
Abstract
This study reports the synthesis of a series of new 2-amino-3-cyano-8-methyl-4-substituted-5,6,7,8-tetrahydroquinolines along with some derived fused-ring systems. Ten compounds have shown remarkable cytotoxic activity against human colon carcinoma HT29, hepatocellular carcinoma HepG2 and Caucasian breast adenocarcinoma MCF7 cell lines. Six compounds showed considerable broad-spectrum cytotoxic activity among which two proved to be the most active derivatives. Likewise, seven compounds from the series were found to exhibit significant antimicrobial activity and three of them proved to be the most active candidates. Two alkylthio-pyrimido quinolines are suggested as possible antimicrobial and anticancer candidates in the present series.
Introduction
The ongoing efforts of research on the treatment of malignancy are focused on the discovery of novel products interacting with novel biological targets. Among the wide variety of heterocycles that have been explored for developing pharmaceutically important molecules, pyridines and fused pyridine ring systems have received much attention since they are proved to be biologically versatile compounds possessing a variety of activities. A wide range of chemotherapeutic activities have been ascribed to pyridine derivatives including antimicrobialCitation1–3, antiamoebicCitation1, antiparasiticCitation4 and antiviralCitation5,Citation6 activities. As far as the antineoplastic potential is concerned, pyridine derivatives were reported to contribute to a variety of anticancer activity including cytotoxicCitation7,Citation8, antiproliferativeCitation9 and CDK kinase inhibitory activityCitation10.
Cyanopyridylureas have been claimed for their properties in treating hyperproliferative and angiogenesis disorders. The 3-cyano-2,6-dihydropyridine is a potent inhibitor of dihydrouracil dehydrogenase and its co-administration with 1-ethoxymethyl-5-fluorouracil enhances the antitumor effectCitation11. Furthermore, Piritrexim (2,4-diamino-6-(2,5-dimethoxybenzyl)-5-methylpyrido[2,3-d]pyrimidine), a second-generation antineoplastic drug, having a pyridine ring among their structures was reported to inhibit the enzyme dihydrofolate reductase. Recently, it has been reported that some fused pyridines emerged as potential inhibitors of protein kinase BCitation12 and HIF 1-α prolyl hydroxylaseCitation13, which play a major role in cancer cell division. During our ongoing studies aimed at the discovery of new structure leads endowed with diverse chemotherapeutic activities, much concern has been raised on the antimicrobial and antitumor potentials of some pyridine derivativesCitation14–17. The obtained results prompted further structure modification of the disubstituted-2(1H)-pyridinone scaffold by fusing it with a cyclohexane ring leading to the synthesis of new tetrahydroquinoline analogs. The substitution profile of the main tetrahydroquinoline ring was attempted to comprise some counterparts that would confer different electronic, lipophilic and steric environment, which would influence the targeted biological activities. It was, therefore, considered worthwhile to synthesize some tetrahydroquinoline-derived fused-ring systems as interesting structural variation to improve the anticipated chemotherapeutic profile.
Experimental
Chemistry
Melting points were determined on a Gallenkamp melting point apparatus and are uncorrected. The microwave reactor used was CEM Discover 300 W. The infrared (IR) spectra were recorded on Shimadzu FT-IR 8400S IR spectrophotometer using the KBr pellet technique. 1H and 13C NMR spectra were recorded on a Bruker DPX-400 FT NMR spectrometer using tetramethylsilane as the internal standard and DMSO-d6 as a solvent (chemical shifts in δ, ppm). Splitting patterns were designated as follows: s, singlet; d, doublet; m, multiplet and q, quartet. Elemental analyses were performed on a 2400 Perkin Elmer Series 2 analyzer and the found values were within ±0.4% of the theoretical values. Follow up of the reactions and checking the homogeneity of the compounds were made by TLC on silica gel pre-coated aluminum sheets (Type 60 F254, Merck) and the spots were detected by exposure to UV-lamp at λ 254. The analysis data are reported in and .
Table 1. Physicochemical and analytical data of compounds 1–28.
Table 2. Physicochemical and analytical data of the scheme 2 compounds 29–51.
2-Amino-3-cyano-8-methyl-4-substituted-5,6,7,8-tetrahydroquinolines (1–6)
A mixture of the 2-methylcyclohexanone (1.12 g, 0.01 mol), the appropriate aldehyde (0.01 mol), malononitrile (0.66 g, 0.01 mol) and anhydrous ammonium acetate (6.2 g, 0.08 mol) in absolute ethanol (50 mL) was refluxed for 3–6 h. The reaction mixture was cooled and the resulting precipitate was filtered, washed with water, dried and crystallized with the appropriate solvent. IR (cm−1): 3210–3450 (NH2), 2214–2226 (CN). 13C NMR (δ ppm) for 1 (R = C6H5): 20.8 (CH3), 21.3, 27.12, 30.5, 36.6 (cyclohexyl C), 117.0 (CN), 91.8, 122.4, 152.9, 157.4, 166.1 (pyridine C), 123.9, 129.6, 132.4, 135.8 (Ar C). 2 (R = 4-BrC6H4): 20.2 (CH3), 21.1, 27.02, 30.9, 36.5 (cyclohexyl C), 116.8 (CN), 89.7, 120.3, 153.1, 157.2, 165.8 (pyridine C), 123.2, 129.8, 132.0, 135.3, (Ar C). 3 (R = 4-CH3OC6H4): 20.6 (CH3), 22.1, 27.6, 31.2, 38.4 (cyclohexyl C), 56.2 (OCH3), 116.8 (CN), 89.7, 121.3, 152.8, 157.7, 165.3 (pyridine C), 114.2, 127.8, 131.5, 162.4 (Ar C). 4 (R = 4-CH3C6H4): 20.2(CH3), 21.2 (CH3), 21.4, 27.1, 30.7, 36.5 (cyclohexyl C), 116.9 (CN), 90.1, 120.6, 154.6, 157.2, 165.3 (pyridine C), 128.0, 129.3, 133.4, 138.6 (Ar C). 5 (R = 2-Thienyl): 20.3 (CH3), 21.5, 28.2, 31.2, 37.81 (cyclohexyl C), 116.8 (CN), 92.3, 122.3, 148.8, 158.5, 165.3 (pyridine C), 127.4, 127.6, 128.8, 136.9, (thiophene ring C). 6 (R = 1-Methyl-pyrrol-2-yl): 21.9 (CH3), 32.4 (NCH3), 21.9, 27.28, 31.5, 37.6 (cyclohexyl C), 117.3 (CN), 90.5, 121.8, 153.9, 156.6, 164.8 (pyridine C), 110.9, 112.2, 122.9, 124.1 (pyrrole C).
