
Abstract
Crocus sativus L. is known in herbal medicine for the various pharmacological effects of its components, but no data are found in literature about its biological properties toward Helicobacter pylori, Plasmodium spp. and Leishmania spp. In this work, the potential anti-bacterial and anti-parasitic effects of crocin and safranal, two important bioactive components in C. sativus, were explored, and also some semi-synthetic derivatives of safranal were tested in order to establish which modifications in the chemical structure could improve the biological activity. According to our promising results, we virtually screened our compounds by means of molecular modeling studies against the main H. pylori enzymes in order to unravel their putative mechanism of action.
Introduction
One of the most important microbial infections affecting the human stomach is due to Helicobacter pylori, a Gram-negative bacterium that inhabits the inner mucosa, causing not only gastritis but also gastroduodenal ulcers and, in the more severe stages, tumors. One of the reasons for the failure of H. pylori eradication in many countries is the increasing antibiotic resistanceCitation1. Among the new strategies for the treatment of H. pylori infection, it can be mentioned the inhibition of ureaseCitation2,Citation3, an enzyme that permits the survival of H. pylori in the harsh acidic environment of the stomach and its colonization of the gastric mucosa, by neutralization of the gastric acidity thanks to the ammonia production from host urea. Several natural and synthetic compounds such as sulforaphane, an isothiocyanate extracted from crucifersCitation4, allyl isothiocyanatesCitation5, flavonoids and their corresponding reductive derivativesCitation6, quercetin and its analoguesCitation7 and some synthetic thiosemicarbazonesCitation3 are effective urease inhibitors. Instead, caffeic acid phenethyl ester, one of the principal components of propolis, was found to be a competitive inhibitor of H. pylori peptide deformylase (HpPDF), an enzyme that catalyzes the removal of the formyl group from the N-terminus of nascent polypeptide chains, which is essential for H. pylori survivalCitation8. Other targets for the treatment of H. pylori infection are as follows: (1) the type II dehydroquinase (DHQ2), the third enzyme of the shikimic acid pathwayCitation9; (2) glutamate racemase, which provides d-glutamate for the construction of N-acetylglucosamine-N-acetylmuramic acid peptidoglycan subunitsCitation10; (3) H. pylori β-hydroxyacyl-ACP (FabZ), an important enzyme involved in the bacterial type II fatty acid synthetic pathwayCitation11,Citation12; (4) H. pylori phosphopantetheine adenylyltransferase (HpPPAT), which catalyzes the penultimate step in coenzyme A biosynthesisCitation13; (5) the class II fructose-bisphosphate (FBP) aldolase, an enzyme involved in microbial glycolysis and gluconeogenesisCitation14; and (6) NADPH oxidaseCitation15.
Among the parasitic infections, malaria has always attracted attention because of the increasing resistance of Plasmodium falciparum and Plasmodium vivax parasites to traditional drugs; therefore, nowadays research is focused on the development of drugs and vaccines with the aim of targeting not only the blood stage of infection but also the pre-erythrocytic and sexual stagesCitation16. Beside artemisinin and quinine (found in Artemisia annua and Cinchona spp., respectively), and their semi-synthetic derivatives that represent the most important drugs in current therapies, additional natural products content in Amazonian plants possess anti-plasmodial effects against P. falciparum and P. vivaxCitation17. It is noteworthy that crude plant extracts could have a better anti-plasmodial activity than the individual components at the same dose. This effect is due to the pharmacodynamic synergy that could be found between Cinchona alkaloids, and/or to the pharmacokinetic interactions such as those between natural products in A. annua tea, making artemisinin more rapidly absorbed than the single drugCitation18.
