
Abstract
Previously, a glycoglycerolipid isolated from marine algae was reported to be a potent and selective inhibitor of the human Myt1 kinase, an enzyme involved in cell cycle regulation with great potential as an anti-cancer target. Based on that report, a lot of research effort has been invested by several working groups to synthesize and derivatize this compound. However, reliable assay data confirming the inhibitory potential and the mechanism of action of these glycoglycerolipids are missing so far. Here, based on experimental data and theoretical considerations, we show that the aforesaid glycoglycerolipid 1,2-dipalmitoyl-3-(N-palmitoyl-6′-amino-6′-deoxy-α-d-glucosyl)-sn-glycerol is not an inhibitor of the human Myt1 kinase.
Introduction
Myt1 is a cell cycle regulating kinase with great potential as a drug targetCitation1. However, it cannot be sufficiently addressed by inhibitors so farCitation2. In 2005, a report by Zhou et al. described the identification of a marine natural product as a potent and selective Myt1 kinase inhibitorCitation3. The most potent identified compound was the glycoglycerolipid 1,2-dipalmitoyl-3-(N-palmitoyl-6′-amino-6′-deoxy-α-d-glucosyl)-sn-glycerol (GGL1, see ). It was found to inhibit Myt1 potently (IC50 = 0.12 µg/ml, equals 124 nM) without affecting two other kinases (Akt and Chk1)Citation3.
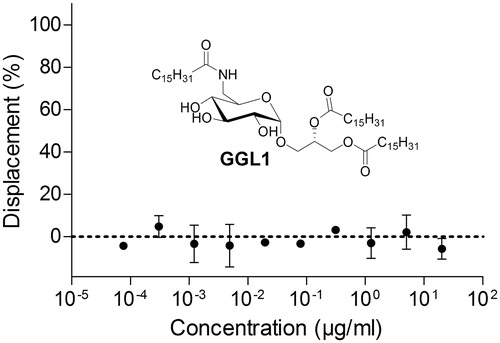
Figure 1. The reported Myt1 glycoglycerolipid inhibitor GGL1 did not have any effect in a fluorescence anisotropy-based Myt1 kinase binding assay. Anisotropies were normalized against positive controls (10 µM dasatinib; displacement 100%) and vehicle (1% DMSO; displacement 0%). Data represent means ± SEM (n = 3).
That report gained a lot of attention and several research groups, including ours, synthesized and derivatized the compound for further biological evaluationCitation4–8. However, presumably caused by the restrictive substrate acceptance of this kinaseCitation9, reliable assay data confirming the original report and elucidating the mechanism of action are still missing.
Recently, besides a set of common kinase inhibitors, we tested GGL1 and a set of derivatives in a TR-FRET-based kinase binding assay utilizing full-length Myt1, but no effect could be determined in concentrations up to 250-fold the reported IC50Citation7. As such an absolute discrepancy between functional and binding data is very uncommon in kinase research, further studies were required.
Methods
Unless stated otherwise, all reagents were purchased from Sigma (Schnelldorf, Germany) and were at least of analytical grade. Dasatinib was from LC Laboratories (Woburn, MA).
In this study, Myt1 was used as a full-length protein and as the isolated kinase domain. In protein generation processes, all steps were carried out at +4 °C. Purification procedures were controlled by coomassie-stained SDS-PAGE analysis. Determination of protein concentrations was carried out via BCA-assay as described previously using BSA as protein standardCitation10. Identity of the proteins was, additionally, confirmed via Western blotting experiments (anti-Myt1 and anti-His tag). Solutions containing eluted protein were frozen in EtOH/dry ice and stored at −80 °C.
Generation of full-length Myt1
Full-length Myt1 was expressed in the human cell line HEK293 as described beforeCitation7. Briefly, cells were cultured at 37 °C in Dulbecco's modified Eagle medium (DMEM), supplemented with 10% FCS and GlutaMAX in a humidified incubator. Transfection was carried out with pcDNA3.1-His-Myt1 expression plasmid using lipofectamine 2000 (Invitrogen, Madison, WI) according to recommendations of the manufacturer. Cells were harvested and resuspended in lysis buffer containing 25 mM Tris-HCl pH 7.8,150 mM NaCl, 10 mM MgCl2, 10% glycerol, 1% Triton X-100 and Complete EDTA-free protease inhibitor cocktail (Roche, Basel, Switzerland). Cells were finally lysed by shear stress using a 25 g needle and the lysate shaken on ice for 1 h. After centrifugation (10 000g) for 15 min, the supernatant was loaded onto a HIS-Select column (Sigma, Schnelldorf, Germany) pre-equilibrated with buffer A (50 mM Tris-HCl pH 7.8; 150 mM NaCl; 3 mM MgCl2; 20% glycerol; 0.5% Triton X-100). The column was washed with buffer B buffer A + 5 mM imidazole) and eluted with buffer C (buffer A + 250 mM imidazole). The final eluate was supplemented with DTT (2 mM).
