5. EhHK1 elution profile. The principal peak corresponds to a molecular weight of 53.8 kDa. The second peak represents less than 5% of total area under curve.
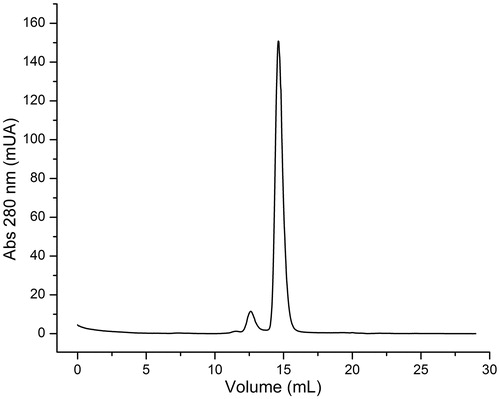
Table 1. EhHK1 kinetic parameters.
Inhibition assays at a concentration of 200 µM of the molecules selected from the virtual screening studies were initially made. shows the inhibition percent of EhHK1 activity induced by the 16 most active compounds; some of their physicochemical properties are also described. The ranking position within the 100 molecules were 59, 4, 19, 25, 98, 77, 32, 54, 29, 14, 17, 23, 30, 31, 33 and 21, respectively, according to their position in . The three most potent molecules (i.e. compounds 2755, 2921 and 11275) were selected for further studies. From the concentration curves, the concentration that inhibits 50% of enzyme activity (I50) was calculated (). Values of 48 µM for disodium 5-nitro-2-[2-(4-nitro-2 sulfonatophenyl)vinyl]benzenesulfonate (2921), 91 µM for 3,5-di(3,4,5-trimethoxybenzylidene)tetrahydro-2H-pyran-4-one (11275) and 96 µM for 5,6,7-trimethoxy-2-(2,3,4-trimethoxybenzylidene)indan-1-one (2755) were obtained. A particular feature of their inhibition profiles is that the curves were sigmoid, suggesting that more than one molecule is needed to inhibit the enzymeCitation28.
Figure 6. EhHK1 Inhibition by different concentrations of compounds 2755, 2921 and 11275. The I50 value was calculated adjusting the data to the equation described in “Materials and methods” section.
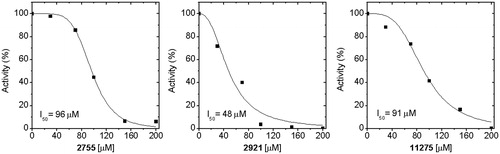
Table 2. EhHK1 inhibition by the 16 most active compounds found through virtual screening.
A detailed structural analysis on the binding mode in EhHK1 () revealed that compound 2755 establishes hydrogen bonds with side chains of Ser318, Lys315, Thr63 and Thr396; additionally, it forms cation–π interaction with Arg320 (). On the other hand, compound 2921 formed hydrogen bonds with side chains of Thr204, Ser430 and with the backbone of Thr63, Ser430 and a cation–π interaction with Arg66 (). Finally, compound 11275 made hydrogen bonds with side chains of Ser318, Ser319, Thr63, Thr204, Thr396 and Arg320 ().
Figure 7. Binding of compounds 2755, 2921 and 11275 (A, C and E, respectively) into EhHK1 predicted ATP binding site. 2D Interactions map among the enzyme and compounds 2755, 2921 and 11275 (B, D and F, respectively). Arrows indicate H-bond formation, and a cross with an aromatic ring means cation–π interaction.
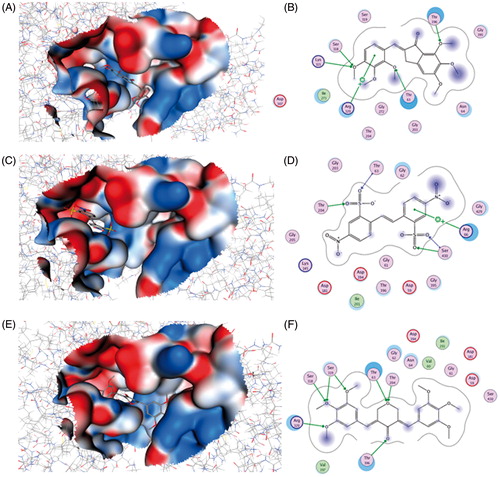
Structurally, the three molecules possess an aromatic ring in each end; compounds 2921 and 11275 are symmetrical whereas compound 2755 shares certain similarity with the ATP structure. Although compound 2921 had the lower binding energy (), it showed the highest inhibitory activity. This suggests that the cation–π interaction with Arg66 and the hydrogen bonds with Ser430, which are absent in the other two compounds, could be important for enzyme inhibition.
Conclusion
To our knowledge, no inhibitor of EhHK1 has been described. Since the inhibitors here described act in the low micromolar range, it is likely that the three compounds could be exploited to find more potent and selective EhHK1 inhibitors and in consequence, lead to the discovery of new chemotherapeutic agents for amebiasis.