Another disease caused by parasite infection is leishmaniasis, which is present especially in poor populations characterized by low healthcare conditions like some disadvantaged regions of South America, Africa and Asia. Leishmaniasis is caused by protozoan parasites whose transmission is through the bite of the phlebotomine sand fly: cutaneous leishmaniasis is mostly due to Leishmania major infection, while visceral leishmaniasis is due to Leishmania donovani, widespread in India and East Africa. Anti-leishmanial drugs used in current therapies, such as meglumine antimoniate, sodium stibogluconate, pentamidine, miltefosine and amphotericin B, cause several side effects and are too expensive for these populations. Hence, in the research of new naturally occurring anti-leishmanial drugs, we can mention chloroform extract of Valeriana wallichii roots, thanks to the caffeic acid bornyl esterCitation19 and natural product-inspired quinazolinone hybridsCitation20. Disuccinyl betulin, diglutaryl dihydrobetulin and disuccinyl dihydrobetulin, derivatives of the natural triterpene betulin present in the Betula spp. cork layer, inhibit the relaxation activity of the enzyme IB type topoisomerase of L. donovani; this effect decreases the intracellular parasites number in macrophagesCitation21.
With the aim of finding new natural or semi-synthetic antimicrobial drugs, we focused our attention on Crocus sativus L. (also called saffron), that is a herbaceous perennial-cormous plant containing in the stigmas a great variety of bioactive compounds: the volatile oil safranal, a carotenoid derivative (crocetin) and its di-glycosidic esters (crocins), picrocrocin and still other components such as anthocyanins, flavonoids, vitamins, amino acids and proteins. In fact, in addition to its use as food additive, saffron is also known in natural medicine for improving memory and learning abilities and for the treatment of sick, spasm, asthma, bronchitis, fever, colds and cardiovascular diseases; furthermore, it also possesses anticancer, antioxidant, hypolipemic and anticonvulsant activitiesCitation22. Despite all these pharmacological effects, few data are available in literature, regarding exclusively the activity of its aqueous and methanolic extracts, about antimicrobial and anti-parasitic activities (Bacillus subtilis and cereus, Staphylococcus aureus and Staphylococcus pyogenes, Escherichia coli, Pseudomonas aeruginosa, Cladosporium herbarum, Brucella melitensis, Candida albicans, Cryptococcus neoformans, Trichophyton rubrum, Aspergillus fumigatus, Pyricularia oryza, and L. major)Citation23,Citation24. Given our interest in this fieldCitation25–28, we explored the potential anti-H. pylori, anti-malarial and anti-leishmanial effects of crocin (1) and safranal (2), which are two important products found in C. sativus, and tested some semi-synthetic derivatives of safranal (3–9, Scheme 1) in order to establish, with the support of computational studies, which modifications in the chemical structures could improve their biological activity to provide new leads for the development of promising antimicrobial and anti-parasitic agents. In addition, we obtained a direct correlation between specific biological activity and active principles of this spice.
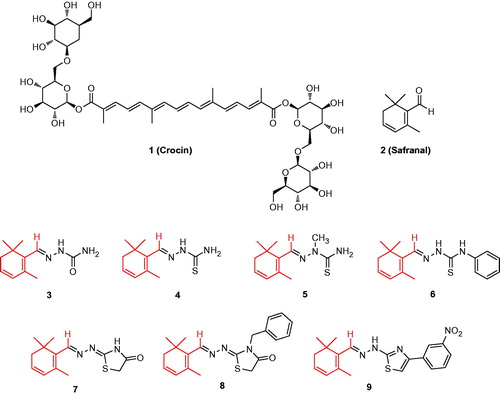
Scheme 1. Structures of bioactive compounds (crocin 1 and safranal 2) of Crocus sativus L. and semi-synthetic safranal derivatives 3–9.
Materials and methods
Chemistry
Commercial samples of crocin (crocin-1, crocetin di-gentiobiose ester) and safranal (>88%) were purchased from Sigma-Aldrich (Milan, Italy). Safranal was further purified by column chromatography on silica gel (230–400 mesh, G60 Merck, ethyl acetate:n-hexane 1:3). The other compounds (3–9) have been synthesized and characterized to ensure purity as reported in the literatureCitation29.
Antimicrobial studies
H. pylori culture
Nineteen clinical strains of H. pylori, isolated from patients with duodenal ulcer or gastritis, and reference strain ATCC 43504 were used for this study. The H. pylori strains were maintained at −80 °C in Wilkins–Chalgren broth with 10% (v/v) horse serum (Seromed) and 20% (v/v) glycerol (Merck) until required for the experiments. Before being used, the bacteria were subcultured twice on Columbia agar base (Difco Laboratories) supplemented with 10% horse serum and 0.25% Bacto yeast extract (Difco). Plates were incubated for 72 h at 37 °C in an atmosphere of 10% CO2 in a gas incubator.
