
Abstract
The synthesis and in vitro evaluation of 40 new 2-phenylisothiazolidin-3-one-1,1-dioxide derivatives are described. The optimization based on biological screening data and molecular modeling resulted in a 10-fold increase in inhibitory activity compared with previously reported inhibitors of this class and led to the identification of 3-{[2-chloro-4-(1,1-dioxido-3-oxoisothiazolidin-2-yl)benzoyl]amino}benzoic acid, a potent inhibitor of human protein kinase CK2 (ІC50 = 1.5 μM).
Introduction
Protein kinase CK2 (CK2) is a ubiquitous serine/threonine protein kinase for hundreds of endogenous substrates. CK2 has been considered to be involved in many diseases including cancers. In our previous studies of 2-phenylisothiazolidin-3-one-1,1-dioxide derivativesCitation1, compound 1 () was identified as CK2 inhibitor (ІC50 = 20 μM). In order to improve its inhibitory properties we have decided to carry out optimization of the lead compound. Using the molecular docking method, compound 1 with CK2 ATP acceptor site was generated and investigated. The visual inspection of the complex allowed us to recognize the substituents, which are involved in the key interactions with amino acid residues of CK2 and to determine the routes for further optimization.
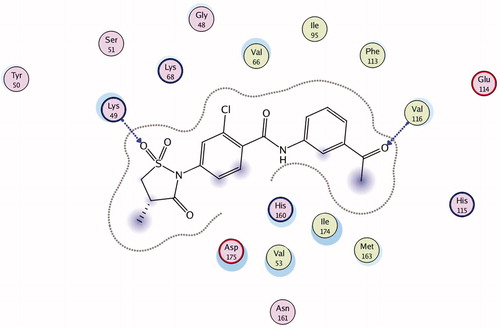
Figure 1. Compound 1 in ATP-binding site of CK2 (2D-model).
Experimental
General
Starting materials and solvents were purchased from commercial suppliers and were used without further purification. 1H NMR spectra were recorded on a Varian VXR 400 instrument (Agilent Technologies, Santa Clara, CA) at 400 MHz. 13C NMR spectra were recorded on a Varian VXR 400 instrument at 100 MHz. Chemical shifts are described as parts per million (δ) downfield from an internal standard of tetramethylsilane, and spin multiplicities are given as s (singlet), d (doublet), dd (double doublet), t (triplet), q (quartet) or m (multiplet). The HPLC-MS analysis was performed using the Agilent 1100 LC/MSD SL (Agilent Technologies, Santa Clara, CA) separations module and Mass Quad G1956B mass detector (Agilent Technologies, Santa Clara, CA) with an electrospray ionization (+ve or −ve ion mode as indicated) and with HPLC performed using Zorbax SB-C18 (Agilent Technologies, Santa Clara, CA), Rapid Resolution HT Cartridge (Agilent Technologies, Santa Clara, CA) 4.6 × 30 mm 1.8-Micron (Agilent P/N:823975-902) i.d. column, at a temperature of 40 °C with gradient elution of 0%–100% CH3CN (with 1 mL/L HCOOH): H2O (with 1 mL/L HCOOH) at a flow rate of 3 mL/min and a run time of 2.8 min. Compounds were detected at 215 nm using a Diode Array G1315B detector. All tested compounds had ≥90% purity as determined by this method. All purified synthetic intermediates had ≥95% purity as determined by this method.
Virtual screening
For virtual screening of a combinatorial library we used DOCK 4.0 package (Irwin “Tack” Kuntz's Group, University of California, San Francisco, CA)Citation2–5. Calculations of ligand geometry were performed using YFF force field developed by the authorsCitation6,Citation7. Partial atomic charges of the ligands were calculated with the Kirchhoff methodCitation8. Minimization of CK2 crystal structure (PDB ID: 2ZJW) was performed with GROMACSCitation9 package (Biophysical Chemistry Department of University of Groningen, AB Groningen, The Netherlands) (steepest descent algorithm, GROMACS force field). Partial atomic charges of the receptor were calculated using AMBER force field. Flexible docking and scoring were performed as described previouslyCitation10.