Generation of the isolated Myt1 kinase domain
The Myt1 kinase domain (comprising amino acids 75–362) was generally expressed and purified as described recentlyCitation11. The corresponding gene sequence of a Myt1 fragment (Myt175-362) was cloned in pET28 plasmide and transformed into E. coli BL21(DE3) by electroporation. After inoculation of 1 l LB media, the cells were cultured at 37 °C until OD600 reached 0.6. Following subsequent cultivation at 22 °C until OD600 = 1.0, expression was induced by addition of IPTG (0.5 mM). Cells were harvested by centrifugation, the pellet resuspended in binding buffer and frozen at −20 °C. Thawed bacterial cells were disrupted by sonication on ice. Subsequently, after another centrifugation step, the supernatant containing the His6-fusion protein was purified by affinity chromatography (1 ml Ni2+ HiTrap column, GE Healthcare Life Sciences, Freiburg, Germany) according to the recommendations of the manufacturer.
Kinase binding assay
The utilized Myt1 kinase binding assay followed a procedure described recentlyCitation11. Assays were conducted in black 96-well half-area microplates (Corning Incorporated, Corning, NY) at an assay volume of 27 µl in sterile-filtered binding assay buffer (50 mM HEPES-NaOH pH 7.5, 10 mM MgCl2, 0.03% CHAPS, 1 mM DTT). A final DMSO concentration of 1% was included in all experiments. Tracer (5 nM) was incubated with Myt175-362 (100 nM) in binding assay buffer at various concentrations of GGL1. After the incubation period, fluorescence anisotropy (485 nm Ex/520 nm Em) was measured using a PolarStar OMEGA plate reader (BMG, Heidelberg, Germany). Samples containing 10 µM dasatinib and vehicle (no compound; 1% DMSO) were carried along as controls and used for normalization purposes. Using this assay, the Myt1 kinase inhibitors dasatinib and PD166285 delivered Ki values of 73.2 nM and 2.02 nM, respectively.
Functional assay
To monitor Myt1 kinase activity, GST-Cdk1 (125 nM; ProQinase, Freiburg, Germany) was incubated with full-length Myt1 (50 nM) and ATP (250 µM) for 2 h at 30 °C in kinase buffer consisting of 50 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 40 mM NaCl, 0.04% Triton X-100, 1 mM DTT, 2 mM molybdate, Complete EDTA-free protease inhibitor cocktail. The kinase reaction was terminated by addition of 20 µl stop-solution (100 mM EDTA, pH 7.5). Protein was isolated by trichloroacetic acid (TCA) precipitation following standard protocols, dried in vacuo and redissolved in SDS-PAGE loading buffer. SDS-PAGE was carried out discontinuously with 4%/12% gels (C = 2.6%). Antibodies used in Western blot experiments were: anti-Cdk1 antibody (#9112, Cell Signaling Technology, Beverly, MA) and anti-phospho-Cdk1(Tyr15) antibody (#9111, Cell Signaling Technology, Beverly, MA). Detection was carried out by enhanced chemiluminescence (Amersham Western Blot analysis system, GE Healthcare Life Sciences).
Critical micelle concentration (CMC) assay
A fluorimetric CMC assay based on insertion of 1,6-diphenyl-1,3,5-hexatriene (DPH) was carried out as described beforeCitation12. The same microplates as for kinase binding studies were used at an assay volume of 60 µl. Fluorescence was measured using a NOVOstar plate reader (BMG) at filter settings of 355 nm Ex/460 nm Em. CMC values obtained for the positive control Triton X-100 were in accordance with literature values.
Results and discussion
To confirm our results with the kinase binding assay utilizing full-length Myt1, another orthogonally acting binding assay approach was taken. Recently, we described a fluorescence anisotropy-based Myt1 kinase binding assay that uses the Myt1 kinase domain as a model protein, delivering data in excellent agreement with full-length dataCitation11. The assay proved suitable for the identification of novel Myt1 inhibitors in screening processesCitation13. Using this assay, we tested GGL1 in concentrations up to 30 µg/ml, equal to 250-fold the reported IC50. Due to the poor solubility of GGL1, it was not possible to use higher concentrations.