9-Methyl-5-substituted-6,7,8,9-tetrahydro-3H-pyrimido[4,5-b]quinolin-4-ones (7–11)
A mixture of the appropriate compound 1–5 (0.01 mol) and formic acid (5 mL) was heated in a boiling water bath for 30 min. After cooling, the reaction mixture was poured onto ice-cold water; the precipitated solid was filtered, washed with water and crystallized from ethanol. IR (cm−1): 3258–3150 (NH), 1695–1715 (C=O). 13CNMR (δ ppm) for 7 (R = C6H5): 21.2 (CH3), 21.4, 27.2, 31.0, 36.4 (cyclohexyl C), 119.6, 126.8, 129.0, 129.1, 135.4, 138.4, 152.2, 162.1, 163.4, 171.5 (ArC), 170.2 (CO). 8 (R = 4-BrC6H4): 20.8 (CH3), 21.6, 27.5, 31.2, 36.8 (cyclohexyl C), 118.6, 126.4, 129.2, 129.3, 136.4, 138.2, 153.2, 162.6, 163.0, 170.3 (ArC), 169.2 (CO). 9 (R = 4-CH3OC6H4): 20.7 (CH3), 21.7, 27.4, 31.8, 39.0 (cyclohexyl C), 56.1 (OCH3), 115.2, 127.4, 131.5, 135.2, 138.5, 152.6, 162.7, 163.0, 171.0 (Ar C), 170.5 (CO). 10 (R = 4-CH3C6H4): 21.0 (CH3), 22.1 (CH3), 21.6, 27.3, 30.4, 36.8 (cyclohexyl C), 118.4, 126.5, 129.1, 129.2, 135.6, 138.6, 152.7, 162.4, 163.2, 171.6 (ArC), 169.7 (CO). 11 (R = 2-thienyl): 21.3 (CH3), 22.2, 27.8, 31.4, 37.6 (cyclohexyl C), 120.3, 122.4, 125.3, 127.4, 135.8, 142.4, 148.4, 162.2, 163.0, 171.7 (Ar C), 170.1 (CO).
2,9-Dimethyl-5-substituted-6,7,8,9-tetrahydro-3H-pyrimido[4,5-b]quinolin-4-ones (12–16)
A mixture of the appropriate compound 2–6 (0.01 mol), acetic anhydride (5 mL) and concentrated H2SO4 (0.5 mL) was heated in a boiling water bath for 10 min then cooled, poured into ice-cold water and treated with 20% NaOH solution until the pH is alkaline (pH 11). The crude solid product was filtered and crystallized from ethanol. IR (cm−1): 3226–3431 (NH), 1707–1715 (C=O). 13CNMR (δ ppm) for 12 (R = 4-BrC6H4): 19.9 (CH3), 20.9 (CH3), 22.3, 27.4, 31.2, 37.4 (cyclohexyl C), 119.8, 123.5, 129.1, 132.3, 135.2, 137.2, 152.4, 162.4, 164.0, 170.6 (ArC), 168.9 (CO). 13 (R = 4-CH3OC6H4): 19.8 (CH3), 20.7 (CH3), 21.7, 27.4, 31.8, 39.0 (cyclohexyl C), 56.2 (OCH3), 114.2, 127.7, 130.5, 135.2, 138.5, 152.6, 162.0, 162.4, 163.9, 171.6 (ArC), 170.2 (CO). 14 (R = 4-CH3C6H4): 19.5 (CH3), 20.1 (CH3), 21.4 (CH3), 22.6, 27.6, 30.5, 37.0 (cyclohexyl C), 119.4, 126.3, 129.2, 129.4, 135.5, 138.2, 152.6, 162.3, 163.6, 170.6 (ArC), 169.8 (CO). 16 (1-Methyl-pyrrol-2-yl): 20.0 (CH3), 21.3 (CH3), 32.4 (NCH3), 23.2, 28.1, 31.9, 37.4 (cyclohexyl C), 113.1, 119.6, 120.2, 122.2, 135.6, 147.2, 153.4, 161.4, 164.4, 171.6 (ArC), 170.7 (CO).
4-Imino-9-methyl-3-phenyl-5-substituted-3,4,6,7,8,9-hexahydro-1H-pyrimido[4,5-b]-quinoline-2-thiones (17–22)
A mixture of the appropriate compound 1–6 (0.01 mol), phenyl isothiocyanate (1.35 g, 0.01 mol) in pyridine (15 mL) was refluxed for 2 h. After cooling, the solid was filtered, washed thoroughly with water, dried and recrystallized from acetic acid. IR (cm−1): 3180–3327 (NH), 1629–1615 (C=N), 1195–1210 (C=S). 13CNMR (δ ppm) for 17 (R = C6H5): 22.2 (CH3), 25.4, 31.2, 32.5, 39.4 (cyclohexyl C), 109.8, 124.5, 124.7, 125.5, 126.8, 128.8, 129.1, 138.3, 139.2, 149.6, 156.8, 163.4 (ArC), 166.0 (C=NH), 179.8 (CS). 18 (R = 4-BrC6H4): 21.9 (CH3), 25.3, 31.4, 32.3, 39.2 (cyclohexyl C), 109.5, 123.2, 124.6, 125.3, 126.8, 128.8, 129.3, 132.3, 137.3, 139.4, 148.9, 156.7, 163.4 (ArC), 165.4 (C=NH), 180.5 (CS). 19 (R = 4-CH3OC6H4): 22.0 (CH3), 26.0, 31.8, 32.5, 39.4 (cyclohexyl C), 56.6 (OCH3), 109.8, 114.4, 124.3, 125.2, 127.6, 128.6, 130.5, 139.7, 148.8, 157.1, 162.3, 163.2 (ArC), 164.7 (C=NH), 178.4 (CS). 20 (R = 4-CH3C6H4): 20.8 (CH3), 22.3 (CH3), 25.6, 30.8, 32.5, 38.8 (cyclohexyl C), 109.8, 124.1, 124.5, 125.4, 126.6, 128.6, 129.5, 135.4, 139.5, 148.8, 157.1, 163.2 (ArC), 164.9 (C=NH), 179.0 (CS). 22 (1-Methyl-pyrrol-2-yl): 20.6 (CH3), 32.4 (NCH3), 25.2, 31.4, 32.1, 38.5 (cyclohexyl C), 108.1, 110.5, 112.2, 124.6, 125.2, 128.8, 129.8, 133.6, 139.6, 145.2, 157.4, 163.4 (ArC), 164.4 (C=NH), 179.6 (CS).
