
Abstract
A series of hydroxy and phenolic compounds have been assayed for the inhibition of two physiologically relevant carbonic anhydrase (CA, EC 4.2.1.1) isozymes, the cytosolic human isozymes I and II. The investigated molecules showed inhibition constants in the range of 1.07–4003 and 0.09–31.5 μM at the hCA I and hCA II enzymes, respectively. In order to investigate the binding mechanisms of these inhibitors, in silico studies were also applied. Molecular docking scores of the studied compounds are compared using three different scoring algorithms, namely Glide/SP, Glide/XP and Glide/IFD. In addition, different ADME (absorption, distribution, metabolism and excretion) analysis was performed. All the examined compounds were found within the acceptable range of pharmacokinetic profiles.
Introduction
The carbonic anhydrases (CAs, EC 4.2.1.1) are zinc-containing metaloenzymes which participate in the maintenance of pH homeostasis in the human body, catalyzing the reversible hydration of carbon dioxide in a two-step reaction, to yield bicarbonate and protonsCitation1–3. So far, 16 CA isozymes have been described, that differ in their subcellular localization and catalytic activity. These isozymes play important roles in different tissuesCitation2–5. CAs constitute interesting targets for the design of pharmacological agents that are useful in the treatment or prevention of a variety of disorders such as glaucoma, acid–base disequilibria, epilepsy and other neuromuscular diseases, altitude sickness, edema and obesityCitation3–9.
Many hydroxy compound derivatives have been widely used as pro-drugs or drugs. For instance, venlafaxine is used as an antidepressant. Another hydroxy group containing molecule, tetracycline, is mainly used for the treatment of bacterial infections. Our group recently investigated the interaction of CA isozymes I, II with salicylic acid derivatives, antioxidant phenolic compounds, organic nitrates, organic sulphate, hydroxy group containing compounds, etc. ()Citation6–12. We wanted to extend these earlier investigations to some hydroxy compounds in order to discover powerful CA inhibitors (CAIs) which might have implications in medicineCitation6–10.
Figure 1. Chemical structures of some carbonic anhydrase inhibitor hydroxy compounds.
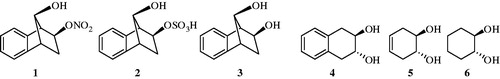
In this work, previously reported synthesized hydroxy including derivatives (i.e. compounds 7 and 8) are tested to check their CA inhibition profiles. These compounds were evaluated for their ability to inhibit human CA I and II enzymes. Inhibition is reported as KI (μM) and the results are the average of at least three independent experiments. Since X-ray structures of these small molecules with CA targets are not yet available, in silico studies are performed for the investigation of their binding mechanisms.
Materials and methods
Chemicals
The CNBr-activated Sepharose 4B, protein assay reagents, p-aminobenzene sulphonamide, l-tyrosine, 4-nitrophenylacetate (4-NPA) and chemicals for electrophoresis were purchased from Sigma-Aldrich Co. (St. Louis, MO). All other chemicals were of analytical grade and obtained from either Sigma or Merck (Darmstadt, Germany).
Synthesis of compounds 7, 8 and 12: Detailed synthetic procedures for the preparation of (1-methyl-4-hydroxy-4-phenylpiperidin-3-yl)(phenyl)methanone (7) and (1-ethyl-4-hydroxy-4-phenylpiperidin-3-yl)(phenyl)methanone (8) can be found in Sharma et alCitation13. Detailed synthetic procedures for the preparation of 1R(S),2R(S)-2-(3-bromo-4,5-dimethoxybenzyl)cyclohexanol (12) can be found in Balaydin et alCitation8.
CA purification assay
Purification of two human CA isozymes (hCA I and hCA II) was previously described with a simple one step method by a Sepharose-4B aniline–sulfanilamide affinity column chromatoghrapyCitation14.
CA activity assay and kinetic studies
Carbonic anhydrase activity was assayed by following the change in absorbance at 348 nm of 4-nitrophenylacetate (4-NPA) to 4-nitrophenylate ion over a period of 3 min at 25 °C using a spectrophotometer (Shimadzu UV–VIS) according to the method described by Verpoorte et alCitation15. The inhibitory effects of compounds 7 and 8 were examined and their inhibitory profiles are compared with previously tested chemicals (). All compounds were tested in triplicate at each concentration used. Control cuvette activity in the absence of inhibitor was taken as 100%. For each inhibitor an Activity %–[Inhibitor] graph was drawn. To determine KI values, three different inhibitor concentrations were tested: In these experiments, 4-NPA was used as substrate at five different concentrations (0.15–0.75 mM). The Lineweaver–Burk curves were drawnCitation16. Regression analysis graphs were drawn for IC50 using inhibition % values by a statistical package (SPSS-for windows; version 10.0) on a computer (Student’s t-test; n: 3).