Biological testing
Selected compounds were tested using in vitro kinase assay. Each test was performed twice in a total reaction volume of 30 μL, containing 6 μg of peptide substrate RRRDDDSDDD (New England Biolabs (UK) Ltd., Herts, UK); 10 units of recombinant human CK2 holoenzyme (New England Biolabs); 50 μM ATP and γ-labeled 32P ATP, diluted to specific activity 100 μCi/μM; CK2 buffer (20 mM Tris-HCl, pH 7.5; 50 mM KCl; 10 mM MgCl2) and inhibitor in varying concentrations. Incubation time was 20 min at 30 °C. The reaction was stopped by adding an equal volume of 10% o-phosphoric acid and the reaction mixture was loaded onto 20-mm discs of phosphocellulose paper (Whatman plc, Kent, UK). Discs were washed three times with 1% o-phosphoric acid solution, air-dried at room temperature, and counted by the Cherenkov method in a beta-counter (LKB). As negative control an equal volume of DMSO was added to the reaction mixture. Quercetin, a known CK2 inhibitorCitation11, was used as an inhibition positive control. The final concentration of quercetin was 0.55 μM (which inhibited CK2 activity by 50%). Percent inhibition was calculated as ratio of substrate-incorporated radioactivity in the presence of inhibitor to the radioactivity incorporated in control reactions, i.e. in the absence of inhibitor. Serial dilutions of inhibitor stock solution were used to determine its IC50 concentration. The IC50 values represent means of triplicate experiments with SEM never exceeding 15%.
Selectivity tests were performed on protein kinases ASK1, JNK3, Met, Aurora А, Rock 1, FGFR1 and Tie2 according to the supplier's recommendations (Merck Millipore, MA).
Chemistry
Benzoyl chloride synthesis (2a, 2d, 2e, 2g, 2h, 2j, 2l), NMR spectra, reaction yields, m.p. temperatures were described in the literatureCitation12. According to the synthetic proceduresCitation12, the corresponding benzoyl chlorides (2b, 2c, 2f, 2k) were synthesized and their spectral properties are presented below.
General procedure for the synthesis of 2-phenylisothiazolidin-3-one-1,1-dioxides (3–39)
The mixture of substituted benzoyl chloride (2a–2l) (1 mmol) and appropriate aminohydroxybenzoic acid (1.1 mmol) was refluxed in acetone (4 mL) for 10 h. Then the solvent was evaporated to dryness and isopropyl alcohol (1 mL) was added. Mixture was heated up to the boiling, cooled down to the room temperature and filtered. Residue was washed by isopropyl alcohol (0.5 mL) and air dried to furnish the corresponding product.
3-(4-methyl-1,1-dioxido-3-oxoisothiazolidin-2-yl)benzoyl chloride (2b)
Yield 87% (white powder); m.p. 161 °C. 1H NMR (CDCl3) δ 1.59 (d, J = 6.8 Hz, 3H, CH3), 3.34–3.61 (m, 2H, CH2), 3.91 (dd, J = 11.7, 6.8 Hz, 1H, CH), 7.54–7.82 (m, 2H, 2CHar), 8.13 (s, 1H, CHar), 8.23 (d, J = 7.3 Hz, 1H, CHar).
3-(4,4-dimethyl-1,1-dioxido-3-oxoisothiazolidin-2-yl)benzoyl chloride (2c)
Yield 90% (white powder); m.p. 144 °C. 1H NMR (CDCl3) δ 1.62 (s, 6 H, 2CH3), 3.64 (s, 2H, CH2), 7.67 (t, J = 7.8 Hz, 1H, CHar), 7.71–7.80 (m, 1H, CHar), 8.15 (s, 1H, CHar), 8.24 (d, J = 7.8 Hz, 1H, CHar).
