
Abstract
Tuberculosis (TB) is still a major health concern worldwide. The increasing incidences of multi-drug-resistant tuberculosis (MDR-TB) necessitate the development of new anti-TB drugs acting via novel mode of action. The search of newer drugs for TB led to the identification of several quinoline-based antimycobacterial agents against both the drug-sensitive and MDR-TB. These agents have been designed by substituting quinoline scaffold with diverse chemical functionalities as well as by modifying quinoline/quinolone-based antibacterial drugs. Several of quinoline/quinolone derivatives displayed excellent antimycobacterial activity and were found free of cytotoxicity. This review highlights the critical aspects of design and structure–activity relationship of quinoline- and quinolone-based antimycobacterial agents.
Introduction
Tuberculosis (TB), caused by Mycobacterium tuberculosis, is one of the oldest life-threatening infectious diseases worldwide. TB affects people of all age groups; however, people with weak immune system are more prone to develop TB. According to World Health Organization, approximately 8.7 million people were affected with TB and 1.4 million died from TB in 2011. Over 95% of deaths occur in countries with lower economyCitation1. Patients with HIV infection and other compromised immune system are more prone to develop TB. At least one-third of the 34 million people with HIV infection worldwide are infected with M. tuberculosis, and they are 21–34 times more likely to develop active TB than people without HIV infectionCitation1. The isoniazid (INH), rifampicin, pyrazinamide and ethambutol are potent first-line antitubercular drugs (). But, in current scenario, the treatment of TB using first-line antitubercular drugs becomes ineffective due to the emergence of multi-drug-resistant tuberculosis (MDR-TB) and extensively drug-resistant strains of Mycobacterium (XDR-TB). Therefore, the development of newer antimycobacterial drugs based on scaffolds having the potential to inhibit the growth of M. tuberculosis is of current interest ().
Figure 1. Structure of first-line antimycobacterial drugs.
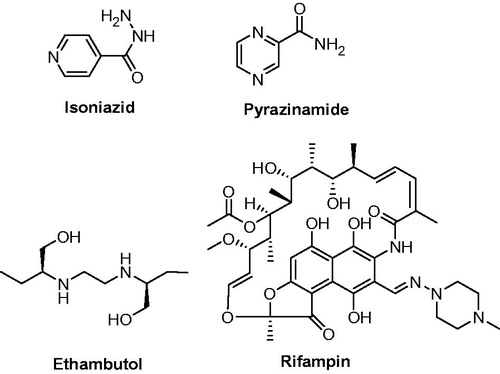
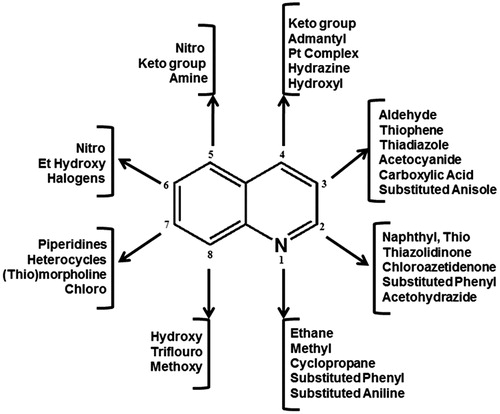
Figure 2. Substitutions on quinoline to develop new antimycobacterial agents.
TMC207 (a diarylquinoline) is a novel antitubercular drug and currently under the Phase II studies for the treatment of patients infected with MDR-TB ()Citation2. TMC207 acts via inhibiting the mycobacterial adenosine triphosphate (ATP) synthase in Mycobacterium and thereby presents a novel mechanism to control the growth of M. tuberculosisCitation3. Potent bactericidal and sterilizing activity of TMC207 against M. tuberculosis and other mycobacterial species makes it an attractive drug candidate to fight against TB. In a Phase II clinical trial on patients with MDR-TB, TMC207 showed significant efficacy after 2 months of treatment and appeared to be safe and well toleratedCitation4. Haagsma et al.Citation5 studied the selectivity of TMC207 towards M. tuberculosis ATP synthase over the eukaryotic homologue and found that TMC207 is free from ATP synthesis-related toxicity in mammalian cells. TMC207, when used alone or in combination with existing antimycobacterial drugs, represented a promising future for the treatment of TBCitation6,Citation7. In 2012, it was approved by U.S. Food and Drug Administration as a new drug to treat MDR-TBCitation8.