Anti-H. pylori activity
MICs were determined by modified broth dilution method as previously describedCitation30. Briefly, an inoculum equivalent to 1 McFarland standard was prepared in Wilkins–Chalgren broth and diluted in MegaCell™ RPMI-1640 medium. Each well was inoculated with H. pylori at a final concentration of approximately 5 × 105 CFU/well. The plates were incubated at 37 °C under microaerophilic conditions (10% CO2 in a gas incubator) and examined after 72 h of incubation. In order to confirm MIC results, a comparative evaluation with agar dilution methodCitation31 was also performed. The MICs obtained for each compounds by microdilution test were the same to those obtained by agar dilution (data not shown).
Plasmodium falciparum cultures and drug susceptibility assay
Plasmodium falciparum cultures were carried out according to Trager and Jensen with slight modificationsCitation32. The CQ-susceptible strain D10 and the CQ-resistant strain W2 were maintained at 5% hematocrit (human type A-positive red blood cells) in RPMI-1640 (EuroClone, Celbio) medium with the addition of 1% AlbuMax (Invitrogen, Milan, Italy), 0.01% hypoxanthine, 20 mM Hepes and 2 mM glutamine. All the cultures were maintained at 37 °C in a standard gas mixture consisting of 1% O2, 5% CO2 and 94% N2. Compounds were dissolved in either water or DMSO and then diluted with medium to achieve the required concentrations (final DMSO concentration <1%, which is non-toxic to the parasite). Drugs were placed in 96-well flat-bottomed microplates (COSTAR) and serial dilutions made. Asynchronous cultures with parasitemia of 1–1.5% and 1% final hematocrit were aliquoted into the plates and incubated for 72 h at 37 °C. Parasite growth was determined spectrophotometrically (OD650) by measuring the activity of the parasite lactate dehydrogenase, according to a modified version of the method of Makler & Hinrichs in control and drug-treated culturesCitation33. The anti-malarial activity is expressed as 50% inhibitory concentrations (IC50); each IC50 value is the mean and standard deviation of at least three separate experiments performed in duplicate.
Evaluation of anti-leishmanial activity
Promastigote stage of Leishmania infantum strain MHOM/TN/80/IPT1 (kindly provided by Dr. M. Gramiccia and Dr. T. Di Muccio, ISS, Roma) and clinical isolate of Leishmania tropica (MHOM/IT/2012/ISS3130) were cultured in RPMI-1640 medium (EuroClone) supplemented with 15% heat-inactivated fetal calf serum (EuroClone), 20 mM Hepes and 2 mM l-glutamine at 22 °C.
To estimate the IC50, the 3-[4.5-dimethylthiazol-2-yl]-2.5-diphenyltetrazolium bromide (MTT) methodCitation34 was used with modifications. Compounds were dissolved in DMSO and then diluted with medium to achieve the required concentrations. Drugs were placed in 96-well round-bottom microplates and seven serial dilutions were made. Amphotericin B was used as the reference anti-Leishmania drug. Parasites were diluted in complete medium to 5 × 106 parasites/mL, and 100 μL of the suspension was seeded into the plates, incubated at 22 °C for 72 h and then 20 µL of MTT solution (5 mg/mL) was added into each well for three hours. The plates were then centrifuged, the supernatants discarded and the resulting pellets dissolved in 100 µL of lysing buffer consisting of 20% (w/v) of a solution of SDS (Sigma), 40% of N,N-dimethylformamide (Merck) in H2O. The absorbance was measured spectrophotometrically at a test wavelength of 550 nm and a reference wavelength of 650 nm. The results are expressed as IC50, which is the dose of compound necessary to inhibit cell growth by 50%; each IC50 value is the mean ± standard deviation of at least three separate experiments performed in duplicate.