2-chloro-5-(4,4-dimethyl-1,1-dioxido-3-oxoisothiazolidin-2-yl)benzoyl chloride (2f)
Yield 94% (white powder); m.p. 147 °C. 1H NMR (CDCl3) δ 1.61 (s, 6H, 2CH3), 3.58 (s, 2H, CH2), 7.53–7.74 (m, 2H, 2CHar), 8.12 (s, 1H, CHar).
2-chloro-4-(4,4-dimethyl-1,1-dioxido-3-oxoisothiazolidin-2-yl)benzoyl chloride (2k)
Yield 91% (white powder); m.p. 154 °C. 1H NMR (CDCl3) δ 3.31 (t, J = 7.6 Hz, 2H, CH2), 3.82 (t, J = 7.6 Hz, 2H, CH2), 7.53 (d, J = 8.3 Hz, 1H, CHar), 7.64 (s, 1H, CHar), 8.19 (d, J = 8.8 Hz, 1H, CHar).
General procedure for the synthesis of 2-phenylisothiazolidin-3-one-1,1-dioxides (40–42)
The suspension of substituted benzoyl chloride (2h) (2.0 mmol) in tetrahydrofuran (1 mL) was added to the solution of appropriate aliphatic amino acid (3.0 mmol) and sodium hydroxide (240 mg, 6.0 mmol) in water (4 mL). Solution was stirred for 0.5 h and acidified by concentrated hydrochloric acid (0.5 mL). The residue was filtered and air dried to furnish the corresponding product. The spectra of the two most active compounds are presented below. The spectra of other compounds of the combinatorial library are available online as Supplemental Information.
3-{[4-(1,1-dioxido-3-oxoisothiazolidin-2-yl)benzoyl]amino}benzoic acid (21)
Yield 64% (white powder); m.p. > 250 °C. LC-MS m/z 375 [M+H+], Rt = 1.07 min. 1H NMR (DMSO-d6) δ 3.26 (t, J = 7.1 Hz, 2H, CH2), 4.02 (t, J = 7.1 Hz, 2H, CH2), 7.43 (t, J = 7.7 Hz, 1H, CHar), 7.50 (d, J = 7.9 Hz, 2H, CHar), 7.69 (d, J = 7.5 Hz, 1H, CHar), 8.07 (d, J = 7.9 Hz, 1H, CHar), 8.13 (d, J = 7.9 Hz, 2H, CHar), 8.38 (br. s., 1H, CHar), 10.44 (s, 1H, NH).
3-{[2-chloro-4-(1,1-dioxido-3-oxoisothiazolidin-2-yl)benzoyl]amino}benzoic acid (31)
Yield 80% (white powder); m.p. > 250 °C. LC-MS m/z 409 [M+H+], Rt = 1.07 min. 1H NMR (DMSO-d6) δ 3.28 (t, J = 7.3 Hz, 2H, CH2), 4.08 (t, J = 7.3 Hz, 2H, CH2), 7.41–7.55 (m, 2H, 2CHar), 7.59 (s, 1H, CHar), 7.70 (d, J = 7.3 Hz, 1H, CHar), 7.82 (d, J = 8.3 Hz, 1H, CHar), 7.89 (d, J = 8.3 Hz, 1H, CHar), 8.39 (s, 1H, CHar), 10.84 (s, 1H, NH), 13.05 (br. s., 1H, COOH).
Results and discussion
The structural optimization of 2-phenylisothiazolidin-3-one-1,1-dioxide derivates was successfully carried out by analyzing the binding mode of the previously described most potent compound of this classCitation1. For this purpose, we have investigated the complex “compound 1 – CK2”, which was attained by molecular docking methods. The substituents’ influence on inhibitory activity has been predicted based on visual inspection of the complex ().
The binding of compound 1 with ATP-binding site of CK2 is presumably taking place via hydrophobic interaction with five amino acid residues of the kinase and two hydrogen bonds. The first hydrogen bond is formed between 3-acetylphenyl substituent of the compound and the CK2 hinge region (Val 116). The formation of intermolecular interactions between the ligand and this particular CK2 active site is one of the prerequisites for ligand’s activity. Particularly this type of interaction is intrinsic for most protein kinase inhibitors, which was proved by numerous data of the X-ray structural analysisCitation13.