Figure 3. Structure of quinoline, floroquinolones and TMC207.
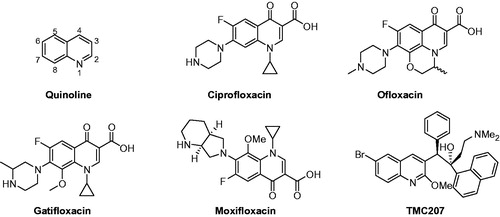
The identification of TMC207 as a new class of antimycobacterial drug with a novel mode of action attracted the attention of medicinal chemists to explore quinoline as a potential scaffold to develop antimycobacterial drugs (). Also, the fluoroquinolone class of antibacterial drugs like ciprofloxacin, ofloxacin, gatifloxacin and moxifloxacin has been used effectively as second-line drugs for the treatment of MDR-TB (). These drugs target Mycobacterium DNA gyrase and are prescribed in combination with first-line and other second-line antimycobacterial drugsCitation9. This further augmented the probability of finding new quinoline-based antimycobacterial drugs. Therefore, numerous quinoline derivatives and analogs have been synthesized and evaluated for their efficacy against drug-sensitive and multi-drug-resistant Mycobacterium. Some studies have come out with the identification of effective quinoline-based antimycobacterial agents without cytotoxicity. This article will highlight the design and structure–activity relationship (SAR) aspects of quinoline-based novel antimycobacterial agents.
Substituted quinolines
2,3-Disubstituted quinolines
Mistry et al. synthesized novel quinoline-based azetidinone (1) and thiazolidinone (2) analogs and evaluated them for antimycobacterial activity. The SAR in both series of compounds showed that 4-chloro- and 4-fluoro-substituted analogs possess better antimycobacterial activity (62.5 µg/mL against M. tuberculosis H37Rv; Rifampicin: 40 µg/mL) than 2/3-chloro-, 2/3-fluoro-, nitro- and methyl-substituted analogs (125–500 µg/mL). A 2-amino-5-methyl-thiazole-substituted thiazolidonone analog also showed comparable antimycobacterial activity (3, MIC: 62.5 µg/mL)Citation10,Citation11. Two series of phenoxy-linked quinoline (4) and bisquinoline (5) derivatives were synthesized and screened in vitro for their activity against M. tuberculosis H37Rv. The study showed that bisquinoline series of compounds possesses better antimycobacterial activity (5, MIC: 1.1–43 µM) compared to quinoline derivatives (4, MIC: 11.8–55.6 µM). Among all the compounds, the 5a and 5b were found to be the most active. The in vitro cytotoxicity (IC50) of 5a and 5b on mouse embryonic fibroblast cell line (NIH 3T3) was reported as 333 and 887 µM, respectively, indicating that 37a and 37b are not cytotoxicCitation12.