Molecular modeling studies
Docking experiments were performed using the X-ray crystallographic structures of six new strategic targets for the treatment of H. pylori infection, downloaded from the Protein Data Bank site (PDB)Citation35 (Table S1). The treatment of X-ray enzyme structures was carried out by “Protein Preparation Wizard”Citation36 function: non-protein residues were removed, hydrogen atoms were added and finally, energy minimization was performed until the root-mean squared deviation (RMSD) of all heavy atoms was within 0.3 Å X-ray PDB model. Both E and Z isomers of the 3D semisynthetic derivatives 4, 5 and 9 were prepared, before the docking simulations, using the latest version of the software Maestro GUI 9.7 in the Schrodinger Suite 2014Citation37. For each molecule, several protonated and tautomeric forms were calculated by means of the “LigPrep” moduleCitation38 and used as starting points for the molecular recognition studies. Glide 6.2 SPCitation39 protocol was applied to dock the flexible 3D ligands within the active sites of every rigid target. The “Receptor Grid Generation” panel was used to definite a cube of length 25 Å as binding region, centered on the former positions of the removed X-ray co-crystallized inhibitors, into each catalytic site. Low values of RMSD between experimental and docked structures, after re-docking procedure, have been shown the good ability of Glide protocol to accurately predict the ligand binding conformations (as shown in Table S1). Ligand-binding energies (dG Bind) in Kcal/mol were estimated by “Prime MM-GBSA” utilityCitation40. With regard to FabZ and HpPDF enzymes, the most stable conformations of docked compounds were submitted to a molecular dynamic (MD) simulation of 1.0 ns carried out by Desmond packageCitation41 and were also analyzed using “Simulation Interactions Diagram” panel. All figures were generated by means of the Pymol 1.7.0.0 version molecular graphic systemCitation42.
Results and discussion
Starting from the commercially available natural product safranal (2), purified by column chromatography, we previously synthesized semicarbazone (3), thiosemicarbazone (4–6), thiazolidinone (7,8) and hydrazothiazole (9) derivatives. For these products, the safranal nucleus was maintained to establish the impact of the chemical modifications, such as the increasing steric hindrance and the different substituents, on the tested biological activity.
Concerning the biological activity of compounds 1–9 against H. pylori strains (), safranal (2) showed an MIC50 value of 32 µg/mL against some strains while its semi-synthetic derivatives 4, 5 and 9 showed an effective anti-bacterial activity; and for some strains, they are markedly more active than the reference drugs, metronidazole (>32 µg/mL) and clarithromycin (≥32 µg/mL). In detail, the two thiosemicarbazonic derivatives (4 and 5) proved their activity in the range of 4–8 µg/mL (MIC50 and MIC90, respectively), while the most effective compound is the hydrazothiazole 9, which contains a phenyl moiety substituted with a nitro group at C3, with a biological activity ranging from 2 to 4 µg/mL (MIC50 and MIC90, respectively), suggesting that the presence of the hydrazone moiety could be important for the explication of the anti-bacterial effect.
Table 1. Anti-bacterial activity (µg/mL) of compounds 1–9 and reference drugs against Helicobacter pylori strains.
With the purpose to elucidate a putative mechanism of action for the active semi-synthetic derivatives of safranal (4, 5 and 9), a virtual screening against some H. pylori targets was performed. Therefore, six enzyme crystal structures were considered for the docking simulations downloaded from the PDBCitation35: DHQ2, FabZ, FBP aldolase, glutamate racemase, HpPDF and urease represent, in fact, new strategic targets for the treatment of the resistant H. pylori infection. Protein X-ray models were selected, for the structure-based virtual screening, considering low values of resolution and the presence of a co-crystallized inhibitor (Table S1).
No crystallographic models of HpPPAT are deposited in the PDB, for this reason, it has not been considered for the molecular recognition studies. For each selected target, both E and Z isomers of the 3D molecules were docked into the binding pocket by means of Glide 6.2 SPCitation39 protocol, and the binding energy values (dG Bind in Kcal/mol), for the best produced ligand-protein complexes, were estimated by Prime-MMGBSA utilityCitation40 considering as final score the average of the two isomers values.
The hydrazothiazole 9 showed the highest dG Bind value of −81.94 Kcal/mol for the FabZ enzyme, conversely the thiosemicarbazonic derivatives 4 and 5, that displayed also a good accommodation into the binding pocket of this target, gave the best results for the HpPDF with the respective dG bind values −54.32 and −56.49 Kcal/mol (Table S2).
