
Abstract
Many cancer cells have high expression of ornithine decarboxylase (ODC) and there is a concerted effort to seek new inhibitors of this enzyme. The aim of the study was to initially characterize the inhibition properties, then to evaluate the cytotoxicity/antiproliferative cell based activity of N-ω-chloroacetyl-l-ornithine (NCAO) on three human cancer cell lines. Results showed NCAO to be a reversible competitive ODC inhibitor (Ki = 59 µM) with cytotoxic and antiproliferative effects, which were concentration- and time-dependent. The EC50,72h of NCAO was 15.8, 17.5 and 10.1 µM for HeLa, MCF-7 and HepG2 cells, respectively. NCAO at 500 µM completely inhibited growth of all cancer cells at 48 h treatment, with almost no effect on normal cells. Putrescine reversed NCAO effects on MCF-7 and HeLa cells, indicating that this antiproliferative activity is due to ODC inhibition.
Introduction
Despite advances in the detection and treatment of cancer, it is still one of the most widespread diseasesCitation1,Citation2. Current therapeutic strategies are limited to slowing the advance of this disorder. Therefore, it is essential to continue the search for potential drugs with new strategies for cancer treatmentCitation3–5. One way of initiating research on potential anticancer drugs is by testing their effect on cancer cell lines.
A new strategy for anticancer activity is aimed at inhibiting the synthesis of polyamines (putrescine, spermidine, spermine) (), a group of cell components that have been implicated in the transformation of cells through the regulation of cell proliferation, cell differentiation and apoptosis, and therefore tumor development. High levels of polyamines are frequently seen in a variety of human malignanciesCitation6–9.
Figure 1. Polyamine biosynthetic pathway. Putrescine is synthesized by the decarboxylation of ornithine by l-ornithine decarboxylase (ODC, E.C. 4.1.1.17). Spermidine is formed by the action of spermidine synthase (SPMS, E.C. 2.5.1.16) that links putrescine to an aminopropyl group derived from decarboxylated S-adenosylmethionine (dcAdoMet), a reaction product of S-adenosylmethionine decarboxylase (AdoMetDC, E.C. 4.1.1.50). Spermine is synthesized from spermidine by a similar process by spermine synthase (SPS, E.C. 2.5.1.22). NCAO (N-ω-chloroacetyl-l-ornithine) is a new competitive inhibitor of ODC.
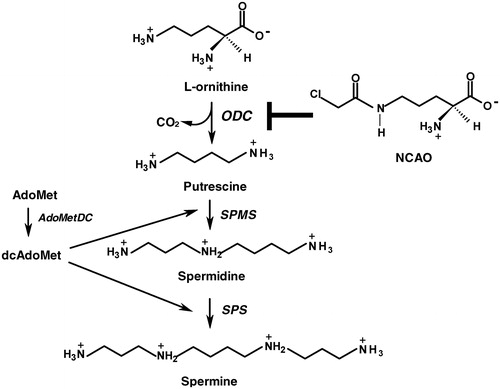
Ornithine decarboxylase (ODC), the first and rate-limiting enzyme in polyamine biosynthesis (), is a promising target for antiproliferative activity in tumors. It plays an important role in tumor progression, cell proliferation and differentiationCitation10,Citation11, and is one of the most highly regulated enzymes in eukaryotic organisms. Factors involved in this regulation include hormones, tumor promoters and growth factorsCitation12–15. It is thought that ODC plays an important role in the stage of carcinogenesis related to tumor promotion, transformation and metastasis of tissues. In accordance with this view is the fact that increased levels of polyamines accompany tumor development in humans and rodentsCitation16–21. Thus, ODC has been identified as a potential target for chemotherapy used to treat cancer and parasitic diseasesCitation4,Citation22,Citation23. Despite this potential, very few ODC inhibitors are in development or on the market, although have been extensively tested in multiple in vitro and in vivo systemsCitation3. Early studies identified a number of compounds as reversible inhibitors of ODC such as α-methyl ornithine and α-hydrazinoornithine, however they were not sufficiently potent or specificCitation3. Starting in 1975, a series of α-substituted ornithine derivatives with either an unsaturation (ethylenyl, ethynyl, allenyl) or a methyl group with a suitable leaving group (chloromethyl, fluoromethyl, difluoromethyl, chlorofluoromethyl, cyanomethyl) was synthesized at the Merrel Dow Research Institute in Strasbourg as enzyme-activated irreversible inhibitorsCitation3. From these series of compounds, α-dl-difluoromethylornithine (DFMO) was selected for development as anticancer drug. A series of putrescine derivatives and putrescine analogues were designed as potential enzyme-activated irreversible inhibitors of ODC, using the same unsaturated substituents. Among these, 2,5-diamino-6-heptyne was most potent but owing to its nephrotoxicity it had to be abandoned. Another ornithine analogue, dl-α-(fluoromethyl)-3-dehydro-ornithine was studied in detail. However, it was not more effective than DFMO at inhibiting cell proliferation in vitro and in vivoCitation3. Thus, no other inhibitor has demonstrated clear advantages over DFMO. DFMO has been clinically evaluated for activity against several tumor models, including gliomas, melanomas, breast, prostate and cervical cancers however, cancer treatment trials demonstrated little benefitCitation3,Citation4,Citation24,Citation25. The reasons for these less than optimal results most probably include pharmacokinetic parameters and compensatory mechanisms within the tumor cells that allow compensation for loss of a single enzyme's activity. Importantly, DFMO has been approved by the Food and Drug Administration as an affective and non-toxic agent for African trypanosomiasisCitation4,Citation22,Citation23. This promising target deserves greater attention in the search for new anticancer drugs.