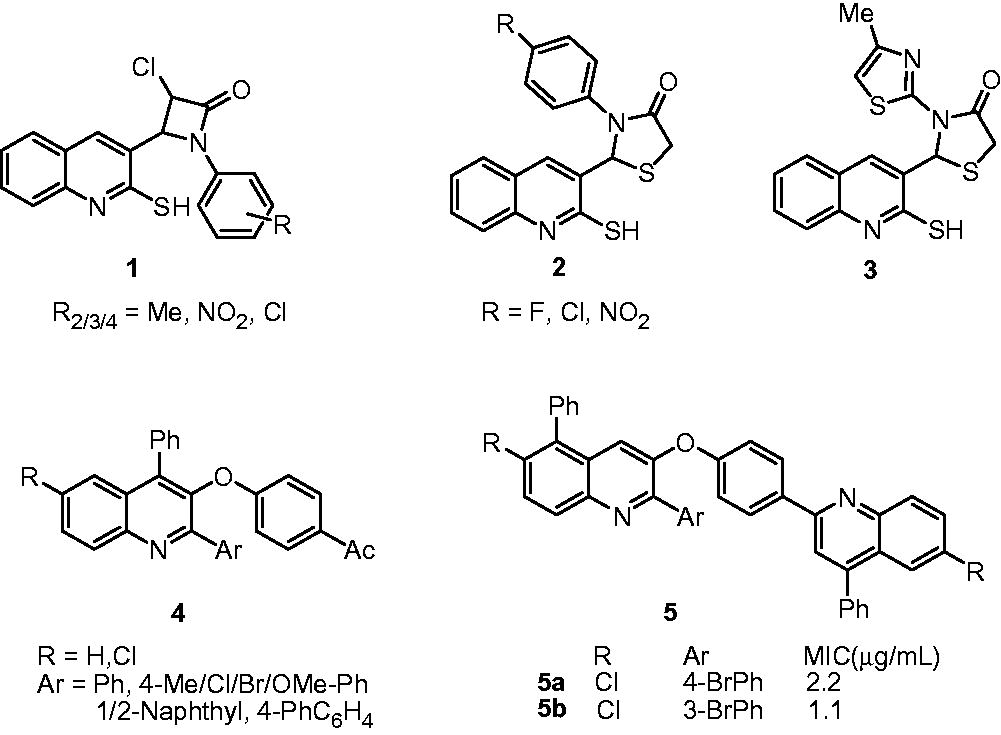
Antimycobacterial activity of 2-aryloxyquinolines (6) and their pyrano[3,2-c]chromene (7) derivatives were assessed by Mungra et al. Among the synthesized compounds, 6a and 7a showed more than 90% growth inhibition at the concentration of 260 and 250 µg/mL, respectivelyCitation13. A series of 3-benzyl-6-bromo-2-methoxy-quinolines (8) and amides of 2-[(6-bromo-2-methoxyquinolin-3-yl)-phenylmethyl]-malonic acid monomethyl ester (9) were synthesized and evaluated for their antimycobacterial activity. Substituted-3-benzyl-6-bromo-2-methoxy-quinoline analogs, that is, 8a, 8b, 8c and 8d, exhibited 94.2%, 99.5%, 92.5% and 100% growth inhibition of M. tuberculosis H37Rv, respectively, at a concentration of 6.25 µg/mL. However, their amide derivatives (9) showed poor activity (less than 22% growth inhibition)Citation14. In the cytotoxicity study on murine macrophage cells, 8c showed 100% cell viability at the concentration of 10 µg/mL. Eswaran et al. reported the synthesis and antimycobacterial activity of 3-(N-substituted urea)quinolines (10) and their cyclized oxazoloquinoline derivatives (11). Some of these compounds displayed good activity (MIC: 1 µg/mL) compared with standard INH and rifampicin (MIC: 1.5 and 0.5 µg/mL respectively). The SARs indicated that the methoxy-phenyl, 3-chloro-phenyl and 4-fluoro-phenyl substituents at R-position are essential for activity. The cyclization of 3-(N-substituted urea)quinolines (10) to oxazoloquinolines (11) also improved the activityCitation15.