Figure 2. Chemical structures of compounds 7–15 and AZA.
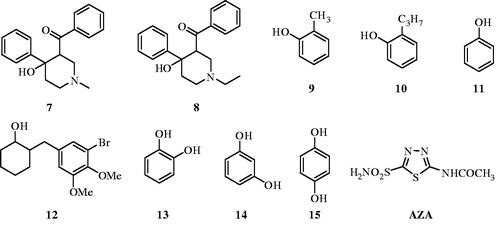
In silico methods
In silico approaches were applied to estimate binding energy values at the ligand-binding domain, critical amino acid residues in binding site, ligand–protein interactions and ADME (Absorption, Distribution, Metabolism and Excretion) profiles of synthesized compounds.
Ligand setting
All chemical structure preparation procedure for the used ligands in this work was performed using MarvinSketchCitation17 software. Consequence ligand preparations steps are follow as: (i) 2D structure of each ligand was sketched, (ii) 3D structures of all ligands were generated using molecular mechanical (MM) geometry optimization approach, (iii) all ligands were then set to the physiological pH (pH 7.4) at the protonation state step and geometry optimization is repeated.
Receptor preparation
Crystal structures of CA I and II are retrieved from Protein Data Bank server (PDB: 2NMX and 3M04)Citation18, respectively. Both structures were submitted in protein preparation procedure using Schrodinger softwareCitation19 which includes: (i) removing of all water molecules from crystal structure (implicit solvation was used), (ii) assignment of bond orders, (iii) adding hydrogen atoms, (iv) setting of physiological pH (pH 7) by protonation states of corresponding amino acid residues using implemented PROPKA softwareCitation20 in Schrodinger molecular modeling package, (v) and finally, restrained minimization of added hydrogen atoms.
Molecular docking simulations
Glide/SP and Glide/XP docking protocolsCitation21 are applied for the prediction of topologies as well as binding energies (i.e. docking scores) of used compounds at the active sites of CA I and CA II enzymes. Active site of the enzyme is defined from the co-crystallized ligands from PDB files. Throughout the docking simulations both partial flexibility and full flexibility around the active site residues are performed by Glide/SP/XP and Induced Fit Docking (IFD)Citation22 approaches. IFD procedure was established in three consequence stages which involves: (i) docking of all ligands into the generated model, (ii) refining of amino acid residues within 4 Å of docked poses, (iii) re-docking of docked ligands against refined protein.
ADME analysis
In order to estimate biological, pharmaceutical and drug similarity of synthesized compounds, ADME prediction analysis was carried out using QikProp programCitation23.
Results and discussion
The rationale of investigating phenols as CAIs is due to the fact that the simple phenol (11) has been shown to be the only competitive inhibitor with CO2 as substrate for the main isoform of CA, i.e. human CA II (hCA II)Citation24. Our group investigated the interactions of a few simple phenols, salicylic acid derivatives, antioxidant phenolic compounds, some natural product polyphenols and phenolic acids with all mammalian isozymes, CA I–XVCitation24–26. Results for these molecules were in low micromolar/submicromolar inhibitions and derived molecules may assist to design isozyme selective CAIs. Indeed, the inhibition profile of various isozymes with this class of agents was diverse, with inhibition constants ranging from the millimolar to the submicromolar for many simple phenolsCitation24–27.
We report here the first study on the inhibitory effects of compounds 7 and 8 on the esterase activity of hCA I and II. Compounds 7–15 and AZA were docked at the binding site of the targets (hCA I and II). Glide SP/XP docking scores of docked inhibitors at hCA I and II targets and corresponding binding interactions are tabulated in . The previous reports by Innocenti et alCitation25 investigated other phenol derivatives (including compounds 13–15) using a stopped flow, CO2 hydration assay for monitoring CA inhibition. Data of show the following regarding inhibition of hCA I and II with compounds 9–12, by an esterase assayCitation8, with 4-nitrophenylacetate (4-NPA) as substrate:
Table 1. KI values obtained from regression analysis graphs for hCA I and hCA II in the presence of different inhibitor concentration (µM).