In the kinase domain binding assay, no displacement of the kinase tracer was determined (). The responses at different concentrations scattered around the baseline (no displacement of the kinase tracer), meaning that no binding to the Myt1 kinase domain was detected. The glycoglycerolipid GGL1 had no effect in this assay.
These results confirm our previous assay data generated in the TR-FRET-based assay.
Generally, two explanations are conceivable: Either this compound unfolds its effect at a site distant to the ATP-binding site (type III inhibitor), or the original publication has to be doubted.
Due to steric reasons caused by three fatty acid chains in the glycoglycerolipid structure, a mechanism of action outside the ATP-binding site is likely, making it type III instead of ATP-competitive. Even though most type III inhibitors do not directly bind to the ATP site, the majority must either alter the active site in a way that displaces the tracer or bind close to the active site. The TR-FRET-based assay used in our previous report was, meanwhile, shown to have excellent performance concerning the identification of allosteric kinase inhibitorsCitation14. Based on the assay data presented, an interaction of GGL1 with Myt1 at a site close to the kinase domain appears to be unlikely. Effects on substrate recognition processes might be responsible for their reported inhibitory potential. To verify or falsify this hypothesis, a functional assay with the native protein substrate was required.
Therefore, we used a similar approach as reported before to monitor the Myt1 kinase activityCitation9. Recombinant GST-Cdk1 was incubated with full-length Myt1 and ATP in the presence or absence of GGL1 (30 µg/ml). Samples containing 10 µM dasatinib, a confirmed Myt1 kinase inhibitorCitation7,Citation15,Citation16, were carried along as inhibitor controls and samples without ATP were used as background controls. Myt1 was present in all samples.
After incubation, the reaction was terminated by addition of EDTA. The protein was isolated by TCA precipitation and prepared for SDS-PAGE and subsequent Western blotting. As primary antibodies, anti-pTyr15 Cdk1 antibody was used to visualize Myt1 kinase activity and anti-Cdk1 total antibody was used as loading control to exclude protein loss caused by work up and handling. Representative western blot results are displayed in .
Figure 2. Representative Western blot results for investigations on the inhibitory properties of the glycoglycerolipid GGL1 (30 µg/ml) on full-length Myt1. Myt1 phosphorylates Cdk1 at Tyr-15 (compare lanes 1 and 2). GGL1 does not alter the extent of phosphorylation (lane 3). Dasatinib (10 µM) was used as an inhibition control and efficiently suppresses Cdk1-phosphorylation (lane 4). Myt1 and Cdk1 were present in all samples.
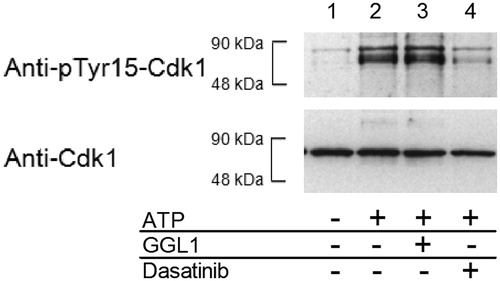
As indicated by the anti-Cdk1 total antibody, an even loading of the samples can be assumed. Therefore, differences in the detected phosphorylation status can be considered a result from the Myt1 kinase reaction. As expected, the control without ATP (lane 1) showed a very low basal phosphorylation of Cdk1, while the kinase reaction (conversion control; containing substrate, co-substrate and kinase; lane 2) drastically altered the phosphorylation status. Addition of GGL1 (lane 3) did not reduce the formation of phospho-Tyr15-Cdk1 compared to the conversion control. The addition of 10 µM dasatinib (lane 4), however, was indeed able to suppress the kinase reaction. The amount of detected kinase reaction product remained close to the basal phosphorylation. The formation of phosphorylated product could not be totally suppressed which is mainly caused by the conditions used in this experiment (large excess of ATP and long incubation periods).
Again, the concentration of GGL1 used here is about 250 times higher than the reported IC50 value. For dasatinib, 10 µM equals approximately 80- to 160-fold the reported IC50 valuesCitation7,Citation15. No relevant functional inhibition occurred in presence of GGL1. Apparently, GGL1 is not able to inhibit Myt1-mediated phosphorylation of Cdk1.