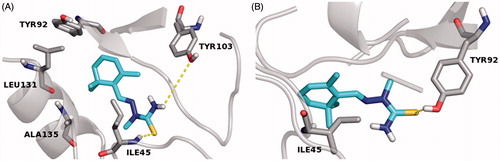
In order to investigate the stereochemical requirements on the basis of a possible inhibition mechanism, a MD of 1.0 ns were also performed in silico, by Desmond packageCitation41, to gain insight into the interactions of E–Z isomers with the FabZ and HpPDF amino acids residues lining the catalytic sites. With regard to the FabZ MD analysis, carried out by the “Simulation Interaction Diagram” panel, the last frames of the semisynthetic isomers revealed a possible target inhibition by a competitive mechanism of action. Moreover, for compound 9, a further investigation suggested that the E isomer is localized inside the binding cavity with the nitrophenyl and the hydrazothiazole (tautomeric form) moieties sandwiched between the entrance residues TYR100 and PRO112′ by π–π interactions. The stabilization of both the 9 E–Z isomers is associated with several hydrophobic contacts formed by residues of the lipophilic tunnel entrance (), by contrast the active compounds 4 and 5 establish only fewer H-bond contacts with HIS58 and GLU72, located near the FabZ active site, and no hydrophobic interactions with the key TYR100 residue (Figures S1 and S2)Citation11. Taking into account the HpPDF MD analysis, all compounds showed the same binding mode within the catalytic site of the target. In agreement with the theoretical data reported in Table S2, the ligand–HpPDF complexes of the derivatives 4 and 5 are stabilized by a great number of hydrophobic/H-bond interactions involving the thiosemicarbazide moiety: in this case, ILE45, TYR92, TYR103, LEU131 and ALA135 residues, forming the lipophilic task, could play an important role for the ligand binding mode ( and ). However, the hydrazothiazole 9 might be a better HpPDF inhibitor than the derivatives 4 and 5: its E–Z isomers make not only hydrophobic but also H-bond contacts by the hydrazonic moiety and one ionic interaction, for the E isomer, by the nitro group of the phenyl substituent (Figure S3).
Figure 1. MD simulation last frame of both E (A) and Z (B) isomers of compound 9 and their hydrophobic contacts into the FabZ binding site. The E–Z isomers are rendered as carbon-pink sticks, the pocket amino acids are labeled as carbon-gray sticks and the target is described as gray cartoon. All non-carbon atoms are colored according to atom types. For the colored version of this Figure, please refer to the online article.
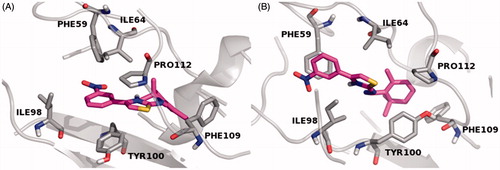
Figure 2. MD simulation last frame of both E (A) and Z (B) isomers of compound 4 and their H-bond/hydrophobic interactions within the HpPDF binding site. The E–Z isomers are rendered as carbon-green sticks, the pocket amino acids are labeled as carbon-gray sticks and the target is described as gray cartoon. All non-carbon atoms are colored according to atom types. For the colored version of this Figure, please refer to the online article.
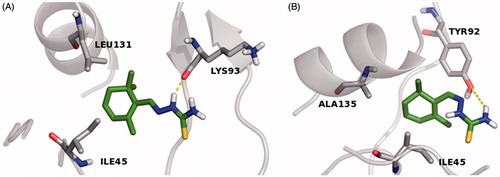
Figure 3. MD simulation last frame of both E (A) and Z (B) isomers of compound 5 and their H-bond/hydrophobic interactions within the HpPDF binding site. The E–Z isomers are rendered as carbon-cyan sticks, the pocket amino acids are labeled as carbon-gray sticks and the target is described as gray cartoon. All non-carbon atoms are colored according to atom types. For the colored version of this Figure, please refer to the online article.
On the basis of these virtual screening studies, we concluded that a probably enzymatic inhibition might be involved in the biological activity of safranal derivatives against the H. pylori strains, and the hydrazothiazole 9 might be considered as a new hit compound for future investigation and enzymatic assays.