1-Benzoyl-3-(3-cyano-8-methyl-4-substituted-5,6,7,8-tetrahydroquinolin-2-yl)-thioureas (23–28)
To a solution of the appropriate derivative 1–6 (0.02 mol) in dry acetone (20 mL), a solution of benzoyl isothiocyanate (3.3 g, 0.02 mol) in dry acetone (10 mL) was added. The resultant solution was heated under reflux for 3 h. The reaction mixture was left overnight, concentrated and allowed to cool. The separated crystalline product was filtered, washed with Et2O and recrystallized from ethanol. IR (cm−1): 3345–3450 (NH), 2210–2220 (CN), 1710–1700 (C=O), 1185–1168 (C=S). 13C NMR (δ ppm) for 23 (R = C6H5): 21.7 (CH3), 25.0, 31.3, 32.4, 39.1 (cyclohexyl C), 91.6, 125.3, 126.8, 127.4, 128.4, 129.1, 129.2, 131.7, 133.5, 138.2, 156.7, 163.2, 166.2 (ArC), 117.6 (CN), 179.5 (CS), 169.9 (CO). 24 (R = 4-BrC6H4): 22.1 (CH3), 25.7, 31.3, 32.4, 37.9 (cyclohexyl C), 91.4, 123.3, 126.8, 127.4, 128.4, 129.1, 131.2, 132.7, 133.5, 137.2, 156.7, 163.2, 166.2 (ArC), 117.2 (CN), 179.1 (CS), 168.7 (CO). 25 (R = 4-CH3OC6H4): 21.8 (CH3), 25.4, 31.5, 32.2, 39.1 (cyclohexyl C), 56.1 (OCH3), 91.3, 114.6, 125.2, 127.3, 128.6, 130.4, 131.8, 133.5, 156.7, 162.5, 163.2, 166.1 (ArC), 118.0 (CN), 179.8 (CS), 169.5 (CO). 26 (R = 4-CH3C6H4): 20.9 (CH3), 22.4 (CH3) 25.6, 31.5, 32.3, 38.0 (cyclohexyl C), 91.7, 125.3, 126.8, 127.3, 128.7, 129.7, 131.8, 133.5, 135.2, 138.3, 156.6, 163.4, 165.7 (ArC), 117.5 (CN), 179.2 (CS), 169.4 (CO). 27 (R = 2-thienyl): 21.8 (CH3), 24.9, 31.1, 32.5, 37.9 (cyclohexyl C), 92.4, 122.7, 125.3, 125.8, 127.2, 127.4, 128.5, 131.8, 133.5, 142.6, 152.4, 163.4, 166.2 (ArC), 118.1 (CN), 180.0 (CS), 168.8 (CO). 28 (1-Methyl-pyrrol-2-yl): 20.6 (CH3), 32.4 (NCH3), 25.2, 31.4, 32.1, 38.5 (cyclohexyl C), 92.4, 110.2, 123.1, 125.8, 127.3, 128.5, 131.9, 133.4, 152.2, 163.1, 165.8 (ArC), 117.7 (CN), 179.6 (CS), 169.7 (CO).
General methods for the preparation of 4-amino-9-methyl-5-substituted-6,7,8,9-tetrahydro-1H-pyrimido[4,5-b]quinoline-2-thiones (29–33) and 4-amino-9-methyl-5-substituted-6,7,8,9-tetrahydro-1H-pyrimido[4,5-b]quinoline-2-ones (34–36)
Method A
A solution of the appropriate thioureido derivative 23–27 (0.02 mol) in a mixture of 1N sodium hydroxide (5.5 mL) and EtOH (10 mL) was heated on a boiling water bath for 30 min. The reaction mixture was filtered, if necessary, allowed to cool and rendered acidic with 10% acetic acid. The product was filtered, washed thoroughly with H2O, dried and crystallized from DMF containing few drops of water. IR (cm−1): 3339–3445 (NH2), 1158–1172 (C=S).
Method B
A mixture of the appropriate derivative 1–5 (0.001 mol) and thiourea (0.4 g, 0.005 mol) or urea (0.3 g, 0.005 mol) was fused at 260–300 °C using sand bath for 1 h. The reaction mixture was allowed to attain room temperature. The resulting solid product was treated with water, then rubbed with ethanol, filtered and crystallized from DMF containing few drops of water.
Method C
A mixture of the appropriate derivative 1–5 (0.001 mol) and thiourea (0.4 g, 0.005 mol) or urea (0.3 g, 0.005 mol) was heated in a microwave reactor at 250 °C for 15 min. Working up of the reaction was carried out exactly as described under method B. IR (cm−1) for compounds: 3320–3432 (NH), 1695–1708 (C=O). 13C NMR (δ ppm) for 29 (R = C6H5): 21.9 (CH3), 25.3, 31.4, 32.6, 39.2 (cyclohexyl C), 109.9, 124.3, 126.9, 129.1, 138.2, 149.4, 157.1, 163.2, 164.7 (ArC), 182.4 (CS). 30 (R = 4-BrC6H4): 22.1 (CH3), 25.4, 31.2, 32.3, 37.6 (cyclohexyl C), 109.5, 123.4, 124.7, 129.2, 132.3, 137.1, 148.9, 157.3, 163.2, 164.4 (ArC), 183.2 (CS). 31 (R = 4-CH3OC6H4): 21.8 (CH3), 25.3, 31.3, 32.4, 38.7 (cyclohexyl C), 56.0 (OCH3), 109.7, 114.4, 124.6, 127.8, 130.3, 149.6, 156.9, 162.5, 163.4, 165.3, (ArC), 182.8 (CS). 32 (R = 4-CH3C6H4): 20.8 (CH3), 22.2 (CH3) 25.4, 31.2, 32.3, 38.2 (cyclohexyl C), 109.9, 124.7, 126.4, 129.8, 135.2, 138.2, 149.5, 157.1, 162.9, 163.7 (ArC), 180.2 (CS). 33 (R = 2-thienyl): 21.9 (CH3), 25.2, 31.3, 32.3, 38.7 (cyclohexyl C), 110.4, 122.2, 125.3, 126.4, 127.5, 142.5, 145.3, 157.2, 163.4, 164 (ArC), 181.3 (CS). 34 (R = C6H5): 21.8 (CH3), 25.1, 30.8, 31.9, 37.9 (cyclohexyl C), 109.5, 124.4, 126.9, 129.0, 138.2, 149.3, 157.2, 163.3, 164.4 (ArC), 163.2 (CO).