A recent studyCitation26 proposed another potential ODC inhibitor. Computational docking simulations indicated that N-ω-chloroacetyl-l-ornithine (NCAO) () should inhibit rat liver ODC with high affinity, and in vivo assays showed its potential properties for inhibiting cell proliferation with low toxicity. NCAO is an ornithine analogue that was originally proposed as a general yeast amino acid permeaseCitation27–29 and a possible Escherichia coli glucosamine-6-phosphate synthase inhibitorCitation30. Based on the recent findings, 30 years of polyamine-related approaches to cancer therapy may finally bear fruit by developing an ODC inhibitor that has good potency, an attractive pharmacokinetic profile, and the ability to overcome cell resistanceCitation3.
Thus, the aim of the present study was to characterize the inhibition of ODC by NCAO through enzyme kinetic analysis. Additionally, evaluation was made of the cytotoxic and antiproliferative activities of this compound against three different human cancer cell lines (MCF-7, HeLa and HepG2) and one immortalized normal epithelial cell line (Vero).
Methods
Chemicals
Putrescine, dithiothreitol, pyridoxal phosphate and l-ornithine were obtained from Sigma-Aldrich (St. Louis, MO). L-[1-l4C]ornithine was obtained from NEN Radiochemicals (Waltham, MA). Cell culture medium components were purchased from Invitrogen, Gibco (Carlsbad, CA). N-ω-chloroacetyl-l-ornithine was prepared by following an established procedureCitation26.
Rat liver ODC
ODC was induced more than 100-fold and prepared from livers of six adult male Wistar rats that had been intraperitoneal injected with thioacetamide (150 mg/kg body weight) 18 h before sacrifice. It was purified about fifteen fold by acid treatment at pH 4.6, as described by Ono et al.Citation31. The protocol for rats was approved by Internal Committee for the Care and Use of Laboratory Animals, Escuela Superior de Medicina-IPN.
Cell lines and cell culture
MCF-7 cells from human breast carcinoma, HeLa cells from human cervical carcinoma, HepG2 cells from human hepatocellular carcinoma, and Vero cells were purchased from the American Type Culture Collection. Vero cells were used as the control. The four cell lines were cultured in DMEM or DMEM/F–12, consisting of l-glutamine supplemented with 10% heat-fetal bovine serum, 25 U/ml penicillin and 25 mg/ml streptomycin, and maintained at 37 °C in a fully humidified atmosphere of 5% CO2 in air. Once cells were grown to around 90–100% confluence, they were sub-cultured by washing with PBS and trypsinized with 1.0 ml 25% trypsin in a 0.53 mM EDTA solution. Then, they were counted by the trypan blue exclusion test to assure that there were 35 × 104 cells seeded in each well (of 24-well plates) under sterile conditions with fresh complete-medium. After being seeded, cells were allowed to attach and grow for 24 h before the addition of NCAO.
Ornithine decarboxylase activity assay
This was done by measuring the release of 14CO2 from l-[1-14C]ornithine as previously describedCitation32 with slight modifications. The standard incubation mixture contained in a final volume of 1 ml: 50 mM phosphate buffer, pH 7.0, 0.8 mM l-ornithine, 5 mM dithiothreitol, 0.1 mM pyridoxal phosphate, 0.1 mM EDTA, and 0.1 μCi l-ornithine-1-14C (40–60 mCi/mmol) and enzyme preparation. Incubation was carried out at 37 °C for 1 h in hermetically sealed flask where previously 110 µl of 10% KOH were placed in filter paper strip supported by the flask cap for trapping the 14CO2 released. The reaction was stopped by injection of 0.5 ml of 2 M perchloric acid and the mixture was incubated for an additional 1 h. The filter paper was then transferred into a scintillation counting vial containing 10 ml of scintillation fluid and counted in a liquid scintillator. Under these conditions, the activity of the ODC preparation from rat liver was 0.32 nmol of CO2/min/mg of protein. For determination of the Ki value, the inhibitor (NCAO) and the cofactor were incubated with the buffer for 5 min at 37 °C, and then the reaction was started by adding the enzyme. The Ki value was determined by analyzing the initial rates at various substrate concentrations in the presence of 0.07 or 0.4 mM NCAO and in the absence of this inhibitor. From each double reciprocal plot, the values for Vmax, Km and were determined by linear regression. The calculation of Ki was performed using the equation described by StojanCitation33. The average Ki value was obtained from the two concentrations of NCAO assayed.
Time-dependent inhibition of ODC by NCAO
We determined the activity of ODC in the presence and absence of 0.40 mM NCAO. The enzyme and inhibitor were preincubated for 30, 60, 90 and 120 min and subsequently ODC activity was determined as described above.
Cytotoxicity test
Cytotoxicity was assayed in wells seeded with MCF-7, HeLa, HepG2 or Vero cells, as described above. NCAO was dissolved in sterile DMEM, sterilized by filtration and added to the wells at the following concentrations: 0.001, 0.010, 0.100, 1.000 and 2.000 mM. The absence of NCAO was used as the control. Each concentration was incubated for three different periods (24, 48 and 72 h), after which time the trypan blue exclusion test was performed according to the method described by Louis and SiegelCitation34 to assess cell viability. This assay was performed in triplicate for each cell line, concentration of NCAO and incubation time. The average value of three assays was expressed as a percentage of the value of the control wells, in which each of the cell lines was cultured without any drug. The concentration of NCAO that reduced cell viability by 50% was then considered the EC50 value.