With regard to the anti-malarial activity of these products (), crocin (1) displayed a biological effect against chloroquine-sensitive D10 strain (IC50 = 18.93 µg/mL). The (thio)semicarbazone derivatives 3, 4 and 5 were active against both chloroquine-sensitive D10 strain and chloroquine-resistant W2 strains (IC50 = 13.26 and 10.28 µg/mL, 10.15 and 13.07 µg/mL, 6.01 and 6.75 µg/mL, respectively), while 4-phenilthiosemicarbazone 6 only inhibited D10 strain (IC50 = 4.92 µg/mL). Unlike the thiazolidinone 7, which was not active, the corresponding N-benzylated compound 8 showed an inhibitory effect of 5.98 and 7.22 µg/mL against D10 and W2 strains, respectively. Furthermore, the hydrazothiazole 9 displayed its activity against both strains (IC50 = 8.53 µg/mL against D10 and 7.22 µg/mL against W2). With regard to the anti-leishmanial effect, thiosemicarbazone 4 displayed an IC50 value of 11.51 µg/mL toward L. tropica, whereas the N-methylated 5 was active not only against L. tropica (IC50 = 14.69 µg/mL) but also against L. infantum (IC50 = 12.50 µg/mL). Thiazolidinone 7 showed an IC50 of 15.64 µg/mL against L. tropica while its derivative 8, a benzylated substituent at the lactamic nitrogen, was more active than the others toward L. tropica (IC50 = 1.98 µg/mL) and also on L. infantum with an IC50 of 6.10 µg/mL. These encouraging results showed that safranal and its semi-synthetic derivatives could be further investigated to develop new anti-H. pylori and anti-parasitic agents.
Table 2. Anti-malarial activity (µg/mL) against chloroquine-sensitive D10 strain and chloroquine-resistant W2 strain and anti-leishmanial activity (µg/mL) of compounds 1–9 and reference drugs.
Conclusions
Many reports have explained the great variety of biological activities possessed by saffron and its natural components, but no data in literature are present with regard to the anti-bacterial activity against H. pylori and the anti-parasitic activity against Plasmodium and Leishmania. Starting from this point, we assessed the anti-H. pylori, anti-malarial and anti-leishmanial effects of two natural products found in C. sativus L., crocin (1) and safranal (2), and of the semi-synthetic safranal derivatives (3–9). The thiosemicarbazonic derivatives 4 and 5 and the (thiazol-2-yl)hydrazonic compound 9 exhibited the best anti-H. pylori activities and a possible mechanism of action, corroborated by molecular modeling studies, might be the inhibition of several targets as FabZ and HpPDF. With regard to the anti-parasitic effect, compounds 5, 6, 8 and 9 possessed moderate anti-malarial activity, whereas the N-benzylated thiazolidinone 8 has proven to be the best anti-leishmanial agent of the series. These biological studies, supported by molecular modeling studies, demonstrated that unsaturated nucleus of safranal could have the pharmacophoric prerequisites to provide new leads for the development of promising agents for the management of H. pylori and parasitic infections caused by Plasmodium and Leishmania.
Supplementary material available online
Supplementary Tables S1 and 2; Figures S1-3 and Supporting information.
Supplemental Material.pdf
Download PDF (114.4 KB)Acknowledgements
The authors wish to thank Dr. Claudio Farina for providing three clinical isolates of Helicobacter pylori used in this study.
Declaration of interest
The authors report no declaration of interest.
This work was supported by Interregional Research Center for Food Safety and Health at the Magna Græcia University of Catanzaro (MIUR PON a3_00359).