Second hydrogen bond is formed between oxygen atom of 4-methylisothiazolidin-3-one 1,1-dioxide substituent and the nitrogen atom of Lys49, which is located deep in the pocket of the binding site. There are no data up to date supporting the importance of binding to this amino acid residue, however, it is obvious that it provides additional stabilization of the complex.
The substituents which take part in the hydrophobic interactions play an important role in the ligand binding with ATP-binding pocket of protein kinases. Also, it is well known that these interactions are important for the stabilization “CK2-inhibitor” complexCitation14. In case of compound 1 there are hydrophobic interactions between five amino acid residues of the enzyme (Leu45, Val53, Val66, Val116 and Ile174) and aromatic rings of the ligand.
Taking into consideration the key interaction of compound 1 with the active site of CK2, to carry out the optimization of 2-phenylisothiazolidin-3-one-1,1-dioxide derivates the following approach was adopted:
To synthesize derivates where acetyl group will be substituted by carboxyl group at meta- and para-position. This substitution is justified by the assumption that additional oxygen atom will increase the probability of the hydrogen bond formation with the hinge region of the ATP-acceptor pocket of CK2. The position of the substituent in the benzene ring may also play an important role affecting the compound activity.
To obtain the derivatives with different positioning of isothiazolidin-3-one-1,1-dioxide cycle in the ring A () to investigate its influence on the hydrogen bond formation with Lys49 residue.
To synthesize the derivates with one, two and without methyl groups in isothiazolidin-3-one-1,1-dioxide cycle. It is to be noted that the methyl groups are not only able to promote the formation of new hydrophobic contacts with amino acid residues in the binding pocket to increase the inhibitor activity but also to form steric hindrances leading to destabilization of the complex.
To synthesize the compounds having additional hydroxyl groups in the ring B. The incorporation of these groups can promote the formation of additional hydrogen bonds in the hinge region of ATP-binding site of CK2.
To synthesize the derivatives with and without chlorine atom in benzene ring A to study chlorine’s influence on the formation of hydrophobic bonds.
Based on the above approach, 40 structural analogues of compound 1 were proposed.
The series of benzoyl chlorides 2a–2l () were obtained as previously describedCitation12. Combinatorial synthesis of the benzamides was carried out according to .
Scheme 1. Combinatorial synthesis of (1,1-dioxido-3-oxoisothiazolidin-2-yl)benzamides.
It should be noticed that hydroxyaminobenzoic acids were present among the amine substrates, which were used for combinatory synthesis. It is well known when substrates containing both free amino- and hydroxy-groups are acylated by acyl chlorides, the mixture of acidification products at nitrogen atom of amino group and oxygen atom of hydroxyl group is produced. To avoid acidification at the phenol-hydroxy group of aminohydroxybenzoic acids we have developed a base-free procedure. Since the versatility of synthetic methodology is important for combinatorial synthesis, the acylation of aminobenzoic acids without hydroxyl groups was carried out under the same conditions as for aminohydroxybenzoic acids. As a result, we obtained the series of 2-phenylisothiazolidin-3-one-1,1-dioxide derivatives 3–39 ( and Supplementary Table 1). To synthesize the compounds with aliphatic acid residues (compounds 40–42, ) the standard method of Schotten–Baumann was usedCitation15. The synthesized derivatives of 2-phenylisothiazolidin-3-one-1,1-dioxide were tested in vitro. The IC50 values are presented in the , and Supplementary Table 1.
Table 1. Compounds 21–39 with para-position of the thiazolidine cycle relative to carboxylamide fragment and the data related to their in vitro screening.
Table 2. Compounds 40–42 containing the fragments of amino acids with aliphatic chains and the data related to their in vitro screening.