2-Alkylthio-4-amino-9-methyl-5-substituted-6,7,8,9-tetrahydro-1H-pyrimido[4,5-b]quinolines (37–47)
To a stirred solution of the proper thione 29–33 (0.02 mol) in a mixture of 1N NaOH (5 mL) and ethanol (2 mL), the selected alkyl or aralkyl halide (0.022 mol) was added. The reaction mixture was stirred at room temperature for 2–3 h and the precipitated product was filtered, washed with aqueous ethanol, dried and crystallized from ethanol. IR (cm−1): 3440–3150 (NH). IR (cm−1) for compounds 38 and 41: 1680–1685 (ketone C=O). 13C NMR (δ ppm) for 37 (R = C6H5; R′ = C2H5): 15.8 (CH3), 21.5 (CH3), 28.1 (CH2), 25.2, 31.3, 32.4, 38.4 (cyclohexyl C), 102.8, 126.9, 127.1, 129.0, 135.2, 138.2, 150.0, 157.9, 162.6, 167.3, 167.6 (ArC). 38 (R = C6H5; R′ = CH2COC6H5): 22.3 (CH3), 44.2 (CH2) 24.9, 31.5, 32.4, 38.3 (cyclohexyl C), 102.2, 126.4, 126.7, 128.4, 128.7, 135.3, 129.2, 132.8, 135.3, 137.1, 138.4, 148.9, 158.3, 162.2, 162.8 (ArC), 196.2 (CO). 39 (R = 4-BrC6H4; R′ = CH3): 18.8 (CH3), 21.7 (CH3), 25.1, 31.2, 32.3, 38.8 (cyclohexyl C), 102.1, 123.7, 129.1, 132.4, 135.3, 137.1, 148.7, 158.4, 162.4, 167.3, 167.7 (ArC). 40 (R = 4-CH3OC6H4; R′ = CH3): 18.9 (CH3), 21.9 (CH3), 25.2, 31.4, 32.2, 38.9 (cyclohexyl C), 56.2 (OCH3), 102.2, 114.7, 127.8, 130.5, 135.3, 150.1, 158.4, 162.5, 162.6, 167.3 (ArC). 41 (R = 4-CH3OC6H4; R′ = CH2COC6H5): 21.8 (CH3), 44.1 (CH2), 25.4, 31.1, 32.2, 38.6 (cyclohexyl C), 56.1 (OCH3) 102.1, 114.6, 127.8, 128.4, 128.7, 130.1, 132.8, 135.4, 137.3, 149.5, 158.2, 162.3, 162.6, 167.3, 167.8 (ArC), 196.4 (CO). 42 (R = 4-CH3OC6H4; R′ = C2H5): 15.5 (CH3), 21.6 (CH3), 28.2 (CH2), 25.4, 31.1, 32.2, 38.6 (cyclohexyl C), 56.1 (OCH3), 102.3, 114.4, 127.9, 130.4, 135.4, 149.9, 158.0, 162.2, 162.5, 167.2, 167.6 (ArC). 44 (R = 4-CH3C6H4; R′ = CH2C6H5): 20.9 (CH3), 22.2 (CH3), 25.0, 31.5, 32.3, 38.1 (cyclohexyl C), 40.7 (CH2), 102.5, 124.3, 126.8, 126.9, 127.6, 128.4, 129.7, 135.2, 135.3, 141.2, 149.9, 158.3, 167.4, 167.7 (ArC).
4-Amino-9-methyl-5-substituted-6,7,8,9-tetrahydro-pyrimido[4,5-b]quinolines(48–51)
A mixture of the appropriate intermediate 1–4 (0.01 mol) and formamide (10 ml) was heated under reflux for 2–3 h. The reaction mixture was cooled and the precipitated solid was collected, washed with cold ethanol and crystallized from acetic acid containing few drops of water. IR (cm−1): 3330–3265 (NH2). 48 (R = C6H5): 22.0 (CH3), 24.7, 31.3, 32.0, 38.4 (cyclohexyl C), 106.8, 126.9, 129.1, 135.3, 138.2, 149.9, 157.3, 158.5, 162.4, 167.7 (ArC). 49 (R = 4-BrC6H5): 21.7 (CH3), 25.3, 31.4, 32.2, 38.6 (cyclohexyl C), 106.1, 123.7, 129.0, 132.3, 135.2, 137.3, 149.7, 157.4, 158.1, 162.4, 167.7 (ArC). 50 (R = 4-CH3OC6H5): 21.9 (CH3), 25.6, 31.4, 32.3, 38.7 (cyclohexyl C), 56.0 (OCH3), 106.7, 114.4, 127.9, 130.3, 135.4, 149.5, 157.2, 158.3, 162.4, 167.4 (ArC).
Biological activity
In vitro MTT cytotoxicity assay
The synthesized compounds were investigated for their in vitro cytotoxic effect via the standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) methodCitation18,Citation19 against a panel of three human tumor cell lines: Caucasian breast adenocarcinoma MCF7, hepatocellular carcinoma HepG2 and colon carcinoma HT29. The procedures were done in a sterile area using a laminar flow cabinet biosafety class II level (Baker, SG403INT, Stanford, ME). Cells were batch cultured for 10 d, then seeded at concentration of 10 × 103 cells/well in fresh complete growth medium in 96-well microtiter plastic plates at 37 °C for 24 h under 5% CO2 using a water-jacketed carbon dioxide incubator (Sheldon, TC2323, Cornelius, OR). Media was aspirated, fresh medium (without serum) was added and cells were incubated either alone (negative control) or with eight different concentrations of the test compounds (100, 50, 25, 12.5, 6.25, 3.125, 1.56 and 0.78 μg/mL). DMSO was employed as a vehicle for dissolution of the tested compounds and its final concentration on the cells was less than 0.2%. Cells were suspended in RPMI 1640 medium (for HepG2 and HT29 cell lines) and DMEM (for MCF 7 cell line), 1% antibiotic-antimycotic mixture (10 000 IU/mL penicillin potassium, 10 000 μg/mL streptomycin sulphate and 25 μg/mL amphotericin B), and 1% l-glutamine in 96-well flat bottom microplate at 37 °C under 5% CO2.
After 24 h of incubation, the medium was aspirated, 40 μL of MTT salt (2.5 μg/mL) was added to each well and incubated for further 4 h at 37 °C under 5% CO2. To stop the reaction and dissolve the formed crystals, 200 μL of 10% sodium dodecyl sulphate in deionized water was added to each well and incubated overnight at 37 °C. The absorbance was then measured using a microplate multiwell reader (Bio-Rad Laboratories Inc., model 3350, Hercules, CA) at 595 nm and a reference wavelength of 620 nm. A statistical significance was tested between samples and negative control (cells with vehicle) using independent t-test by SPSS 11 program. Doxorubicin is used as positive control cytotoxic agent. The results of LC50 (μM), which is the lethal concentration of the compound causing death of 50% of the cells in 24 h, are presented in .
Table 3. 1H NMR data of compounds 1–51.
Table 4. Cytotoxic effects (LC50; μM)a of the active compounds on some human tumor cell lines using the MTT assay.