(i) Against the slow cytosolic isozyme hCA I, compounds 13 and 14 behave as moderate inhibitors, with KI values in the range of 795–4003 μM, similarly to the structurally related compounds 7–11, 15 and acetazolamide (AZA) (KIs of 0.25–13.86 µM). It is interesting to note that the compounds 7–12 and 15 were much better hCA I inhibitors as compared to the corresponding compounds 13 and 14. Kinetic investigations (Lineweaver–Burk plots, data not shown) indicate that similarly to sulfonamides and inorganic anionsCitation5–12,Citation25–30, all the investigated natural compounds act as competitive inhibitors with 4-NPA as substrate (i.e. they bind in different regions of the active site cavity as compared to the substrate). However, the binding site of 4-NPA itself is unknown, but it is presumed to be in the same region as that of CO2, the physiological substrate of this enzymeCitation25–28.
(ii) A better inhibitory activity has been observed with compounds 9–11, 15 and AZA investigated here for the inhibition of the rapid cytosolic isozyme hCA II (). Structure–activity relationship (SAR) is thus quite sharp for this small series of hydroxylic compounds: the 4-hydroxy-4-phenylpiperidin-3-yl)(phenyl)methanone containing compounds 7 and 8 are ineffective leads, with two dihydroxy moieties containing compounds 13 and 14 is already a submicromolar hCA II inhibitor. The best hCA II inhibitor in this series of derivatives were compounds 9–11 and 15, with a KI of 0.09–0.38 µM. It must be stressed that KIs measured with the esterase method are always in the micromolar range because hCA I and II are weak esterasesCitation25–28.
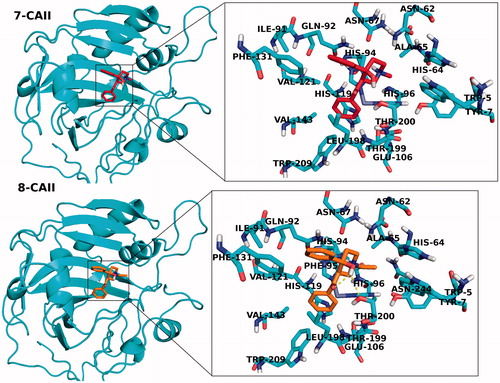
In addition to partial flexible docking simulation, flexible receptor docking at the active site was also carried out to enhance fitting treatment of amino acid residues within 4 Å of the ligand. The IFD results are tabulated in for both CA I and II receptors (top docking scores were considered). Revealed binding energies from IFD simulations are lower (i.e. higher docking scores) than the results of Glide/SP and XP results. In the case of compounds 7 and 8 (synthesized inhibitors), lowest binding energies against both CA I and II receptors were connected to these ligands. 2D and 3D ligand-binding interaction diagrams of docking poses of both inhibitors into the ligand binding site of CA I and II receptors are represented in . These ligand diagrams clearly identify key amino acid residues which participate in ligand-binding interactions for both receptors. Hydrophobicity and polarity of binding pockets were identified for each complex in the diagrams. H-bonding and π–π stacking interactions were displayed by red and blue lines, respectively. Zinc metal ion in all complexes was located in the active site and the neighborhood of inhibitor. His94 amino acid residue was constrained in all complexes, which plays a critical role in CA I and II inhibitory processes. In the case of CA I, Gln92 amino acid residue forms a H-bond with hydroxyl group of compounds 7 and 8. In the case of CA II, Thr200 has participated in ligand–receptor interaction and form H bonds with hydroxyl groups of compounds 7 and 8. and display 3D docking poses of ligands 7 and 8 at the binding pockets of CA I and II, respectively. In order to determine active sites and orientations of ligands, positions of the docked ligand were zoomed graphically for each complex and the corresponded residues around 4 Å of ligands were labeled. According to the superimposition data of the complexes, in individual CA I and II receptors, both inhibitors have shared the similar positions in ligand-binding pockets.
Figure 3. 2D docking poses of compounds 7 and 8 into the hCA I and II receptors.
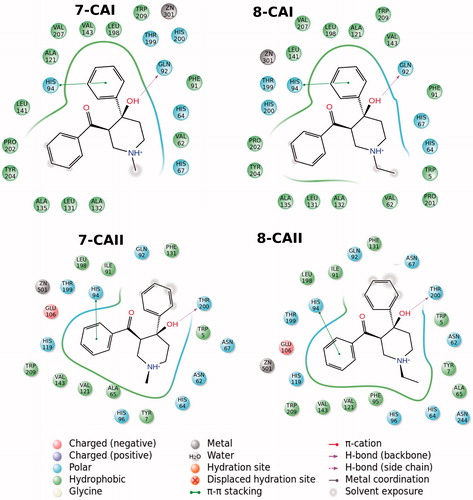
Figure 4. 3D docking poses of compounds 7 and 8 into the hCA I receptor. Key amino acid residues were determined in the 3D positions in active sites. Superimposition of docked compounds 7 and 8 into the hCA I (lower panel).