According to the tests, the biological activity is present in 14 out of 40 synthesized compounds (34.14%). Besides, they inhibit protein kinase CK2 more efficiently than their predecessor, compound 1, i.e. ІC50 < 20 μM. The most potent derivative is compound 31, 3-{[2-chloro-4-(1,1-dioxido-3-oxoisothiazolidin-2-yl)benzoyl]amino}benzoic acid (ІC50 = 1.5 μM). Its binding selectivity was checked with the usage of two serine/threonine (ASK1, Aurora А1) and one tyrosine protein kinase (FGFR1). The results of in vitro selectivity test are shown in . It was found that compound 31 in vitro does not inhibit the activity of all abovementioned kinases, i.e. it exhibits selectivity towards inhibition of CK2.
Table 3. Residual activity of protein kinases (%) with added inhibitor (10 μM) and ATP (100 μM).
Using biochemical test data and molecular docking we have investigated structure activity relationship (SAR) for synthesized compounds.
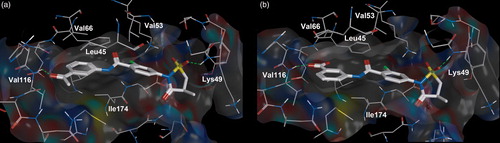
As it was predicted, the replacement of acetyl group (compound 1) with the carboxyl group (compound 35) increases the inhibitor activity (ІC50 values for compounds 1 and 35 are 20.0 and 3.6 μM, respectively). According to the docking data, the ligands have similar disposition in CK2 ATP-binding site (). So, the difference in the compound activities can be explained by the fact that carboxyl group as opposed to acetyl group is more efficient to form the hydrogen bond with the hinge region of protein kinase CK2 (Val116). It is supported by the fact that methyl ester (39) is not active (ІC50 > 30 μM) compared with acid (36) (ІC50 = 6.3 μM).
Figure 2. Compound 1 (a) and compound 35 (b) bound to the active site of the CK2 catalytic subunit. The complex has been obtained with molecular docking. Intermolecular hydrogen bonds are shown as dotted lines.
Since the presence of the acidic residue provides the increase of ligand biological activity, it was reasonable to synthesize and test the derivatives, which contain aliphatic carboxylic acid instead of aromatic one (, compounds 40–42). The aim of this modification was to determine the optimal acid substituent length, which could promote the formation of hydrogen bonds with the hinge region of ATP-acceptor site. In particular, it was important to understand the role of aromatic ring in stabilization of the complex “ligand-CK2”. According to biochemical test results, none of the compounds having aliphatic acid residue inhibit the enzyme. It can be explained by the fact that the absence of aromatic ring makes impossible the rigid ligand fixation caused by the hydrophobic interactions with Val66, Val116 and Ile174 and, as a result, the formation of hydrogen bonds with amino acid residues becomes less probable.
Interestingly, the dioxoisothiazolidine ring positioning turned out to be extremely important. Compounds 3–20 (Supplementary Table 1) and 21–39 () contain the abovementioned substituents at meta- and para-position of ring A correspondently. All meta-dioxoisothiazolidine derivates were not active (ІC50 >30 μM), whereas the activity of their para-substituted analogs was much higher in the range of 1.5–20 μM. This regularity may be explained by total compound length, which is affected by changing substituents’ position.
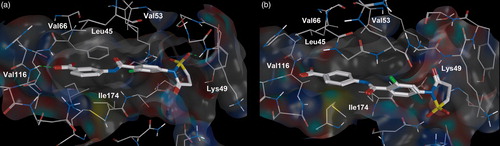
It was found that meta-substituted analog is more active by comparing the carboxyl group substitution at para- and meta-positions of the benzene ring. Thus, para-substituted compound 34 is not active meanwhile compound 31 – 3-{[2-chloro-4-(1,1-dioxido-3-oxoisothiazolidin-2-yl)benzoyl]amino}benzoic acid, which has carboxyl group in meta-position is highly potent (ІC50 = 1.5 μM). As it turned out, para-substitution in benzene ring causes conformationally disfavored orientation of the compound in the CK2 binding site. As a result all hydrogen bonds and key hydrophobic interactions with amino acid residues, Val66, Phe113 and Ile174 (), disappear.
Figure 3. Compound 31 (a) and compound 34 (b) bound to the active site of the CK2 catalytic subunit. The complex has been obtained with molecular docking. Intermolecular hydrogen bonds are shown as dotted lines.