In vitro antibacterial and antifungal activities
All of the newly synthesized compounds were evaluated for their in vitro antibacterial activity against Staphylococcus aureus (ATCC 6538), Bacillus subtilis (ATCC 6633) and Micrococcus luteus (ATCC 21881) as examples of Gram-positive bacteria and Escherichia coli (ATCC 25922), Pseudomonas aeruginosa (ATCC 27853) and Klebsiella pneumonia (clinical isolate) as examples of Gram-negative bacteria. They were also evaluated for their in vitro antifungal potential against Candida albicans (ATCC 10231) and Aspergillus niger (recultured) fungal strains. Each 100 mL of sterile molten agar (at 45 °C) received 1 mL of 6 h broth culture and then the seeded agar was poured into sterile Petri dishes. Cups (8 mm in diameter) were cut in the agar. Each cup received 0.1 mL of the 1 mg/mL solution of the test compounds. The plates were then incubated at 37 °C for 24 h or, in case of C. albicans, for 48 h. A control using DMSO without the test compound was included for each organism. Ampicillin trihydrate and clotrimazole were used as reference drugs. The results were recorded for each tested compound as the average diameter of inhibition zones of bacterial or fungal growth around the discs in mm. The minimal inhibitory concentrations (MIC) of the most active compounds were measured using the twofold serial broth dilution methodCitation20. The test organisms were grown in their suitable broth: 24 h for bacteria and 48 h for fungi at 37 °C. Twofold serial dilutions of solutions of the test compounds were prepared using 200, 100, 50, 25, 12.5 and 6.25 µg/mL. The tubes were then inoculated with the test organisms; each 5 mL received 0.1 mL of the above inoculums and were incubated at 37 °C for 48 h. Then, the tubes were observed for the presence or absence of microbial growth. The MIC values in µg/mL of the prepared compounds are listed in .
Table 5. Minimal inhibitory concentrations [MIC, µg/mL (μM)] of the tested compounds.
Results and discussion
Chemistry
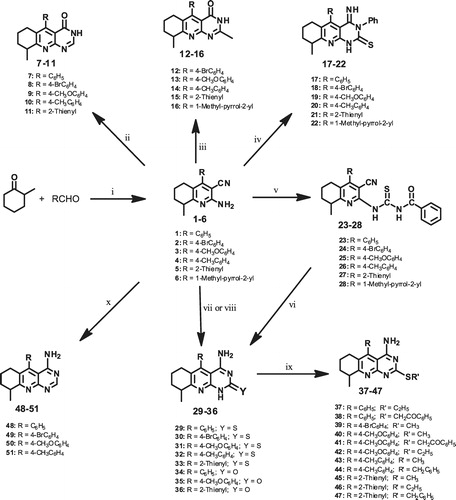
The synthetic strategies adopted for the preparation of the intermediate and target compounds are described in . The key intermediates in this scheme are the 2-amino-3-cyano-8-methyl-4-substituted-5,6,7,8-tetrahydroquinolines 1–6. These compounds were easily synthesized via one-pot multicomponent reaction by treating an appropriate aromatic aldehyde and 2-methylcyclohexanone, with an excess of ammonium acetate and malononitrile in boiling ethanolCitation21,Citation22. This reaction is of considerable importance since it is easy to perform, gives high yields, and is less time consuming compared with the traditional two-step procedure that involves the formation of the chalcone via Claisen-Schmidt condensation followed by cyclocondensation with malononitrile and ammonium acetateCitation23,Citation24.
Scheme 1. Reagents and reaction conditions: (i) 2-methylcyclohexanone (0.01 mol), approp. aldehyde (0.01 mol) malononitrile (0.01 mol), NH4OAc (0.08 mol) in Abs. EtOH (50 mL), reflux 3–6 h; (ii) approp. starting compd. (0.01 mol), formic acid (5 mL), heated in boiling water bath 30 min; (iii) approp. starting compd. (0.01 mol), Ac2O (5 mL), conc. H2SO4 (0.5 mL), heated in boiling water bath 10 min; (iv) approp. starting compd. (0.01 mol), PhNCS (0.01 mol) in pyridine (15 mL), reflux 2 h; (v) approp. starting compd. (0.02 mol), PhCONCS (0.02 mol), dry acetone (10 mL), reflux 3 h; (vi) approp. thioureido deriv. (0.02 mol), 1N NaOH (5.5 mL), EtOH (10 mL), heated in boiling water bath 30 min; (vii) approp. starting compd. (0.001 mol), urea or thiourea (0.005 mol), fused at 260–300 °C, sand bath 1 h; (viii) approp. starting compd. (0.001 mol), urea or thiourea (0.005 mol), heated in microwave reactor at 250 °C, 15 min; (ix) approp. thione (0.02 mol), 1N NaOH (5 mL), EtOH (2 mL), alkyl or aralkyl halide (0.022 mol), stirred at r.t., 2–3 h and (x) approp. starting compd. (0.01 mol), HCONH2 (10 mL), reflux, 2–3 h.
The IR spectra of these compounds revealed absorption bands at 3210–3450 characteristic for the NH2 and at 2214–2225 cm−1 attributed to the CN group. 1H-NMR of this series showed beside the aromatic protons at δ 7.11–7.42 a doublet of three protons intensity at δ 1.13–1.26 ppm (J = 10.0 Hz) for the CH3 group, a D2O exchangeable proton singlet of two proton intensity at δ 5.00–5.42 due to the NH2 group, as well as three multiplets at δ 1.53–1.70, 2.29–2.38 and 2.73–2.84 ppm for the cyclohexyl moiety. The structures were further supported from their 13C NMR spectral data that showed the expected number of aliphatic and aromatic carbons.