Cell proliferation
For measuring cell growth, cells were seeded at a concentration of 1.5 × 104 to 5 × 104 per well (in 24-well plates), and then incubated in 0.5 ml medium containing 5% FBS and 1% penicillin/streptomycin. After 1 day, cells were incubated in the presence of different concentrations of NCAO, or the absence of this inhibitor, for the indicated times. The treated cells were harvested with trypsin on the indicated post-treatment day, and then stained with trypan blue. The number of cells was counted by using a hemocytometerCitation35.
For putrescine reversal experiments, cells were also seeded at a concentration of 1.5 × 104 to 5 × 104 per well and incubated for 1 day. Cells were then incubated for 24 h in the presence of 10 µM putrescine, 1.0 mM NCAO, 1.0 mM NCAO + 10 µM putrescine, or without adding any compound. Then cells were harvested with trypsin, stained with trypan blue and counted as aforementioned.
Statistical analysis
Statistical analysis was performed using SigmaPlot 11 (Systat Software, Inc., San Jose, CA). All data are shown as the mean ± SEM. Statistical significance was determining by using one-way analysis of variance followed by Dunn's test. The differences were considered significant at p < 0.05.
Results
Inhibition of rat liver ODC activity
The inhibitory effect of NCAO on rat liver ODC was concentration-dependent (). In order to characterize the mode of inhibition, double reciprocal equations of substrate concentration (mM−1) and initial velocity were plotted. Enzyme kinetics was determined by evaluating the effect of 0.07 and 0.40 mM NCAO and comparing these values to that found with the control (without NCAO). In each case a linear relationship can be appreciated on the graph, and the three lines intersect on the y-axis, meaning that they have the same Vmax but a different Km. The greater the concentration of the inhibitor, the higher the value of Km found (), which shows that the inhibition is competitive. The Km value for the control (without NCAO) was 2.5 × 10−4 M, and the disassociation constant value (Ki) was 5.9 × 10−5 M (). This indicates that the receptor has greater affinity for the inhibitor than the natural substrate.
Figure 2. Kinetic analysis of the inhibition of N-ω-chloroacetyl-l-ornithine on rat liver ODC. (A) Concentration-dependent effect. (B) Double-reciprocal plot of the inhibition kinetic (Lineweaver–Burk method). (▾) without inhibitor, (○) with 0.07 mM, (•) with 0.4 mM inhibitor. Initial velocities were calculated taking the maximum activity without the inhibitor as 100%.
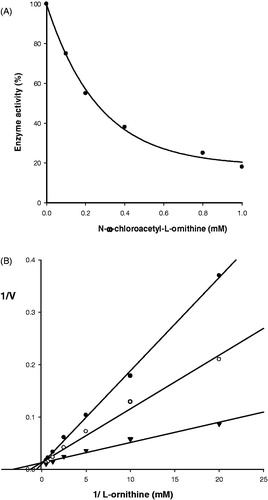
In order to determine whether the inhibitor binds covalently, we incubated it with the enzyme and found that the inhibition was concentration- but not time-dependent. This indicates that the inhibitor binds reversibly rather than covalently (data not shown).
Cell cytotoxicity
Concentrations from 0.001 to 2 mM of NCAO were tested for 24, 48 and 72 h. The viability of all cancer cells was significantly reduced in a concentration-dependent manner by the treatment with this inhibitor (), with the HepG2 cell line showing the greatest sensitivity. Contrarily, with Vero cells there was 80% survival even at the highest concentration of the inhibitor, indicating almost no cytotoxic effect. For HepG2 and MCF-7 cells, the EC50 at 24 h of treatment was 91.4 ± 10 µM and 630 ± 50 µM, respectively, and for HeLa cells was 1070 ± 80 µM (). At a concentration of 100 µM NCAO, MCF-7 and HepG2 cancer cell lines started to respond to treatment at 24 h (), and all MCF-7 and HepG2 cells were killed at 48 h. However, at this same concentration of NCAO, HeLa cells displayed a viability of 40% at 48 h, requiring 72 h of incubation before these cells were all killed. Contrarily, at 72 h Vero cells retained at least 80% cell viability. The EC50 at 72 h of treatment was 10.1 ± 0.6 µM, 17.4 ± 0.9 µM and 15.8 ± 0.8 µM for HepG2, MCF-7 and HeLa cells, respectively ().
Figure 3. Effect of N-ω-chloroacetyl-l-ornithine on cell viability of different cancer cell lines. Cells were cultured in absence (control) or presence of various concentrations of NCAO, during 24, 48 and 72 h. Data presented are means ± SEM of triplicate determinations. *p < 0.05.