Addition of hydroxyl groups to a ring B has an ambiguous influence on the ligand activity. The presence of ОН-group in ortho-position of meta-carboxyphenyls leads to the drop of activity (21 and 22; 31 and 32; 35 and 36) and even to total disappearance of the activity (pairs of compounds: 28 and 29; 24 and 25). In one case, an introduction of the hydroxyl group at para-position does not make an essential influence on inhibitor properties of the compounds (e.g. for the compounds 35 and 37 ІC50 values are 3.6 and 3.7 μM, correspondingly), in other cаse it leads to the significant decrease in activity (compounds 24 and 26). The opposite situation has been observed in case of para-substituted carboxyphenyls. Thus, the introduction of OH-group to ortho-position leads to the sudden rise in activity (compound 34 is inactive, compound 33 is active, ІC50 3 μM). Unfortunately, using molecular docking we were unable to explain the relationship between the position of ОН-groups and ligand inhibitory activity.
Next, we have examined the influence of hydrophobic substituents in the ring A on the ligand activity. Introduction of a chlorine atom in carboxyphenyl ring without hydroxyl group leads either to the 4-fold enhancement of compound inhibitory properties (compound 26, ІC50 is 16 μM and compound 37, ІC50 is 3.7 μM) or does not have any significant influence on the activity (21 and 31, 24 and 35). At the same time the presence of chlorine in the compounds containing ОН-group in carboxyphenyl substituent increases inhibitory activity of the ligands (22 and 32, 23 and 33, 24 and 35, 25 and 36, 27 and 39).
The presence of methyl substituents in dioxoisothiazolidine ring has a clear influence on 2-phenylisothiazolidin-3-one-1,1-dioxides inhibitory properties. It was found that compound activity decreased with an increase in the number of methyl groups. It can be seen in the following compound rows: 21–24–28; 22–25–29; 23–27–30; 31–35; 33–38. Probably, the addition of methyl groups forms steric constraints which prevent tight ligand fixation in the binding pocket.
The binding mode of compound 31 predicted by molecular docking is similar to the interactions of compounds 18E and 19E with CK2, three-dimensional structures of these complexes were determined by X-ray crystallographyCitation16. All three compounds form hydrogen bonds with kinase hinge-region by the oxygen atom, employing carboxylic acid oxygen in the case of compound 31 and oxygen of the phenoxy group in the case of compounds 18E and 19E. Additionally, compounds 18E and 19E form hydrogen bonds with Lys68 side chain, while compound 31 forms hydrogen bond with Lys49 main chain. Considering the inhibitory mechanism of compounds 31, 18E and 19E, the pharmacophore-related properties of these structurally different ligands were investigated. It was established previously that the pharmacophore of CK2 inhibitors should have hydrophobic aromatic moieties to enable efficient binding with the amino acid residues of CK2-active site and for stacking with Phe113. Also, it should contain appropriate groups to form hydrogen bonds with Val116, Glu114 in CK2 hinge region and with Lys68, Asp175 in phosphate-binding regionCitation17. Moreover, the total distance between the pharmacophoric points forming hydrogen bonds with Val116, Glu114 and Lys68 should not exceed 8.5 Å. The pharmacophore analysis of compounds 18E, 19E and 31 showed that these ligands fit the requirements for CK2 inhibitors outlined above. The increase in potency for compounds 18E and 19E could arise from interactions with the hydrophobic pocket in the vicinity of Phe113 and the additional intramolecular hydrogen bonds between ligands and Glu114, Lys68. Compound 34 is not appropriate to the requirement of ligand optimal length and it is not active.
Thereby, we were able to increase the ligand inhibitory activity by 13-fold (compound 31) compared to the parent compound 1. The replacement of acetyl group at benzene ring by the carboxyl group was the most important modification.