Reaction of the 2-aminohydroquinoline derivatives 1–5 with formic acid afforded the targeted 9-methyl-5-substituted-6,7,8,9-tetrahydro-3H-pyrimido[4,5-b]quinolin-4-ones (7–11). The IR spectra of these compounds were characterized by the absence of the CN group absorption and the presence of new sharp absorption bands at 1695–1715 cm−1 due to the new C=O groups. Moreover, the 1H-NMR spectra () showed beside the aromatic protons a doublet of three protons intensity in the range δ 1.31–1.38 ppm for the CH3 group as well as three multiplets at δ 1.53–1.70, 2.29–2.38 and 2.83–2.84 ppm corresponding to the protons of the cyclohexyl moiety. Further confirmation of the structure is supported by their 13C NMR spectral data (see Experimental section) that exhibited beside the expected number of aliphatic and aromatic carbons, signals at δ 169.2–170.5 ppm for the CO. Treatment the 2-aminohydroquinoline derivatives 2–6 with acetic anhydride in presence of few drops of concentrated sulphuric acid gave the corresponding 2,9-dimethyl-5-substituted-6,7,8,9-tetrahydro-3H-pyrimido[4,5-b]quinolin-4-ones 12–16. The IR spectra of these pyrimido-quinolin-4-one derivatives lacked the CN bands that were present in the starting quinolines and instead exhibited a carbonyl absorption bands at 1707–1715 cm−1. The 1H-NMR spectra showed new singlets at δ 2.45–2.31 ppm due to the new CH3 group introduced as well as the three multiplets for cyclohexyl moiety at δ 1.51–1.75, 2.24–2.36 and 2.72–2.88 ppm. Likewise, their 13C NMR spectral data exhibited beside the expected number of aliphatic and aromatic carbons, a new CH3 singlet at δ 19.5–20.2 ppm in addition to a CO signal at δ 168.9–170.7. Reacting the starting aminohydroquinolines 1–6 with the phenyl isothiocyanate in the presence of pyridine gave 4-imino-9-methyl-3-phenyl-5-substituted-3,4,6,7,8,9-hexahydro-1H-pyrimido[4,5-b]quinoline-2-thiones 17–22. Their IR spectra revealed two characteristic absorption bands at 1615–1629 cm−1 and 1195–1210 cm−1 due to the (C=N) and (C=S) groups, respectively. The 1H-NMR spectra showed along with multiplets of the cyclohexyl moiety a doublet of three protons at δ 1.34–1.42 ppm for the CH3 group. The structures of the above pyrimido[4,5-b]quinoline-2-thiones 17–22 were further substantiated by their 13C NMR spectra that exhibited two characteristic signals at 178.0–180.0 and δ 164.4–166.0 ppm for the CS and C=NH groups, respectively, in addition to the expected number of aliphatic (cyclohexyl) and aromatic carbons. On the other hand, when compounds 1–6 were reacted with benzoyl isothiocyanate in dry acetone, the corresponding substituted thioureido derivatives 23–28 were formed in good yields. The IR spectra of these compounds maintained the characteristic CN absorptions 2210–2220 cm−1 in addition to the two new bands at 1700–1710 and 1168–1185 cm−1 attributed to the C=O and C=S groups, respectively. The 13C NMR spectra of these compounds showed three signals at δ 117.2–118.1, 179.1–180.3 and 168.7–169.9 ppm for the CN, CS and C=NH groups, respectively. Moreover, the structure of the quinoline derivative 28 was also supported by X-ray crystallographyCitation25 (). Cyclization of compounds 23–27 was done by heating them with sodium hydroxide to form 4-amino-9-methyl-5-substituted-6,7,8,9-tetrahydro-1H-pyrimido[4,5-b]quinoline-2-thiones 29–33. It is worth mentioning that direct condensation of 1–5 either with thiourea or urea was attempted as another efficient procedure for a one-step synthesis of the target compounds 29–33 and 34–36, respectively. The IR spectra of these derivatives (29–33) were characterized by the disappearance of the cyano and the carbonyl absorptions and the appearance of broad absorption bands at 3339–3445 cm−1 due to amino group as well as a CS absorption at 1158–1172 cm−1. The 1H-NMR spectra showed beside the aromatic protons at δ 6.78–7.42, a doublet of three protons at intensity of δ 1.33–1.36 ppm for the CH3 group. Further confirmation for the structure was done from their 13C NMR spectral data that exhibited beside the expected number of aliphatic and aromatic carbons, a signal at δ 162.3–163.5 or at δ 182.4–184.6 for the CO (in compounds 29–33) and CS (in compounds 34–36), respectively. Thioakylation of the 2-thione derivatives 29–33 could be affected by treating these compounds with a variety of alkyl or aralkyl halides in the presence of sodium hydroxide to produce the alkylthio derivatives 37–47. Their IR spectra exhibited broad absorption bands at 3262–3338 cm−1 due to the amino group. The 1H-NMR spectra showed beside the aromatic protons at δ 6.84–7.62, a doublet of three protons with intensity at δ 1.30–1.35 ppm for the CH3 group, three multiplets for the cyclohexyl moiety as well as a broad singlet at δ 7.89–8.12 for the NH2 group, in addition the methylthio derivatives 39, 40, 43 and 45 that exhibited a singlet at δ 2.45–2.48 for the S-CH3 group while the ethylthio derivatives 37, 42 and 46 afforded a triplet of three proton intensity at δ 1.23–1.26 (J = 8.0 Hz) and a quartet of two proton intensity at δ 2.96–3.21 (J = 8.0 Hz) due to the CH3 and CH2 groups, respectively. Further confirmation for the structure was achieved by their 13C NMR spectral data that lacked the CS signals that existed in the original compounds and exhibited instead an S-CH3 signal at δ 18.5–19.2 in case of the S-CH3 derivatives and two signals at δ 15.5–15.8 and δ 27.8–28, in case of the S-ethyl compounds. Furthermore, heating the key intermediates 1–4 with formamide resulted in the formation of the targeted 4-amino-9-methyl-5-substituted-6,7,8,9-tetrahydro-pyrimido[4,5-b]quinolines 48–51. The IR spectra of these pyrimido[4,5-b]quinolines derivatives were characterized by the disappearance of the cyano groups absorptions and the appearance of broad absorption bands at 3265–3330 cm−1 due to the amino group. Their 1H-NMR spectra showed beside the aromatic protons at δ 6.77–7.45, a distinct doublet for three protons at δ 1.33–1.37 ppm for the CH3 group along with three multiplets at of the cyclohexyl moiety and a broad singlet at δ 7.92–8.22 for the NH2 group. Further structural assignments were done by their 13C NMR spectral data.
Figure 1. 1-Benzoyl-3-[3-cyano-8-methyl-4-(1-methyl-1H-pyrrol-2-yl)-5,6,7,8-tetrahydroquinolin-2-yl]thiourea.
![Figure 1. 1-Benzoyl-3-[3-cyano-8-methyl-4-(1-methyl-1H-pyrrol-2-yl)-5,6,7,8-tetrahydroquinolin-2-yl]thiourea.](/cms/asset/6ca507ba-b294-4853-94cd-4640502c2f5c/ienz_a_787421_f0002_b.jpg)
In vitro MTT cytotoxicity assay
Twenty-one analogs (1, 4, 5, 7, 8, 11, 12, 15, 16, 24, 25, 28, 30, 33, 39, 40, 42, 45, 46, 48 and 49) were selected to be evaluated for their in vitro cytotoxic effect via the standard MTT method against a panel of three human tumor cell lines: Caucasian breast adenocarcinoma MCF7, hepatocellular carcinoma HepG2 and colon carcinoma HT29. The results of LC50 (μM), which is the lethal concentration of the compound causing death of 50% of the cells in 24 h, are presented in .