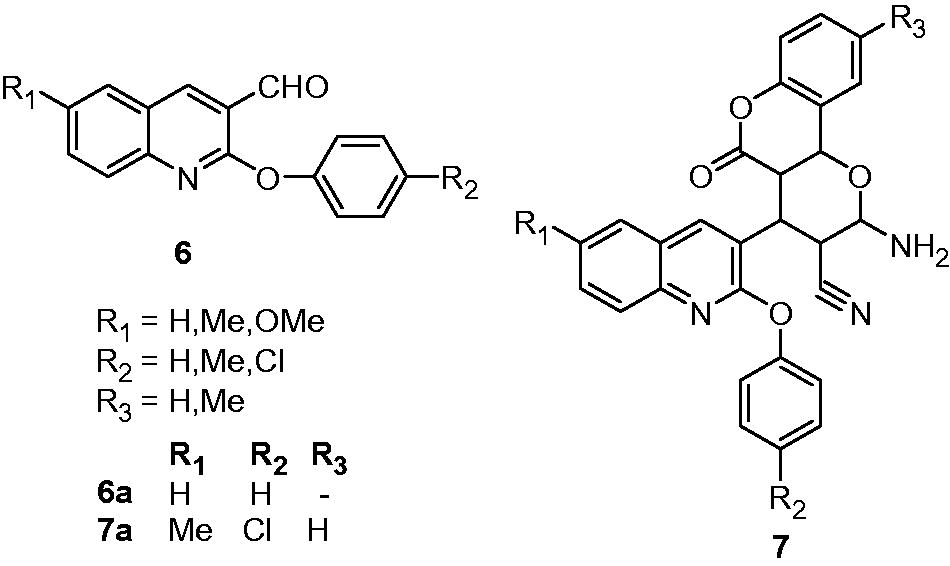
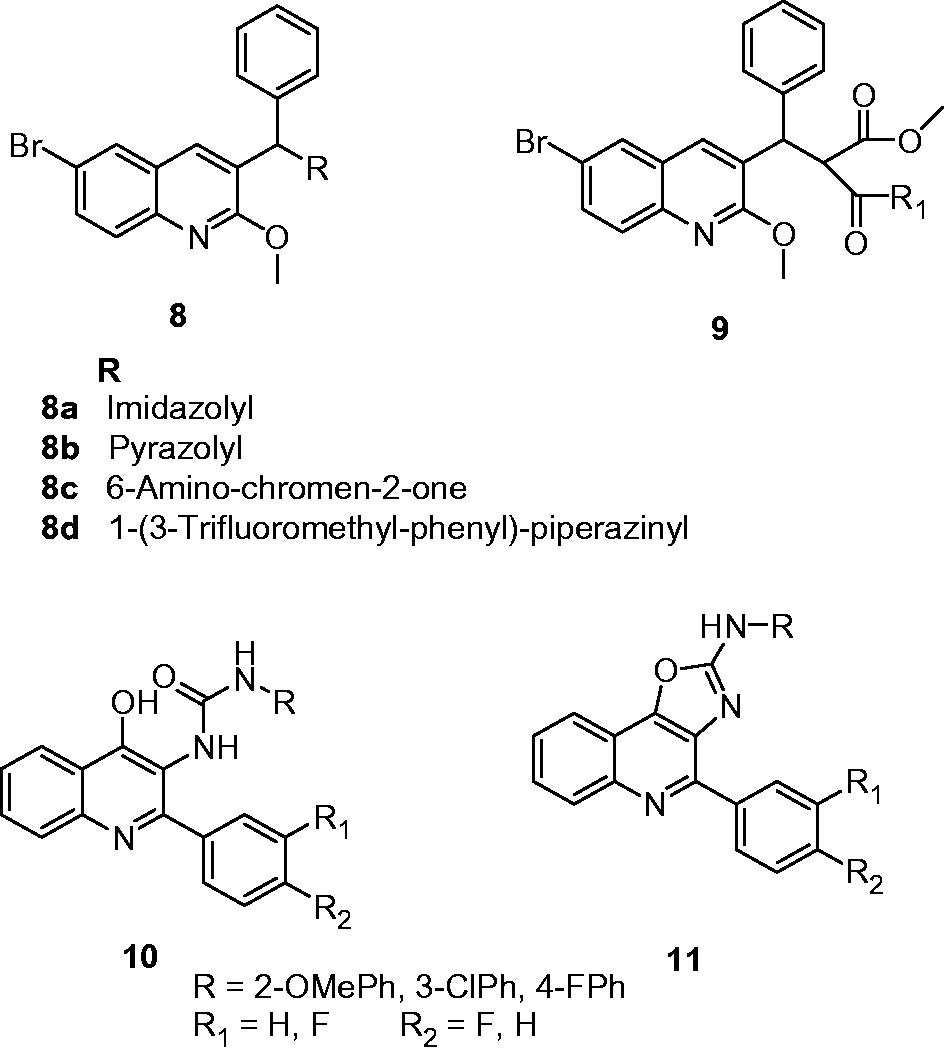
2,4-Disubstituted quinolines
Nayyar et al. reported synthesis, antimycobacterial activity and 3D-QSAR studies on two series of 4-(adamantan-1-yl)-2-substituted quinolines, namely 4-adamantan-1-yl-quinoline-2-carboxylic acid N′-alkylhydrazides and N′,N′-dialkylhydrazides (12) and 4-adamantan-1-yl-quinoline-2-carboxylic acid alkylidenehydrazides (13). On activity evaluation against M. tuberculosis H37Rv, the compounds of these series displayed moderate-to-good antimycobacterial activity in the MIC range of 1.0–6.25 µg/mL compared with INH (MIC: 1 µg/mL). For 4-adamantan-1-yl-quinoline-2-carboxylic acid N′-alkylhydrazides and N′,N′-dialkylhydrazides (12), the SAR suggested that five- and six-membered ring or aryl substituents at R1 and a hydrogen at R2 are favorable for the activity. In 4-adamantan-1-yl-quinoline-2-carboxylic acid alkylidenehydrazides (13), the substitution with small aryl/heteroaryl rings exhibited better activity than other bulkier substituents. The comparative molecular field analysis (CoMFA) indicated that the branched chain and cycloalkyl substituents are favored on the hydrazine nitrogen (12) and phenyl/quinoline substituents are