Conclusions
The main direction for structural optimization of 2-phenylisothiazolidin-3-one-1,1-dioxides has been proposed. Forty compounds of this class have been synthesized and their biological activity has been evaluated. Due to successful optimization, activity of obtained compound has been improved >10-fold as compared with previously reported inhibitors of the same family. The relationship “chemical structure-biological activity” of 2-phenylisothiazolidin-3-one-1,1-dioxides has been investigated, the binding mode of the synthesized compounds with CK2 ATP-acceptor site has been proposed. It has been established that carboxyphenyl substituent has the most powerful influence on the compound activity: oxygen atoms of the carboxyl group are required for the formation of hydrogen bonds with the hinge region of CK2 active site; phenyl aromatic ring is responsible for tight ligand fixation due to hydrophobic interactions with Val66, Val116 and Ile174.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Supplementary Material
Download PDF (170.9 KB)Related Research Data
References
- Chekanov MO, Synyugin AR, Lukashov SS, Yarmoluk SM. The synthesis of 2-phenylisothiazolidine-3-one dioxide derivatives as inhibitors of protein kinase CK2. Ukr Bioorg Acta 2009;7:62–4
- Shoichet BK, Stroud RM, Santi DV, et al. Structure-based discovery of inhibitors of thymidylate synthase. Science 1993;259:1445–50
- Bodian DL, Yamasaki RB, Buswell RL, et al. Inhibition of the fusion-inducing conformational change of influenza hemagglutinin by benzoquinones and hydroquinones. Biochemistry 1993;32:2967–78
- Ring CS, Sun E, McKerrow JH, et al. Structure-based inhibitor design by using protein models for the development of antiparasitic agents. Proc Natl Acad Sci USA 1993;90:3583–7
- Ewing TJ, Makino S, Skillman AG, Kuntz ID. DOCK 4.0: search strategies for automated molecular docking of flexible molecule databases. J Comput Aided Mol Des 2001;15:411–28
- Yakovenko OY, Oliferenko AA, Golub AG, et al. The new method of distribution integrals evaluations for high throughput virtual screening. Ukrainica Bioorganica Acta 2007;1:52–62
- Yakovenko OY, Li YY, Oliferenko AA, et al. Ab initio parameterization of YFF1, a universal force field for drug-design applications. J Mol Model 2012;18:663–73
- Yakovenko OY, Oliferenko AA, Bdzhola VG, et al. Kirchhoff atomic charges fitted to multipole moments: implementation for a virtual screening system. J Comput Chem 2008;29:1332–43
- Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Mod 2001;7:306–17
- Prykhod'ko AO, Yakovenko OY, Golub AG, et al. Evaluation of 4H-4-chromenone derivatives as inhibitors of protein kinase CK2. Biopolym Cell 2005;21:1–6
- Meggio F, Pagano M, Moro S, et al. Inhibition of protein kinase CK2 by condensed polyphenolic derivatives. An in vitro and in vivo study. Biochemistry 2004;43:12931–6
- Synyugin АR, Chekanov MO, Lukashov SS, Yarmoluk SM. Synthesis of (1,1-dioxido-3-oxoisothiazolidin-2-yl)benzenamides. Ukr Bioorg Acta 2008;6:28–32
- Backes AC, Zech B, Felber B, et al. Small-molecule inhibitors binding to protein kinases. Part I: exceptions from the traditional pharmacophore approach of type I inhibition. Expert Opin Drug Discov 2008;3:1409–25
- Sarno S, Salvi M, Battistutta R, et al. Features and potentials of ATP-site directed CK2 inhibitors. Biochim Biophys Acta 2005;1754:263–70
- Greenstein JP, Winitz M. Chemistry of the amino acids. New York – London – Sydney – Toronto: John Wiley and Sons; 1961
- Prudent R, Moucadel V, Nguyen CH, et al. Antitumor activity of pyridocarbazole and benzopyridoindole derivatives that inhibit protein kinase CK2. Cancer Res 2010;70:9865–74
- Morshed MN, Muddassar M, Pasha FA, Cho SJ. Pharmacophore identification and validation study of CK2 inhibitors using CoMFA/CoMSIA. Chem Biol Drug Des 2009;74:148–58