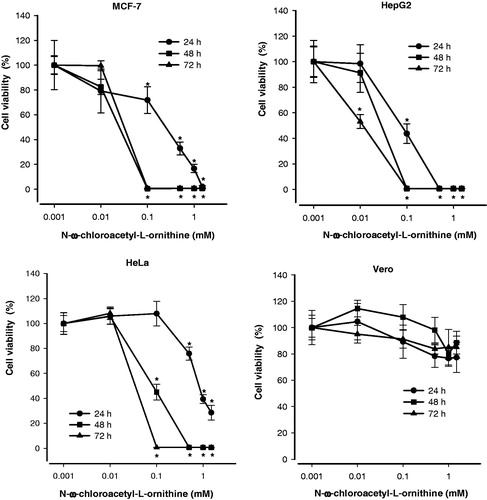
Table 1. Cytotoxic effect of N-ω-chloroacetyl-l-ornithine against cell viability of different cancer cell lines exposed 24 and 72 h.
According to these results, the cytotoxic effect of NCAO on all cancer cell lines was concentration- and time-dependent.
Cell growth inhibition
NCAO inhibited growth by cells of the three cancer cell lines, with HepG2 being the most sensitive. Contrarily, normal Vero cells were resistant to growth inhibition even at the highest concentration (2000 µM) of the inhibitor (). Two of the cancer cell lines, MCF-7 and HepG2, had all died at 48 h of treatment with 500 µM NCAO (). This same effect was observed with HeLa cells at 72 h of treatment with 500 µM NCAO. The inhibitory effect of NCAO on cell growth for all cancer cell lines was concentration- and time-dependent.
Figure 4. Effect of N-ω-chloroacetyl-l-ornithine treatment on the cell proliferation of different cancer cell lines. Cells were cultured in absence (control) or presence of various concentrations of NCAO. Data presented are means ± SEM of triplicate determinations. *p < 0.05.
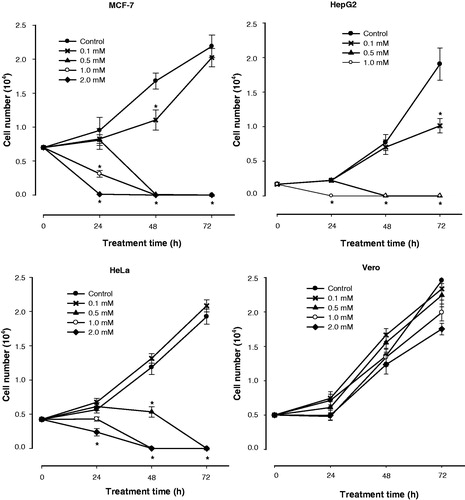
For the MCF-7 and HeLa cancer cell lines, the growth inhibition caused by NCAO treatment was reverted by 10 µM putrescine (showing 76.4 and 50% recovery, respectively) (). This indicates that NCAO had an inhibitory effect on ODC. On the other hand, HepG2 cancer cells showed very little recovery with the putrescine treatment, indicating either cell death or another mechanism of antiproliferative activity, or both. Interestingly, putrescine produced the opposite effect on Vero cells, increasing cell death slightly but not significantly.
Table 2. Protective effect of putrescine against the growth inhibition of human cancer cell lines produced by treatment for 24 h with 1.0 mM N-ω-chloroacetyl-l-ornithine.
Discussion
Since polyamines play an important role in a variety of processes, including cell proliferation, the inhibition of polyamine biosynthesis represents a potential target for cancer interventionCitation10,Citation11,Citation36,Citation37. ODC, rate-limiting enzyme in polyamine biosynthesis, is highly expressed in many cancer cells. Consequently, there has been a great effort to discover compounds that can inhibit ODC activity in tumor cells Citation3,Citation13,Citation38–42.
Previously, our work group reported the synthesis and study of N-ω-iodoacetyl-l-ornithine (NIAO) as an ODC inhibitorCitation43,Citation44. This compound, directed to the active site of ODC, showed irreversible inhibition of this enzyme, with a half-life of 15 min and Ki = 2.08 mM. However, because the iodoacetyl group is a highly reactive alkylating agent, its toxicity made pharmacological studies irrevelantCitation45. Therefore, a compound was sought with a less reactive alkylating group, leading to the design and synthesis of NCAO. In vivo tests showed low toxicityCitation26.
The present study demonstrates the inhibitory action of NCAO on three human tumor cell lines. The kinetics study reveals that NCAO inhibited rat liver ODC, and that this inhibition was competitive with respect to the substrate, l-ornithine. These experimental results agree with the computational docking simulations done by Correa-Basurto et al.Citation26, who reported that NCAO binds to the active site of ODC with high affinity. The docking procedure showed that NCAO can interact with PLP and Cys328 in the active site of the enzyme. However, the theoretical Ki was 4.40 µM and the experimental value 59 µM, showing a 13.4-fold difference between in silico and in vitro determinations. This could be, in part, because docking simulations do not consider several biological factors, such as ionic strength and the presence of an explicit solvent.
Moreover, the experimental binding affinity of NCAO to ODC is 4.2-fold greater than its affinity to the substrate, l-ornithine (the Km and Ki were 250 µM and 59 µM, respectively). Therefore, under intracellular conditions NCAO should be highly successful in competing with l-ornithine for the ODC active site, resulting in the inhibition of this enzyme.
The Ki of NCAO obtained in the present study proved to be 1.5-fold greater than the Ki previously reported for DFMO (39 µM)Citation46, the most widely used inhibitor of ODC. However, despite having a high affinity to ODC, DFMO has proved to have poor transport into cells, as evidenced by cell-based assays and animal modelsCitation3,Citation11.