References
- Cui R, Zhou L. Helicobacter pylori infection: an overview in 2013, focus on therapy. Chin Med J 2014;127:568–573
- Ardekani LS, Gargari SL, Rasooli I, et al. A novel nanobody against urease activity of Helicobacter pylori. Int J Infect Dis 2013;17:e723–8
- Aslam MA, Mahmood SU, Shahid M, et al. Synthesis, biological assay in vitro and molecular docking studies of new Schiff base derivatives as potential urease inhibitors. Eur J Med Chem 2011;46:5473–9
- Fahey JW, Stephenson KK, Wade KL, Talalay P. Urease from Helicobacter pylori is inactivated by sulforaphane and other isothiocyanates. Biochem Biophys Res Commun 2013;435:1–7
- Shin IS, Masuda H, Naohide K. Bactericidal activity of wasabi (Wasabia japonica) against Helicobacter pylori. Int J Food Microbiol 2004;94:255–61
- Xiao ZP, Peng ZY, Dong JJ, et al. Synthesis, structure-activity relationship analysis and kinetics study of reductive derivatives of flavonoids as Helicobacter pylori urease inhibitors. Eur J Med Chem 2013;63:685–95
- Xiao ZP, Wang XD, Peng ZY, et al. Molecular docking, kinetics study, and structure-activity relationship analysis of quercetin and its analogous as Helicobacter pylori urease inhibitors. J Agric Food Chem 2012;60:10572–7
- Cui K, Lu W, Zhu L, et al. Caffeic acid phenethyl ester (CAPE), an active component of propolis, inhibits Helicobacter pylori peptide deformylase activity. Biochem Biophys Res Commun 2013;435:289–94
- Paz S, Tizón L, Otero JM, et al. Tetrahydrobenzothiophene derivatives: conformationally restricted inhibitors of type II dehydroquinase. Chem Med Chem 2011;6:266–72
- Basarab GS, Hill P, Eyermann CJ, et al. Design of inhibitors of Helicobacter pylori glutamate racemase as selective antibacterial agents: incorporation of imidazoles onto a core pyrazolopyrimidinedione scaffold to improve bioavailabilty. Bioorg Med Chem Lett 2012;22:5600–7
- Zhang L, Kong Y, Wu D, et al. Three flavonoids targeting the beta-hydroxyacyl-acyl carrier protein dehydratase from Helicobacter pylori: crystal structure characterization with enzymatic inhibition assay. Protein Sci 2008;17:1971–8
- Chen J, Zhang L, Zhang Y, et al. Emodin targets the beta-hydroxyacyl-acyl carrier protein dehydratase from Helicobacter pylori: enzymatic inhibition assay with crystal structural and thermodynamic characterization. BMC Microbiol 2009;9:91
- Cheng CS, Jia KF, Chen T, et al. Experimentally validated novel inhibitors of Helicobacter pylori phosphopantetheine adenylyltransferase discovered by virtual high-throughput screening. PLoS One 2013;8:e74271
- Fonvielle M, Coinçon M, Daher R, et al. Synthesis and biochemical evaluation of selective inhibitors of class II fructose bisphosphate aldolases: towards new synthetic antibiotics. Chemistry 2008;14:8521–9
- Cho SO, Lim JW, Kim H. Red ginseng extract inhibits the expression of MCP-1 and iNOS in Helicobacter pylori-infected gastric epithelial cells by suppressing the activation of NADPH oxidase and Jak2/Stat3. J Ethnopharmacol 2013;150:761–4
- Kappe SH, Vaughan AM, Boddey JA, Cowman AF. That was then but this is now: malaria research in the time of an eradication agenda. Science 2010;328:862–6
- Pohlit AM, Lima RB, Frausin G, et al. Amazonian plant natural products: perspectives for discovery of new antimalarial drug leads. Molecules 2013;18:9219–40
- Rasoanaivo P, Wright CW, Willcox ML, Gilbert B. Whole plant extracts versus single compounds for the treatment of malaria: synergy and positive interactions. Malar J 2011;10:S1–4
- Glaser J, Schultheis M, Hazra S, et al. Antileishmanial lead structures from nature: analysis of structure-activity relationships of a compound library derived from caffeic acid bornyl ester. Molecules 2014;19:1394–410