The obtained data revealed that the three tested human tumor cell lines exhibited variable degree of sensitivity profiles towards ten of the tested compounds: 24, 25, 28, 30, 33, 39, 40, 42, 45 and 46. Among these, compounds 39, 40 and 45 showed strong activity against the human colon carcinoma HT29 cell line with LC50 values of 7.9, 10.1 and 11.5 μM, respectively. Moreover, a remarkable cytotoxic potential was displayed by compounds 42 and 46 against the same cell line (15.4 and 18.2 μM). Compounds 24, 25 and 28 revealed almost similar cytotoxicity profile against colon carcinoma HT29 with LC50 values of 23.6, 28.4 and 30.3 μM, respectively. However, compounds 30 and 33 were able to exhibit moderate activity against the same cell line with LC50 value range of 45.6 and 50.4 μM. Furthermore, the growth of the human hepatocellular carcinoma HepG2 cell line was found to be moderately inhibited by eight of the active compounds 24, 30, 33, 39, 40, 42, 45 and 46 with LC50 values ranging from 20.2 to 50.3 μM. Among these, the highest cytotoxic activity was displayed by compounds 39 and 40 (LC50 values of 20.2 and 24.6 μM, respectively). On the other hand, human breast cancer MCF 7 was proved to be the least sensitive among the cell lines tested as it was affected by only seven of the test compounds. However, an outstanding growth inhibition potential was shown by compounds 39, 40, 42, 45 and 46 as evidenced from their LC50 values (2.0, 2.8, 8.7, 3.8 and 9.4 μM, respectively). The remaining two active compounds, namely 24 and 33, showed moderate to mild activity against the same cell line with LC50 values of 49.2 and 40.4 μM, respectively. Further interpretation of the results revealed that compounds 39, 40, 42, 45 and 46 showed considerable broad spectrum of cytotoxic activity against the three tested human tumor cell lines. In particular, compound 39 proved to be the most active member in this study with a broad spectrum of activity against the tested cell lines, with special effectiveness against the human colon carcinoma HT29 and human breast cancer MCF 7 cell lines (LC50 values 7.9 and 2.0 μM, respectively). A close examination of the structure of the active compounds showed that the 4-bromophenyl counterpart at position 4 of the tetrahydroquinoline skeleton is the most favourable substituent when compared with other analogs. Moreover, the thioalkyl substituent at position 2 as in compounds 39, 40, 42, 45 and 46 was responsible for the high activity displayed by these analogs. Thiomethyl analogs, 39 and 40, displayed better profile when compared with a thioethyl derivative as in case of compound 42. Although compound 30 showed some activity because it carries a 4-bromophenyl substituent, but it lacks optimal activity owing to the presence of 2-thione and/or the absence of a thioalkyl substituent at position 2. Substitution at position 2 with a thiourea moiety or cyclization of these thiourea derivatives resulted in moderate to weakly active compounds 24, 25, 28, 30 and 33 whereas cyclization of the intermediate quinoline derivatives (1–6) with formic acid, acetic anhydride and formamide led to complete abolishment of the cytotoxic activity.
In vitro antibacterial and antifungal activities
As revealed from MIC data recorded in [MIC expressed in both concentration units μg/mL (μM)], 24 of the 51 newly synthesized compounds displayed variable inhibitory effects on the growth of the tested Gram-positive and Gram-negative microorganisms with pronounced activity against S. aureus and E. coli bacterial strains. In addition, some members exhibited moderate antifungal activity against C. albicans, whereas, all the tested compounds lacked antifungal activity against Aspergillus niger.
Among the tested Gram-positive bacterial strains, two organisms, namely S. aureus and B. subtilis, showed relatively high sensitivity towards the tested compounds. In this view, compound 39 was almost equipotent to ampicillin (MIC 6.25 μg/mL) against S. aureus, whereas the analogs 40, 42 and 46 (MIC 12.5 μg/mL) were 50% less active than ampicillin. Compounds 39 and 40 showed half the activity of ampicillin (MIC 25 μg/mL) against B. subtilis. However, these two compounds were able to produce a distinctive growth inhibitory profile against E. coli that was equipotent to ampicillin (MIC 6.25 μg/mL), whereas compounds 42, 45 and 46 showed 50% less activity than ampicillin (MIC 12.5 μg/mL) against the same organism. Meanwhile, the tested P. aeruginosa strain showed moderate sensitivity towards most of the active compounds, particularly compound 39 (MIC 12.5 μg/mL) which was equipotent to ampicillin.
Eleven compounds 18, 19, 30, 31, 33, 39, 40, 41, 42, 45 and 46 have displayed a significant growth inhibitory potential against C. albicans, among which compounds 39 and 46 (MIC 12.5 μg/mL) were the most active members when compared with clotrimazole (MIC 6.25 μg/mL). A close examination of the structures of the active compounds revealed that their antimicrobial activity is strongly related to the nature of the substituents at positions C-2 and C-4, together with the nature of the rings fused with the quinoline system. In general, it could be seen that potential antibacterial activity was connected with the electron-withdrawing group (R = 4-BrC6H4) at C-4, whereas moderate activity was displayed by the electron-donating (R = 4-CH3OC6H4) group at the same carbon. In this context, hexahydroquinoline, the key precursors 2, 3, 4, 5 and 6 showed moderate antimicrobial potential, with particular effectiveness against S. aureus and E. coli. Tetrahydropyrimido[4,5-b]quinolin-4(3H)-ones 7–11 obtained as a result of formic acid-induced cyclization led to the complete abolishment of the activity. However, acetic anhydride-derived cyclized products introduced a methyl group at position 2. In these series, two compounds, 12 and 13, were found to retain antimicrobial activity where the substituent R at position 4 is either 4-bromophenyl or 4-methoxyphenyl, although these two compounds are noticeably less active than the parent compounds 2 and 3. On the other hand, phenylisothiocyanate-derived cyclization gave rise to four active compounds 18, 19, 21 and 22 among which the bromo-derivative 18 was the most active one. Furthermore, reaction of the key intermediates, hexaquinoline derivatives with benzoylisothiocyanate, produced a series of compounds, out of which 24 and 25 showed slightly improved antimicrobial profiles when compared with 2 and 3. The cyclization of the thiourea derivatives, 23–27, to the corresponding 2-thiones, 29–33, however, did not change the biological profile significantly. Moreover, alkylation of the quinoline-2-thiones 29–33 resulted in an obvious enhancement of the antimicrobial activity as well as the spectrum as can be seen in 39, 40, 42, 45 and 46. It could be clearly recognized that the activity of these analogs is connected with the aryl substituent in position 4 as well as to the substituent linked to position 2. Within this series, compound 39 (R = 4-Br–C6H4) was the most active member as it displayed a fourfold increase in the antimicrobial activity against S. aureus, E. coli and P. aeruginosa (MIC 6.25, 6.25 and 12.5 μg/mL, respectively), together with a remarkable antifungal activity (12.5 μg/mL), when compared with the parent compound 2.