Since docking studies have shown that the chlorine atom of NCAO can interact with Cys328 in the ODC active siteCitation26, we decided to determine whether NCAO is an irreversible inhibitor of ODC. No irreversible inhibition was found for up to 2 h. It is possible that the alkylating or acylating reaction requires more time. On the other hand, NIAO was previously found to irreversibly inhibit ODC. This difference is consistent with the distinct reactivity of the chloroacetyl group of NCAO and the iodoacetyl group of NIAO.
Since in silico and in vitro studies showed a possible inhibitory role of NCAO on ODC activity, we assessed the effect of this compound on three cell lines derived from human carcinomas with overexpressed ODC at least double compared to normal non-cancerous cells (MCF-7, HeLa and HepG2 cells)Citation10,Citation47–52. We selected the non-tumorigenic Vero lineage as control cell lineCitation53,Citation54.
Administration of NCAO resulted in decreased cell viability and increased growth inhibition in a dose- and time-dependent manner, according to the trypan blue assay. Whereas Vero cells proved to be almost unaffected, all the cancer cell lines showed significant inhibition, indicating a selective activity of NCAO. Hence, NCAO should show low toxicity with in vivo assays. Furthermore, these results confirm the importance of the high expression of ODC found in many types of neoplastic cellsCitation10,Citation11,Citation39. Contrarily, in normal cells ODC is tightly regulated by other growth controlsCitation15.
In the present study, NCAO showed a potent in vitro cytotoxic effect on MCF-7, HepG2 and HeLa tumor cell lines, with an average EC50 of 14.5 µM after 72 h drug treatment. In contrast, DFMO – the most representative ODC inhibitor – was found to provoke a cytostatic more than cytotoxic effect on the majority of tumors evaluated, including gliomas, melanomas and brain, breast, prostate and cervical cancer cell linesCitation4. The EC50 of DFMO varies in the millimolar range after 48–144 h (2–6 d) of treatment. For example, after a 48 h treatment the EC50 was 3.0 mM in L1210 cells (mouse leukemia)Citation55, 0.5 mM in L5178 cells (mouse lymphoma)Citation55, 2.5 mM in B16 cells (mouse melanoma)Citation56 and 33.3 mM in BE(2)-C cells (human neuroblastoma)Citation57. On the other hand, the EC50 was higher than 0.5 mM after 72 h treatment of LN229 cells (human glioblastoma)Citation58.
If one compares the cytotoxic effect caused by NCAO and DFMO in the cancer cell lines tested in this study, it becomes evident that NCAO outperforms DFMO in all cancer cell lines. For HeLa cells, DFMO has an EC50 of 0.8 mM after 120 h of treatmentCitation59, whereas NCAO has an EC50 of 15.8 µM after only 72 h treatment. NCAO that is produced a faster cytotoxic effect that was 50 times more potent than DFMO. For MCF-7 cells, Glikman et al.Citation60 showed that the EC50 is between 10 and 100 µM after 144 h of treatment with DFMO. On the other hand, the EC50 for NCAO is 17.5 µM after only 72 h of treatment. NCAO also caused a greater cytotoxic effect than DFMO on HepG2 cells. The EC50 of the NCAO was 10.1 µM after 72 h of treatment, whereas DFMO caused no cytotoxic effect after 72 h treatment at 100 µMCitation61. As can be apreciated, NCAO is over 10 times more effective than DFMO in HepG2 cells. According to the aforementioned, NCAO presents much better cytotoxic properties than DFMO. It is unlikely that this markedly enhanced cytotoxic activity is related to the affinity of the inhibitor for the enzyme, since the Ki, (59 µM) is very similar to that for DFMO (39 µM). The difference can be due to improved cellular uptake.
The active and passive transport systems of tumor cells have a greater capacity than those of normal cells. Tumor cells need this greater capacity to constantly supply the large quantity of nutrients needed to support their characteristic unabated growthCitation62–64. Whereas the lack of effectivenes of DFMO as a chemotherapeutic agent seems to be due to its poor transport into the cellsCitation11, NCAO apparently utilizes the efficient transport capacity of tumor cells.
Indeed, the differences in NCAO cytotoxicity observed between the three human cancer cell lines employed in the present study after 24 h treatment may be caused, at least in part, by differences in cellular uptake through amino acid transport systems (ATSs). NCAO is an amino acid derivative that may be incorporated into the cells by several ATSs that have enhanced expression in cancer cells. The principal ATSs in cancer cells are ASCT2, LAT1 and ATB0,+, which have the appropriate characteristics for transporting NCAO because they have broad substrate specificity and participate in the transport of several drugsCitation62,Citation64,Citation66–68. Furthermore, a variety of drug transporters are expressed in HepG2 cells, including OATPs (organic anion transporting polypeptides)Citation69,Citation70 and OCTs (organic cation transport)Citation71,Citation72. These transporters are highly expressed in HepG2 cells but hardly expressed in HeLa and MCF-7 cellsCitation71,Citation73. It is likely that differences in the transport by passive diffusion along with differences in the expression of the ATSs, OATPs and OCTs in MCF-7, HeLa and HepG2 may be responsible for their different sensitivities towards NCAO after 24 h treatment. However, it is difficult to make quantitative conclusions about the individual contributions to the NCAO transport into these cancer cells. It is noteworthy that after 72 h of treatment, almost no differences were found in the sensitivity of cell lines to NCAO.