favored on hydrazone nitrogen (13). Also, small electronegative substituents are conducive for the activity. Compound 13a was identified as the most potent compound which exhibited 99% inhibition with the MIC of 1 µg/mL, equivalent to INH (MIC: 1 µg/mL; 99% inhibition)Citation16.
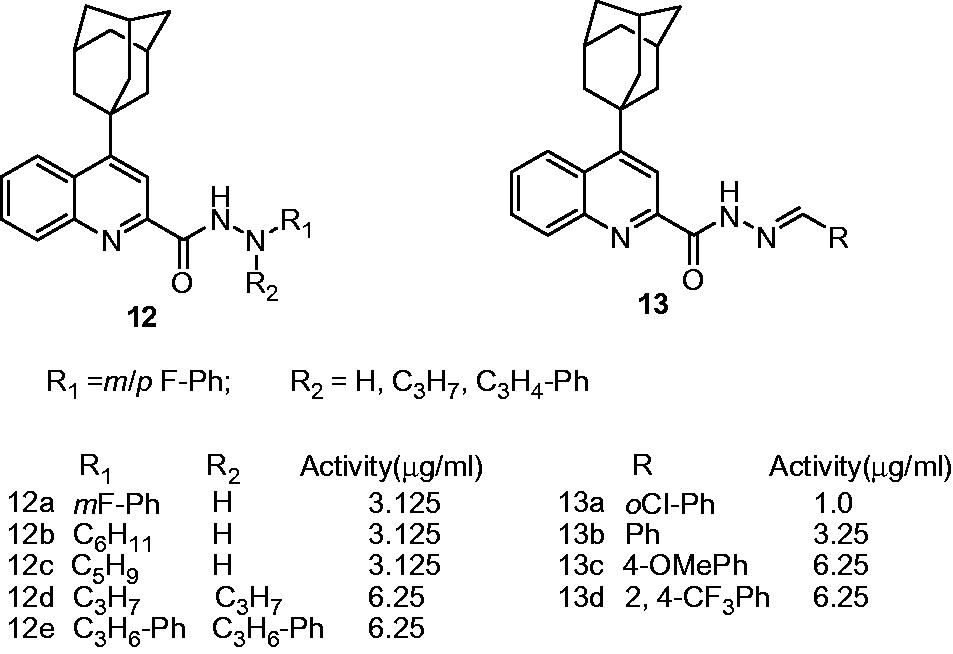
The further development of 4-(adamantan-1-yl)-2-substituted quinolines led to the development of four series of antimycobacterial compounds, namely: {1-[N′-(4-adamantan-1-yl-quinoline-2-carbonyl)hydrazinocarbonyl]alkyl carbamic acid tert-butyl esters (14); 4-adamantan-1-yl-quinoline-2-carboxylic acid N′-(2-aminoalkyl)hydrazides (15); methyl 2-[4-(adamantan-1-yl)-2-quinonylcarboxamido]alkanoates (16) and N2-[1-hydrazinocarbonylalkyl]-4-(adamantan-1-yl)-2-quinolinecarboxamides (17)Citation17. In this study, 14a, 14b, 14c, 16a and 16b were identified as potent analogs and showed 99% inhibition of M. tuberculosis H37Rv at the concentration of 3.125 µg/mL. Compound 17a displayed best activity (99% inhibition) against drug-sensitive M. tuberculosis H37Rv strain at 1.00 µg/mL, comparable to standard drug INH used in this study (INH: 99% inhibition at MIC of 1 µg/mL). The SAR of series 14 and 15 indicated that the presence of