- Sharma M, Chauhan K, Shivahare R, et al. Discovery of a new class of natural product-inspired quinazolinone hybrid as potent antileishmanial agents. J Med Chem 2013;56:4374–92
- Chowdhury S, Mukherjee T, Sengupta S, et al. Novel betulin derivatives as antileishmanial agents with mode of action targeting type IB DNA topoisomerase. Mol Pharmacol 2011;80:694–703
- Bathaie SZ, Mousavi SZ. New applications and mechanisms of action of saffron and its important ingredients. Crit Rev Food Sci Nutr 2010;50:761–86
- (a) Sengul M, Yildiz H, Gungor N, et al. Total phenolic content, antioxidant and antimicrobial activities of some medicinal plants. Pak J Pharm Sci 2009;22:102–106; (b) Zheng CJ, Li L, Ma WH, et al. Chemical constituents and bioactivities of the liposoluble fraction from different medicinal parts of Crocus sativus. Pharm Biol 2011;49:756–63; (c) Motamedi H, Daabpou E, Gholipour M, et al. In vitro assay for the anti-brucella activity of medicinal plants against tetracycline-resistant Brucella melitensis. J Zhejiang Univ Sci B 2010;11:506–11
- Yousefi E, Eskandari A, Gharavi MJ, Khademvatan S. In vitro activity and cytotoxicity of Crocus sativus extract against Leishmania major (MRHO/IR/75/ER). Infect Dis – Drug Targets 2014;14:56–60
- Chimenti F, Bizzarri B, Bolasco A, et al. Synthesis and in vitro selective anti-Helicobacter pylori activity of N-substituted-2-oxo-2H-1-benzopyran-3-carboxamides. Eur J Med Chem 2006;41:208–12
- Chimenti F, Bizzarri B, Bolasco A, et al. A novel class of selective anti-Helicobacter pylori agents 2-oxo-2H-chromene-3-carboxamide derivatives. Bioorg Med Chem Lett 2007;17:3065–71
- Chimenti F, Bizzarri B, Bolasco A, et al. Synthesis, selective anti-Helicobacter pylori activity, and cytotoxicity of novel N-substituted-2-oxo-2H-1-benzopyran-3-carboxamides. Bioorg Med Chem Lett 2010;20:4922–6
- Chimenti F, Bizzarri B, Bolasco A, et al. Synthesis and anti-Helicobacter pylori activity of 4-(coumarin-3-yl)thiazol-2-ylhydrazone derivatives. J Heterocyclic Chem 2010;47:1269–74
- De Monte C, Carradori S, Chimenti P, et al. New insights into the biological properties of Crocus sativus L.: chemical modifications, human monoamine oxidases inhibition and molecular modelling studies. Eur J Med Chem 2014;82:164–71
- Sisto F, Scaltrito MM, Russello G, et al. Antimicrobial susceptibility testing of Helicobacter pylori determined by microdilution method using a new medium. Curr Microbiol 2009;58:559–63
- National Committee for Clinical Laboratory Standards. Methods for antimicrobial susceptibility testing of anaerobic bacteria. Approved standard M11-A6. 6th ed. Villanova, PA: National Committee for Clinical Laboratory Standards; 2004
- Trager W, Jensen JB. Human malaria parasites in continuous culture. Science 1976;193:673–5
- Makler MT, Hinrichs DJ. Measurement of the lactate dehydrogenase activity of Plasmodium falciparum as an assessment of parasitemia. Am J Trop Med Hyg 1993;48:205–10
- Sereno D, Lemesre JL. Use of an enzymatic micromethod to quantify amastigote stage of Leishmania amazonensis in vitro. Parasitol Res 1997;83:401–3
- Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res 2000;28:235–42
- Schrödinger Suite 2013 Protein Preparation Wizard; Epik version 2.7, Impact version 6.2, Prime version 3.4. New York (NY): Schrödinger, LLC; 2013
- Maestro, version 9.7. New York (NY): Schrödinger, LLC; 2014
- LigPrep, version 2.9. New York (NY): Schrödinger, LLC; 2014
- Glide, version 6.2. New York (NY): Schrödinger, LLC; 2014
- Prime version 3.4. New York (NY): Schrödinger, LLC; 2014
- (a) Desmond Molecular Dynamics System, version 3.7. New York (NY): D. E. Shaw Research; 2014. (b) Maestro-Desmond Interoperability Tools, version 3.7. New York (NY): Schrödinger; 2014
- The PyMOL Molecular Graphics System, Version 1.7.0.0. New York (NY): Schrödinger, LLC