Addition of 10 µM putrescine to cancer cells with or without NCAO demonstrated that the NCAO inhibition of proliferation could be reversed with the MCF-7 and HeLa lines, but was scarcely affected with the HepG2 cell line. This strongly suggests that with the former two cancer cell lines, the antiproliferative phenotype of NCAO is a direct result of inhibiting the enzyme target (ODC) leading to a decreased pool of polyamines and cell death. On the other hand, NCAO apparently functions by other mechanisms in the HepG2 cell line.
Conclusions
The present study shows that NCAO, an analogue of l-ornithine, is a reversible competitive inhibitor of rat liver ODC with a Ki value in a micromolar range (59 µM). Besides, NCAO exert cytotoxic and antiproliferative effects in neoplastic human cancer cell lines (HeLa, MCF-7 and HepG2) compared to normal cells (Vero). Both effects were concentration and time-dependent being more efficient against HepG2 cell line. The EC50,24 h was 1070, 630 and 91 µM for HeLa, MCF-7 and HepG2 cells, respectively. The EC50,72 h was 15.8, 17.5 and 10.1 µM for HeLa, MCF-7 and HepG2 cells, respectively. NCAO at 500 µM completely inhibited growth of all cancer cells at 48 h treatment, with almost no effect on normal cells. For the MCF-7 and HeLa cancer cell lines, the growth inhibition caused by NCAO treatment was reverted by 10 µM putrescine (showing 76.4 and 50% recovery, respectively). This indicates that NCAO had an inhibitory effect on ODC. Further research is needed to gain greater insights into the mechanism of antineoplastic activity produced by this compound.
Declaration of interest
The authors report no declarations of interest. This work was partially supported by research grants from the Secretaría de Investigación y Posgrado of the Instituto Politécnico Nacional (SIP-IPN), Mexico. J.G.T.F. and L.R.P. are fellows of the COFAA-IPN and of the SNI-CONACYT. V.A.F. and L.A.F. are fellows of the COFAA-IPN. M.M.M.E. and A.L.V.G. are fellows of the CONACYT and PIFI in the IPN.
References
- Ferlay J, Shin HR, Bray F, et al. GLOBOCAN 2008, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 10. Lyon, France: International Agency for Research on Cancer; 2008. Available from: http://globocan.iarc.fr [last accessed 22 Oct 2013]
- Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin 2011;6:69–90
- Seiler N. Thirty years of polyamine-related approaches to cancer therapy. Retrospect and prospect. Part 1. Selective enzyme inhibitors. Curr Drug Targets 2003;4:537–64
- Casero RA Jr, Marton LJ. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat Rev Drug Discov 2007;6:373–90
- Iyer AK, Singh A, Ganta S, Amiji MM. Role of integrated cancer nanomedicine in overcoming drug resistance. Adv Drug Deliv Rev 2013 65:1784–802
- LaMuraglia GM, Lacaine F, Malt RA. High ornithine decarboxylase activity and polyamine levels in human colorectal neoplasia. Ann Surg 1986;204:89–93
- Luk GD, Casero RA Jr. Polyamines in normal and cancer cells. Adv Enzyme Regul 1987;26:91–105
- Paz EA, Garcia-Huidobro J, Ignatenkos NA. Polyamines in cancer. Adv Clin Chem 2011;54:45–70
- Soda KJ. The mechanisms by which polyamines accelerate tumor spread. Exp Clin Cancer Res 2011;30:95. doi: 10.1186/1756-9966-30-95
- Shantz LM, Levin VA. Regulation of ornithine decarboxylase during oncogenic transformation: mechanisms and therapeutic potential. Amino Acids 2007;33:213–23
- Nowotarski SL, Woster PM, Casero RA Jr. Polyamines and cancer: implications for chemotherapy and chemoprevention. Expert Rev Mol Med 2013;15:e3. doi: 10.1017/erm.2013.3
- Russell D, Snyder SH. Amine synthesis in rapidly growing tissues: ornithine decarboxylase activity in regenerating rat liver, chick embryo, and various tumors. Proc Natl Acad Sci USA 1968;60:1420–7
- Pegg AE, Shantz LM, Coleman CS. Ornithine decarboxylase as a target for chemoprevention. J Cell Biochem Suppl 1995;22:132–8
- Pegg AE, Feith DJ, Fong LY, et al. Transgenic mouse models for studies of the role of polyamines in normal, hypertrophic and neoplastic growth. Biochem Soc Trans 2003;31:356–60
- Pegg AE. Regulation of ornithine decarboxylase. J Biol Chem 2006;281:14529–32
- Verma AK. Inhibition of tumor promotion by dl-alpha-difluoromethylornithine, a specific irreversible inhibitor of ornithine decarboxylase. Basic Life Sci 1990;52:195–204
- Kubota S, Kiyosawa H, Nomura Y, et al. Ornithine decarboxylase overexpression in mouse 10T1/2 fibroblasts: cellular transformation and invasion. J Natl Cancer Inst 1997;89:567–71
- Peralta Soler A, Gilliard G, Megosh L, et al. Polyamines regulate expression of the neoplastic phenotype in mouse skin. Cancer Res 1998;58:1654–9
- Shantz LM, Pegg AE. Ornithine decarboxylase induction in transformation by H-Ras and RhoA. Cancer Res 1998;58:2748–53