Conclusion
The present paper describes the synthesis of 2-amino-3-cyano-8-methyl-4-substituted-5,6,7,8-tetrahydroquinolines as well as derived fused-ring systems. Ten compounds showed remarkable cytotoxic efficiency against human colon carcinoma HT29, hepatocellular carcinoma Hep-G2 and Caucasian breast adenocarcinoma MCF7 cell lines. Compounds 24, 39, 40, 42, 45 and 46 showed considerable broad-spectrum cytotoxic activity among which 39 and 40 proved to be the most active derivatives. A close examination of the structure of the active compounds showed that the 4-bromophenyl counterpart of the tetrahydroquinoline skeleton is the most favourable substituent when compared with other analogs. Moreover, the thioalkyl substituent at position 2 as in compounds 39, 40, 42 and 45 was responsible for the high activity displayed by these analogs. However, the cyclization of the intermediate quinoline derivatives (1–6) with formic acid, acetic anhydride and formamide led to complete abolishment of activity. Likewise compounds 18, 19, 39, 40, 42, 45 and 46 were found to exhibit moderate antimicrobial activity and compounds 39, 40 and 46 proved to be the most active candidates. Compounds 39 and 40 could be considered as possible antimicrobial and anticancer candidates that deserve further investigation and derivatization in order to explore the scope and limitation of their biological activities.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Acknowledgements
The authors are deeply thankful to the authorities of the Bioassay-Cell Culture Laboratory, Drug Discovery Unit, National Research Centre, Cairo, Egypt, for their efforts in performing the MTT cytotoxicity assay.
References
- Goda FE, Badria FA. Synthesis and biological evaluation of certain new substituted pyrido[2,3-D]pyrimidin-4(1H)-one and pyrido[2,3-D]triazolo[3,4-B]pyrimidine analogs. Saudi Pharmaceutical J 2005;13:64–72
- Srivastava BK, Solanki M, Mishra B, et al. Synthesis and antibacterial activity of 4,5,6,7-tetrahydro-thieno[3,2-c]pyridine quinolones. Bioorg Med Chem 2007;15:1924–9
- Aanandhi MV, George S, Vaidhyalingam V. Synthesis and antimicrobial activities of 1-(5-substituted-2-oxoindolin-3-ylidene)-4-(substituted pyridin-2-yl)thio-semicarbazide. ARKIVOC 2008;2008(xi):187–94
- Goebel T, Ulmer D, Projahn H, et al. In search of novel agents for therapy of tropical diseases and human immunodeficiency virus. J Med Chem 2008;51:238–50
- Gudmundsson KS, Johns BA, Wang Z, et al. Synthesis of novel substituted 2-phenylpyrazolopyridines with potent activity against herpesviruses. Bioorg Med Chem 2005;13:5346–61
- Allen SH, Johns BA, Gudmundsson KS, et al. Synthesis of C-6 substituted pyrazolo[1,5-a]pyridines with potent activity against herpesviruses. Bioorg Med Chem 2006;14:944–54
- Kamal A, Khan MNA, Reddy KS, et al. Synthesis of a new class of 2-anilino substituted nicotinyl arylsulfonylhydrazides as potential anticancer and antibacterial agents. Bioorg Med Chem 2007;15:1004–13
- Perez-Rebolledo A, Ayala JD, de Lima GM, et al. Structural studies and cytotoxic activity of N(4)-phenyl-2-benzoylpyridine thiosemicarbazone Sn(IV) complexes. Eur J Med Chem 2005;40:467–72
- Poreba K, Opolski A, Wietrzyk J, et al. Synthesis and antiproliferative activity in vitro of new derivatives of 3-aminopyrazolo[3,4-b]pyridine. Part 1. Reaction of 3-aminopyrazolo[3,4-b]pyridine with 1,3-,1,4-diketones and α, β-unsaturated ketones. Arch Pharm Pharm Med Chem 2001;334:219–23
- Huang S, Lin R, Yu Y, et al. Synthesis of 3-(1H-benzimidazol-2-yl)-5-isoquinolin-4-ylpyra-zolo[1,2-b]pyridine, a potent cyclin dependent kinase 1 inhibitor. Bioorg Med Chem Lett 2007;17:1243–5
- Tatsumi K, Yamauchi T, Kiyono K, et al. 3-Cyano-2,6-dihydroxypyridine (CNDP), a new potent inhibitor of dihydrouracil dehydrogenase. J Biochem (Tokyo) 1993;114:912–18
- Zhu G-D, Gong J, Gandhi VB, et al. Design and synthesis of pyridine–pyrazolopyridine-based inhibitors of protein kinase B/Akt. Bioorg Med Chem 2007;15:2441–5
- Warshakoon NC, Wu S, Boyer A, et al. Design and synthesis of a series of novel pyrazolopyridines as HIF 1-α prolyl hydroxylase inhibitors. Bioorg Med Chem Lett 2006;16:5687–90
- Faidallah HM, Khan KA, Asiri AM. Synthesis of some new 2-oxo-1,4-disubstituted-1,2,5,6-tetrahydrobenzo[h]quinoline-3-carbonitriles and their biological evaluation as cytotoxic and antiviral agents. J Chem Sci 2012;124:625–31
- Rostom SAF, Faidallah HM, Al-Saadi MSM. A Facile synthesis of some 3-cyano-1,4,6-trisubstituted -2-(1H)-pyridinones and their biological evaluation as anticancer agents. Med Chem Res 2011;20:1260–72
- Faidallah HM, Rostom SAF, Asiri AM, et al. 3-Cyano-8-methyl-2-oxo-1,4-disubstituted-1,2,5,6,7,8-hexahydroquinolines: synthesis and biological evaluation as antimicrobial and cytotoxic agent. J Enzyme Inhibition Med Chem 2013; 28:123--30
- Faidallah HM, Rostom SAF, Khan KA, Basaif SA. Synthesis and characterization of some hydroxypyridone derivatives and their evaluation as antimicrobial agents. J Enzyme Inhibition Med Chem 2013; 28:495--508
- Mosmann T. Rapid colourimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63
- Denizot F, Lang R. Rapid colourimetric assay for cellular growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods 1986;22:271–7
- Scott AC. In: Collee JG, Duguid JP, Fraser AG, Marmion BP, eds. Mackie & McCartney practical medical microbiology. 13th ed. New York: Churchill Livingstone; 1989:161–81
- Khaksar S, Yaghoobi M. A concise and versatile synthesis of 2-amino-3-cyanopyridine derivatives in 2,2,2-trifluoroethanol. J Fluorine Chem 2012;142:41–4
- Wan Y, Yuan R, Zhang F-R, et al. One-pot synthesis of N2-substituted 2-amino-4-aryl-5,6,7,8-tetrahydroquinoline-3-carbonitrile in basic ionic liquid [bmim]OH. Synth Commun 2011;41:2997–3015
- Paul S, Gupta R, Loupy A. Improved synthesis of 2-amino-3-cyanopyridines in solvent free conditions under microwave irradiation. J Chem Res (Synop) 1998;330–1
- Elkholy YM, Morsy MA. Facile synthesis of 5,6,7,8-tetrahydropyrimido [4,5-b]-quinoline derivatives. Molecules 2006;11:890–903
- Asiri AM, Faidallah HM, Al-Youbi AO, et al. 1-Benzoyl-3-[3-cyano-8-methyl-4-(1-methyl-1H-pyrrol-2-yl)-5,6,7,8-tetra-hydro-quinolin-2-yl]thiourea. Acta Crystallogr Sect E: Struct Rep. Online 2011;E67:o2430