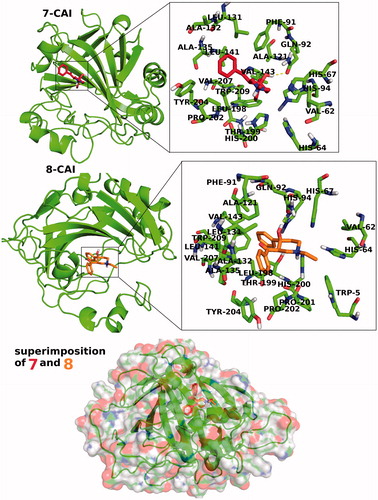
Figure 5. 3D docking poses of compounds 7 and 8 into the CA II receptors. Key-amino acid residues were determined in the 3D positions.
In order to examine the biological and pharmaceutical characteristics of synthesized molecules, different ADME tests such as hydrophobic solvent accessible surface area (SASA), hydrophilic SASA, central nervous system (CNS) activity, blockage of hERG K+ channel activity and percentage of human oral absorption were performed for all compounds. The results are tabulated in . All the compounds were located in an acceptable range on the base of these estimations. In addition, their standard drug similarity was predicted, and five approved similar drugs (based on used ligand similarity indices of >80%) for individual inhibitors has been reported (). In order to examine the CA I and II inhibitory capacity of approved drugs from drug similarity analysis, several drugs were randomly selected and docked into the receptors, the results are tabulated in .
Table 2. Predicted ADME properties of synthesized compounds.
Table 3. Drug similarity using QikProp algorithm.
Table 4. Docking scores of approved drugs within hCA I and II enzymes (docking scores are in kcal/mol).
In a recent study, it was reported that simple compounds such as catechol (13) and resorcinol (14)Citation25 as CA inhibitors do not involve sulfonamide, sulfamate or related functional groups. These compounds can be used as starting point for a new class of CA inhibitors that may have advantages for patients with sulfonamide allergiesCitation25,Citation31. The sulfonamide zinc-binding group is thus superior to the thiol one (from the thioxolone hydrolysis product) for generating CA inhibitors with a varied and sometimes isozyme-selective inhibition profile against the mammalian enzymes. However, it is critically important to explore further classes of potent CAIs in order to detect compounds with a different inhibition profile as compared to the sulfonamides and their bioisosteres and to find novel applications for the inhibitors of these widespread enzymes.
Conclusions
Compounds used in this study affect the activity of CA isozymes due to the presence of the different functional groups (i.e. OH, phenyl, benzyl and cyclohexyl) present in their aromatic and nonaromatic scaffolds. In addition to the well-known sulfonamides and sulfamates, in this study another class of CAIs is presented. Indeed, some hyroxylic compounds investigated here showed effective hCA I and II inhibitory activity, in the low micromolar range, by the esterase method which usually gives KIs an order of magnitude higher as compared to the CO2 hydrase assayCitation24–32. These findings point out that substituted hydroxylic compounds may be used as leads for generating potent CAIs eventually targeting other isoforms which have not been assayed yet for their interactions with such agents.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Pastorekova S, Parkkila S, Pastorek J, Supuran CT. Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J Enzyme Inhib Med Chem 2004;19:199–229
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81
- Scozzafava A, Carta F, Supuran CT. Secondary and tertiary sulfonamides: a patent review (2008–2012). Expert Opin Ther Patents 2013;23:2003–13
- Masini E, Carta F, Scozzafava A, Supuran CT. Antiglaucoma carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Patents 2013;23:705–16
- Ozdemir ZO, Senturk M, Ekinci D. Carbonic anhydrase inhibitors: Inhibition of mammalian isoforms I, II and VI with thiamine and thiamine-like molecules. J Enzyme Inhib Med Chem 2013;28:316–19
- Ekinci D, Kurbanoglu NI, Salamci E, et al. Carbonic anhydrase inhibitors: inhibition of human and bovine isoenzymes by benzenesulphonamides, cyclitols and phenolic compounds. J Enzyme Inhib Med Chem 2012; 27:845–8
- Ekinci D, Cavdar H, Talaz O, et al. NO-releasing esters show carbonic anhydrase inhibitory action against human isoforms I and II. Bioorg Med Chem 2010;18:3559–63