- Hurta RA. Altered ornithine decarboxylase and S-adenosylmethionine decarboxylase expression and regulation in mouse fibroblasts transformed with oncogenes or constitutively active Mitogen-Activated Protein (MAP) kinase kinase. Mol Cell Biochem 2000;215:81–92
- Hardin MS, Mader R, Hurta RA. K-FGF mediated transformation and induction of metastatic potential involves altered ornithine decarboxylase and S-adenosylmethionine decarboxylase expression-role in cellular invasion. Mol Cell Biochem 2002;233:49–56
- Heby O, Persson L, Rentala M. Targeting the polyamine biosynthetic enzymes: a promising approach to therapy of African sleeping sickness, Chagas' disease, and leishmaniasis. Amino Acids 2007;33:359–66
- Birkholtz LM, Williams M, Niemand J, et al. Polyamine homoeostasis as a drug target in pathogenic protozoa: peculiarities and possibilities. Biochem J 2011;438:229–44
- Vlastos AT, West LA, Atkinson EN, et al. Results of a phase II double-blinded randomized clinical trial of difluoromethylornithine for cervical intraepithelial neoplasia grades 2 to 3. Clin Cancer Res 2005;11:390–6
- Messing E, Kim KM, Sharkey F, et al. Randomized prospective phase III trial of difluoromethylornithine vs placebo in preventing recurrence of completely resected low risk superficial bladder cancer. J Urol 2006;176:500–4
- Correa-Basurto J, Rodríguez-Páez L, Aguiar-Moreno ES, et al. Computational and experimental evaluation of ornithine derivatives as ornithine decarboxylase inhibitors. Med Chem Res 2009;18:20–30
- Larimore FS, Roon RJ. Possible site-specific reagent for the general amino acid transport system of Saccharomyces cerevisiae. Biochemistry 1978;17:431–6
- Woodward JR, Kornberg HL. Membrane proteins associated with amino acid transport by yeast (Saccharomyces cerevisiae). Biochem J 1980;192:659–64
- Woodward JR, Kornberg HL. Changes in membrane proteins associated with inhibition of the general amino acid permease of yeast (Saccharomyces cerevisiae). Biochem J 1981;196:531–6
- Auvin S, Cochet O, Kucharczyk N, et al. Synthesis and evaluation of inhibitors for Escherichia coli glucosamine-6-phosphate synthase. Bioorg Chem 1991;19:143–51
- Ono M, Inoue H, Suzuki F, Takeda Y. Studies of ornithine decarboxylase from the liver of thioacetamide treated rats. Purification and some properties. Biochem Biophys 1972;284:285–97
- Hayashi S. Ornithine decarboxylase (rat liver). In: Tabor H, Tabor CW, eds. Methods in enzymology. Vol. 94. Polyamines. New York (NY): Academic Press Inc.; 1983:154–8
- Stojan J. Enzyme inhibitors. In: Smith J, Simons C, eds. Enzymes and their inhibition. Boca Raton (FL): CRC Press; 2005:149–69
- Louis KS, Siegel AC. Cell viability analysis using trypan blue: manual and automated methods. Methods Mol Biol 2011;740:7–12
- Li S, Liang Y, Wu M, et al. The novel mTOR inhibitor CCI-779 (temsirolimus) induces antiproliferative effects through inhibition of mTOR in Bel-7402 liver cancer cells. Cancer Cell Int 2013;13:30
- Pegg AE. Mammalian polyamine metabolism and function. IUBMB Life 2009;6:880–94
- Pegg AE, Casero RA Jr. Current status of the polyamine research field. Methods Mol Biol 2011;720:3–35
- Wallace HM, Fraser AV, Hughes A. A perspective of polyamine metabolism. Biochem J 2003;376:1–14
- Wallace HM, Fraser AV. Inhibitors of polyamine metabolism. Amino Acids 2004;26:353–65
- Wu F, Grossenbacher D, Gehring H. New transition state-based inhibitor for human ornithine decarboxylase inhibits growth of tumor cells. Mol Cancer Ther 2007;6:1831–9
- Wu F, Gehring H. Structural requirements for novel coenzyme-substrate derivatives to inhibit intracellular ornithine decarboxylase and cell proliferation. FASEB J 2009;23:565–74
- Silva TM, Andersson S, Sukumaran SK, et al. Norspermidine and novel Pd(II) and Pt(II) polynuclear complexes of norspermidine as potential antineoplastic agents against breast cancer. PLoS One 2013;8:e55651
- Trujillo JG, Ceballos G, Yanez R, Joseph-Nathan P. Regioselective synthesis of (+)-S-2-amino-5-iodoacetamidopentanoic and (+)-S-2-amino-6-iodoacetamidohexanoic acids. Synth Comm 1991;21:683–91
- Rodríguez-Páez L, Neri CD, Baeza RI, Wong RC. La δ-N-yodoactil ornitina y la ε-N-yodoacetil lisina como inhibidores irreversibles dirigidos al sitio activo de la ornitina descarboxilasa. An Esc Nac Cienc Biol Mex 1998;44:99–115
- Trujillo-Ferrara J, Koizumi G, Muñoz O, et al. Antitumor effect and toxicity of two new active-site-directed irreversible ornithine decarboxylase and extrahepatic arginase inhibitors. Cancer Lett 1992;67:193–7
- Sjoerdsma A, Schechter PJ. Chemotherapeutic implications of polyamine biosynthesis inhibition. Clin Pharmacol Ther 1984;35:287–300