A closer look at the only assay used in the original report may help explain the discrepancy compared to our findings. In fact, it reveals some conceptual deficits: A fluorescence polarization immunassay was conducted and Kristjánsdóttir and Rudolph were cited as a referenceCitation17. However, the assay by Zhou et al. was run in a direct instead of a competitive approach using a fluorescently labelled Cdk1-derived peptide substrate. Interestingly, two independent reports suggest that Myt1 does not accept short Cdk1-derived peptides as substratesCitation9,Citation17. For this reason, Kristjánsdóttir and Rudolph had to use a protein substrate instead of a peptide substrate for their final Myt1 assayCitation17. Moreover, direct fluorescence polarization assays carry some inherent limitations. In principle, a labelled substrate is directly phosphorylated and, subsequently, bound by an antibody. The resulting fluorescence polarization will be increased compared to the free (i.e. unbound) probe. According to Equation (1), the measured fluorescence anisotropy r is an additive entity based on the intrinsic anisotropies of the free and bound fluorescent probe (rb and rf) together with their respective fractions (fb and ff)Citation18.
(1)
The unphosphorylated peptide will not be bound by the antibody but, as it is labelled, it would still be present in considerable amounts and interfere with the assay by narrowing the dynamic rangeCitation19. As shown by Wu, substrate conversion of at least 50% is required to overcome this effect and to yield an appropriate assay windowCitation20.
In addition, if it is taken into account that the substrate probe was used at a concentration of 2.5 nM, such a conversion is intrinsically limited by diffusion. Altogether, as no illustrations were included that allow for assessment of the data quality, the assay used to prove the inhibitory effects of GGL1 on Myt1 is not very trustworthy.
As pointed out by Tang, co-author of the original GGL1 report, the inhibition was initially determined using the compound directly as isolated from marine algae. After elucidation of the chemical structure and chemical synthesis of the compound, even the identical assay failed to confirm the inhibitory potency of GGL1Citation21. These facts add some more doubt to the initial results published in 2005 by Zhou et al.
As a possible source of error, we examined assay interference by insertion of the labelled substrate into association colloids formed by GGL1. As this is a generic problem for assays with a fluorescence polarization readoutCitation22, reagents/detergents with a low CMC are to be avoided in the respective assay bufferCitation11,Citation23.
To investigate whether GGL1 might be prone to formation of association colloids at relevant concentrations, DPH inclusion assays were conducted. DPH is a fluorophore which offers a vastly increased quantum yield upon insertion into a lipophilic environment. By monitoring the fluorescence intensity in the presence of different amounts of compound, a sensitive detection of association colloids is possibleCitation12. Using this sensitive method, no formation of association colloids was detected up to 40 µg/ml. Therefore, insertion phenomena are unlikely to be responsible for the false-positive results.
Besides interesting carbohydrate chemistry, the latest publication dealing with these glycoglycerolipids as Myt1 inhibitors attempts to provide preliminary biological dataCitation8. However, although using the well-known ADP-Glo kinase assay platformCitation24, the results reported should be handled cautiously. No validation data such as IC50 values for known inhibitors were included and source/constitution/concentration of critical reagents (e.g. Myt1, peptide substrate) were not mentioned. The compounds were tested at a single concentration without reported replicates and, most importantly, the actual test concentration used was not stated. Staurosporine (concentration not stated) was used as a positive control and exploited the assay window to only 65%. To be noted, a more general issue is raised by the use of staurosporine for this purpose. Independent reports describe Myt1 to be one of the few examples that are insensitive towards these compoundsCitation7,Citation15. Other compounds with confirmed inhibitory potency towards Myt1, such as dasatinib or PD166285, should be preferred.
Conclusions
The present report described all Myt1 test results for GGL1 and its derivatives that have been described so far and expands the knowledge by providing some more assay data. As far as we are concerned, GGL1 did not affect full-length Myt1 in a well-validated kinase binding assay. Also, the kinase domain in another validated Myt1 kinase binding assay was not affected and even a functional assay with full-length Myt1 utilizing the native protein substrate failed to reproduce the initial results obtained by Zhou et al. Altogether, theoretical considerations and experimental data suggest that the glycoglycerolipid 1,2-dipalmitoyl-3-(N-palmitoyl-6′-amino-6′-deoxy-α-d-glucosyl)-sn-glycerol is not an inhibitor of the human Myt1 kinase.
Acknowledgements
We thank research funds from “Kultusministerium des Landes Sachsen-Anhalt” for the doctoral fellowship to C. Göllner.
Declaration of interest
The authors declare that there are no competing interests. This work was supported by the European Regional Development Fund of the European Commission.