- Balaydin HT, Senturk M, Menzek A. Synthesis and carbonic anhydrase inhibitory properties of novel cyclohexanonyl bromophenol derivatives. Bioorg Med Chem Lett 2012;22:1352–7
- Cavdar H, Ekinci D, Talaz O, et al. α-Carbonic anhydrases are sulfatases with cyclic diol monosulfate esters. J Enzyme Inhib Med Chem 2012;27:148–54
- Senturk M, Ekinci D, Goksu S, Supuran CT. Effects of dopaminergic compounds on carbonic anhydrase isozymes I, II, and VI. J Enzyme Inhib Med Chem 2012;27:365–9
- Durdagi S, Sentürk M, Ekinci D, et al. Kinetic and docking studies of phenol-based inhibitors of carbonic anhydrase isoforms I, II, IX and XII evidence a new binding mode within the enzyme active site. Bioorg Med Chem 2011;19:1381–9
- Ceyhun SB, Senturk M, Erdogan O, Kufrevioglu OI. In vitro and in vivo effects of some pesticides on carbonic anhydrase enzyme from rainbow trout (oncorhynchus mykiss) gills. Pest Biochem Physiol 2010;97:177–81
- Sharma VL, Bhandari K, Chatterjee SK. Novel synthesis of 3-benzoyl-4-hydroxy-1 methyl-4 phenylpiperidine. Indian J Chem Section B: Org Chem Includ Med Chem 1991;30B:876–7
- Akin Kazancioglu E, Guney M, Senturk M, Supuran CT. Simple methanesulfonates are hydrolyzed by the sulfatase carbonic anhydrase activity. J Enzyme Inhib Med Chem 2012;27:880–5
- Verpoorte JA, Mehta S, Edsall JT. Esterase activities of human carbonic anhydrases B and C. J Biol Chem 1967;242:4221–9
- Lineweaver H, Burk D. The determination of enzyme dissociation constants. J Am Chem Soc 1934;56:658–66
- Marvin 14.12.15.0, 2014, ChemAxon (http://www.chemaxon.com)
- Aggarwalv M, Boone CD, Kondeti B, McKenna R. Structural annotation of human carbonic anhydrases. J Enzyme Inhib Med Chem 2013;28:267–77
- Madhavi Sastry G, Adzhigirey M, Day T, et al. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 2013;27:221–34
- Bas DC, Rogers DM, Jensen JH. Very fast prediction and rationalization of pKa values for protein–ligand complexes. Proteins Struct Funct Genet 2008;73:765–83
- Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 2004;47:1739–49
- Sherman W, Beard HS, Farid R. Use of an induced fit receptor structure in virtual screening. Chem Biol Drug Des 2006;67:83–4
- QikProp, version 3.6. New York (NY): Schrödinger, LLC; 2013
- Nair SK, Ludwig PA, Christianson DW. Two-site binding of phenol in the active site of human carbonic anhydrase II: structural implications for substrate association. J Am Chem Soc 1994;116:3659–60
- Innocenti A, Vullo D, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: interactions of phenols with the 12 catalytically active mammalian isoforms (CA I–XIV). Bioorg Med Chem Lett 2008;18:1583–7
- Balaydin HT, Durdagi S, Ekinci D, et al. Inhibition of human carbonic anhydrase isozymes I, II and VI with a series of bisphenol, methoxy and bromophenol compounds. J Enzyme Inhib Med Chem 2012;27:467–75
- Senturk M, Gulcin I, Beydemir S, et al. In vitro inhibition of human carbonic anhydrase I and II isozymes with natural phenolic compounds. Chem Biol Drug Des 2011;77:494–9
- Alp C, Ekinci D, Gültekin MS, et al. A novel and one-pot synthesis of new 1-tosyl pyrrol-2-one derivatives and analysis of carbonic anhydrase inhibitory potencies. Bioorg Med Chem 2010;18:4468–74
- Ekinci D, Al-Rashida M, Abbas G, et al. Chromone containing sulfonamides as potent carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:744–7
- Ekinci D, Cavdar H, Durdagi S, et al. Structure–activity relationships for the interaction of 5,10-dihydroindeno[1,2-b]indole derivatives with human and bovine carbonic anhydrase isoforms I, II, III, IV and VI. Eur J Med Chem 2012;49:68–73
- Innocenti A, Maresca A, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: thioxolone versus sulfonamides for obtaining isozymeselective inhibitors? Bioorg Med Chem Lett 2008;18:3938–41
- Supuran CT. Carbonic anhydrases: from biomedical applications of the inhibitors and activators to biotechnologic use for CO2 capture. J Enzyme Inhib Med Chem 2013;28:229–30