- Williams-Ashman HG, Coppoc GL, Weber G. Imbalance in ornithine metabolism in hepatomas of different growth rates as expressed in formation of putrescine, spermidine, and spermine. Cancer Res 1972;32:1924–32
- Russell DH. Ornithine decarboxylase as a biological and pharmacological tool. Pharmacology 1980;20:117–29
- Ichimura S, Hamana K, Nenoi M. Significant increases in steady states of putrescine and spermidine/spermine N1-acetyltranserase mRNA in HeLa cells accompanied by growth arrest. Biochem Biophys Res Commun 1998;243:518–21
- Pyronnet S, Pradayrol L, Sonenberg N. Alternative splicing facilitates internal ribosome entry on the ornithine decarboxylase mRNA. Cell Mol Life Sci 2005;62:1267–74
- Deng W, Jiang X, Mei Y, et al. Role of ornithine decarboxylase in breast cancer. Acta Biochim Biophys Sin 2008;40:235–43
- Verma AJ. Polyamines and cancer. In: Wang J-Y, Casero RA Jr, eds. Polyamine cell signaling. Totowa (NJ): Humana Press; 2010:313–28
- Lundgren DW, Prokay SL. Glucose elevates ornithine decarboxylase expression in vero cells. J Cell Physiol 1988;137:469–75
- Marverti G, Ligabue A, Lombardi A, et al. Modulation of the expression of folate cycle enzymes and polyamine metabolism by berberine in cisplatin-sensitive and -resistant human ovarian cancer cells. Int J Oncol 2013;43:1269–80
- Pera PJ, Kramer DL, Sufrin JR, Porter CW. Comparison of the biological effects of four irreversible inhibitors of ornithine decarboxylase in two murine lymphocytic leukemia cell lines. Cancer Res 1986;46:1148–54
- Sunkara PS, Chang CC, Prakash NJ, Lachmann PJ. Effect of inhibition of polyamine biosynthesis by dl-alpha-difluoromethylornithine on the growth and melanogenesis of B16 melanoma in vitro and in vivo. Cancer Res 1985;45:4067–70
- Samal K, Zhao P, Kendzicky A, et al. AMXT-1501, a novel polyamine transport inhibitor, synergizes with DFMO in inhibiting neuroblastoma cell proliferation by targeting both ornithine decarboxylase and polyamine transport. Int J Cancer 2013;133:1323–33
- Wu F, Gehring H. Structural requirements for novel coenzyme-substrate derivatives to inhibit intracellular ornithine decarboxylase and cell proliferation. FASEB J 2009;23:565–74
- Zou C, Vlastos AT, Yang L, et al. Effects of difluoromethylornithine on growth inhibition and apoptosis in human cervical epithelial and cancerous cell lines. Gynecol Oncol 2002;85:266–73
- Glikman P, Manni A, Demers L, Bartholomew M. Polyamine involvement in the growth of hormone-responsive and -resistant human breast cancer cells in culture. Cancer Res 1989;49:1371–6
- Li M, Li Q, Zhang YH, et al. Antitumor effects and preliminary systemic toxicity of ANISpm in vivo and in vitro. Anticancer Drugs 2013;24:32–42
- Fuchs BC, Bode BP. Amino acid transporters ASCT2 and LAT1 in cancer: partners in crime? Semin Cancer Biol 2005;15:254–66
- Delcros JG, Tomasi S, Duhieu S, et al. Effect of polyamine homologation on the transport and biological properties of heterocyclic amidines. J Med Chem 2006;49:232–45
- Ganapathy V, Thangaraju M, Prasad PD. Nutrient transporters in cancer: relevance to Warburg hypothesis and beyond. Pharmacol Ther 2009;121:29–40
- Palacín M, Estévez R, Bertran J, Zorzano A. Molecular biology of mammalian plasma membrane amino acid transporters. Physiol Rev 1998;78:969–1054
- del Amo EM, Urtti A, Yliperttula M. Pharmacokinetic role of L-type amino acid transporters LAT1 and LAT2. Eur J Pharm Sci 2008;35:161–74
- Shikano N, Ogura M, Okudaira H, et al. Uptake of 3-[125I]iodo-alpha-methyl-L-tyrosine into colon cancer DLD-1 cells: characterization and inhibitory effect of natural amino acids and amino acid-like drugs. Nucl Med Biol 2010;37:197–204
- Kalliokoski A, Niemi M. Impact of OATP transporters on phamacokinetics. Br J Pharmacol 2009;158:693–705
- König J. Uptake transporters of the human OATP family: molecular characteristics, substrates, their role in drug-drug interactions, and functional consequences of polymorphisms. In: Fromm MF, Kim RB, eds. Drug transporters. Handbook of experimental pharmacology. Vol. 201. Berlin, Heidelberg: Springer-Verlag; 2011:1–28
- Fearn RA, Hirst BH. Predicting oral absorption and hepatobiliary clearance: human intestinal and hepatic in vitro cell models. Environ Toxicol Pharmacol 2006;21:168–78
- Nies AT, Koepsell H, Damme K, Schwab M. Organic cation transporters (OCTs, MATEs), in vitro and in vivo evidence for the importance in drug therapy. In: Fromm MF, Kim RB, eds. Drug transporters. Handbook of experimental pharmacology. Vol. 201. Berlin, Heidelberg: Springer-Verlag; 2011:105–67
- Lozano E, Herraez E, Briz O, et al. Role of the plasma membrane transporter of organic cations OCT1 and its genetic variant in modern liver pharmacology. Biomed Res Int 2013;2013:692071