References
- Chow JPH, Poon RYC. The CDK1 inhibitory kinase MYT1 in DNA damage checkpoint recovery. Oncogene 2013;32:4778–88
- Tibes R, Bogenberger JM, Chaudhuri L, et al. RNAi screening of the kinome with cytarabine in leukemias. Blood 2012;119:2863–72
- Zhou BN, Tang S, Johnson RK, et al. New glycolipid inhibitors of Myt1 kinase. Tetrahedron 2005;61:883–7
- Wu HJ, Li CX, Song GP, Li YX. Synthesis of natural α-6-dehydroxy-6-aminoglucoglycerolipids. Chin J Chem 2008;26:1641–6
- Göllner C, Philipp C, Dobner B, et al. First total synthesis of 1,2-dipalmitoyl-3-(N-palmitoyl-6′-amino-6′-deoxy-alpha-d-glucosyl)-sn-glycerol -- a glycoglycerolipid of a marine alga with a high inhibitor activity against human Myt1-kinase. Carbohydr Res 2009;344:1628–31
- Sun Y, Zhang J, Li C, et al. Synthesis of glycoglycerolipid of 1,2-dipalmitoyl-3-(N-palmitoyl-6′-amino-6′-deoxy-α-d-glucosyl)-sn-glycerol and its analogues, inhibitors of human Myt1-kinase. Carbohydr Res 2012;355:6–12
- Rohe A, Göllner C, Wichapong K, et al. Evaluation of potential Myt1 kinase inhibitors by TR-FRET based binding assay. Eur J Med Chem 2013;61:41–8
- Li C, Sun Y, Zhang J, et al. Synthesis of 6′-acylamido-6′-deoxy-alpha-d-galactoglycerolipids. Carbohydr Res 2013;376C:15–23
- Rohe A, Erdmann F, Bäßler C, et al. In vitro and in silico studies on substrate recognition and acceptance of human PKMYT1, a Cdk1 inhibitory kinase. Bioorg Med Chem Lett 2012;22:1219–23
- Smith PK, Krohn RI, Hermanson GT, et al. Measurement of protein using bicinchoninic acid. Anal Biochem 1985;150:76–85
- Rohe A, Henze C, Erdmann F, et al. A fluorescence anisotropy based Myt1 kinase binding assay. Assay Drug Dev Technol 2014;12:136–44
- Chattopadhyay A, London E. Fluorimetric determination of critical micelle concentration avoiding interference from detergent charge. Anal Biochem 1984;139:408–12
- Wichapong K, Rohe A, Platzer C, et al. Application of docking and QM/MM-GBSA rescoring to screen for novel Myt1 kinase inhibitors. J Chem Inf Model 2014;54:881–93
- Lebakken CS, Reichling LJ, Ellefson JM, Riddle SM. Detection of allosteric kinase inhibitors by displacement of active site probes. J Biomol Screen 2012;17:813–21
- Davis MI, Hunt JP, Herrgard S, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotech 2011;29:1046–51
- Hantschel O, Rix U, Superti-Furga G. Target spectrum of the BCR-ABL inhibitors imatinib, nilotinib and dasatinib. Leuk Lymphoma 2008;49:615–19
- Kristjánsdóttir K, Rudolph J. A fluorescence polarization assay for native protein substrates of kinases. Anal Biochem 2003;316:41–9
- Jameson DM, Ross JA. Fluorescence polarisation/anisotropy in diagnostics and imaging. Chem Rev 2010:2685–708
- Roehrl MHA, Wang JY, Wagner G. A general framework for development and data analysis of competitive high-throughput screens for small-molecule inhibitors of protein–protein interactions by pluorescence polarization. Biochemistry 2004;43:16056–66
- Wu G. Assay development: fundamentals and practices. Hoboken (NJ): John Wiley & Sons, Inc., 2010:187–99
- Tang S. Structural and synthetic studies of bioactive natural products. Blacksburg (VA): Virginia Polytechnic Institute and State University; 2005:91–110
- Thorsteinsson MV, Richter J, Lee AL, DePhillips P. 5-Dodecanoylaminofluorescein as a probe for the determination of critical micelle concentration of detergents using fluorescence anisotropy. Anal Biochem 2005;340:220–5
- Munoz L, Selig R, Yeung YT, et al. Fluorescence polarization binding assay to develop inhibitors of inactive p38 alpha mitogen-activated protein kinase. Anal Biochem 2010;401:125–33
- Zegzouti H, Zdanovskaia M, Hsiao KGoueli SA. ADP-Glo: a bioluminescent and homogeneous ADP monitoring assay for kinases. Assay Drug Dev Technol 2009;7:560–72