Declaration of interest
The authors report no declarations of interest.
Partial financial support from CONACYT, México is acknowledged (Grant No. 105532) to ARD. MLSM thanks PROMEP for a fellowship (PROMEP/103.5/11/5723).
Acknowledgements
The authors thank Dr Armando Gómez-Puyou and Dr Mario Pedraza-Reyes for critical reading of the manuscript and helpful suggestions.
References
- WHO/PAHO/UNESCO Report. A consultation with experts on amoebiasis. Epidemiol Bull 1997;18:13–14
- Li E, Stanley SL. Amebiasis. Gastroenterology clinics of North America 1996;25:471–92
- Ximénez C, Morán P, Rojas L, et al. Reassessment of the epidemiology of amebiasis: state of the art. Infect Genet Evol 2009;9:1023–32
- Bruckner DA. Amebiasis. Clin Microbiol Rev 1992;5:356–69
- Katzenstein D, Rickerson V, Braude AI. New concepts of amebic liver abscess derived from hepatic imaging, serodiagnosis and hepatic enzymes in 67 consecutive cases in San Diego. Medicine 1982;61:237–46
- Ali V, Nozaki T. Current therapeutics, their problems, and sulfur-containing-amino-acid metabolism as a novel target against infections by “amitochondriate” protozoan parasites. Clin Microbiol Rev 2007;20:164–87
- Stauffer W, Ravdin JI. Entamoeba histolytica: an update. Curr Opin Infect Dis 2003;16:479–85
- Haque R, Huston CD, Hughes M, et al. Amebiasis. N Eng J Med 2003;348:1565–73
- Petri WA. Therapy of intestinal protozoa. Trends Parasitol 2003;19:523–6
- Bendesky A, Menendez Ostrosky-Wegman DP. Is metronidazole carcinogenic? Mutat Res 2002;511:133–44
- Biagi GL, Barbaro AM, Guerra MC, et al. Quantitative relationship between structure and mutagenic activity in a series of 5-nitroimidazoles. Teratog Carcinog Mutagen 1983;3:429–38
- Roe FJ. Toxicologic evaluation of metronidazole with particular reference to carcinogenic, mutagenic, and teratogenic potential. Surgery 1983;93:158–64
- Saavedra E, Encalada R, Pineda E, et al. Glycolysis in Entamoeba histolytica. Biochemical characterization of recombinant glycolytic enzymes and flux control analysis. FEBS J 2005;272:1767–83
- Cárdenas ML, Cornish-Bowden A, Ureta T. Evolution and regulatory role of the hexokinases. Biochim Biophys Acta 1998;1401:242–64
- Kroschewski H, Ortner S, Steipe B, et al. Differences in substrate specificity and kinetic properties of the recombinant hexokinases HXK1 and HXK2 from Entamoeba histolytica. Mol Biochem Parasitol 2000;105:71–80
- Saavedra E, Marín-Hernández A, Encalada R, et al. Kinetic modeling can describe in vivo glycolysis in Entamoeba histolytica. FEBS J 2007;274:4922–40
- Ortner S, Clark CG, Binder M, et al. Molecular biology of the hexokinase isoenzyme pattern that distinguishes pathogenic Entamoeba histolytica from nonpathogenic Entamoeba dispar. Mol Biochem Parasitol 1997;86:85–94
- Aleshin AE, Kirby C, Liu X, et al. Crystal structures of mutant monomeric hexokinase I reveal multiple ADP binding sites and conformational changes relevant to allosteric regulation. J Mol Biol 2000;296:1001–15
- Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 2006;22:195–201
- Wang J, Cieplak P, Kollman PA. How well does a Restrained Electrostatic Potential (RESP) model perform in calculating conformational energies of organic and biological molecules. J Comput Chem 2001;21:1049–74
- Benkert P, Tosatto SC, Schomburg D. QMEAN: a comprehensive scoring function for model quality assessment. Proteins 2008;71:261–77
- Benkert P, Künzli M, Schwede T. QMEAN Server for Protein model quality estimation. Nucleic Acids Res 2009;37:W510–14
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Del Rev 1997;23:3–25
- Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity a rapid access to atomic charges. Tetrahedron 1980;36:3219–28
- Bradford MM. A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem 1976;72:248–54
- Cáceres AJ, Portillo R, Acosta H, et al. Molecular and biochemical characterization of hexokinase from Trypanosoma cruzi. Mol Biochem Parasitol 2003;126:251–62
- Cáceres AJ, Quiñones W, Gualdrón M, et al. Molecular and biochemical characterization of novel glucokinase from Trypanosoma cruzi and Leishmania spp. Mol Biochem Parasitol 2007;156:235–45
- Téllez-Valencia A, Avila-Ríos S, Pérez-Montfort R, et al. Highly specific inactivation of triosephosphate isomerase from Trypanosoma cruzi. Biochem Biophys Res Commun 2002;295:958–63