t-Boc [–NHCO2C(CH3)3] group imparts higher potency when compared to the compounds with free amino group at the side chain. For example, 14a showed 99% inhibition at 3.125 µg/mL, while 15a showed only 56% inhibition at 6.25 µg/mL. Also, in series 16 and 17, the analogs conjugated with basic heteroaromatic residue like l-histidine and highly cationic l-arginine residue were found more potent when compared to the analogs conjugated with hydrophobic and other residues. All the compounds, namely 14a, 14b, 14c, 16a, 16b and 17a were found to be noncytotoxic up to the concentration of 50 µg/mL on mammalian kidney fibroblast (Vero) cells. The 3D-QSAR study suggested that aromatic amino acid side chains bearing an electropositive group or aliphatic side chains with electronegative substitutions would be favorable for antimycobacterial activity. Also, bulkier groups are detrimental for the activity due to steric effectCitation17.
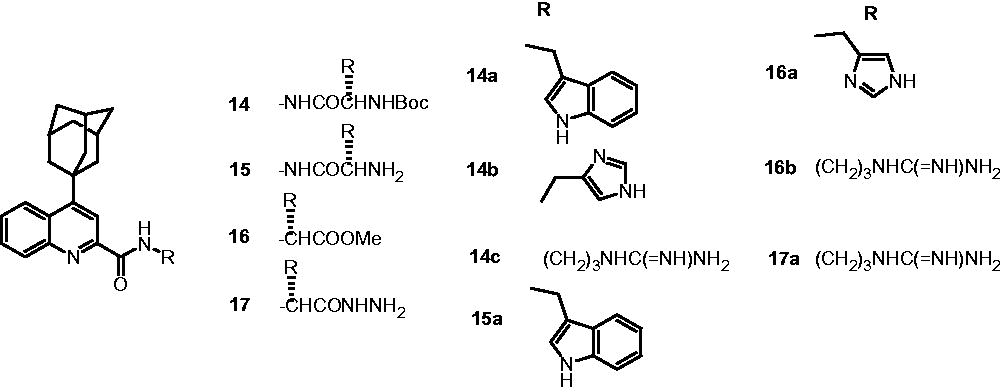
3,4-Disubstituted quinolines
Thomas et al. reported the synthesis and antimycobacterial activities of three series of 4-hydroxy-8-trifluoromethyl-quinoline derivatives, i.e. 4-hydroxy-8-(trifluoromethyl)quinoline-3-carbohydrazone (18), 2-{[4-hydroxy-8-(trifluoromethyl)quinolin-3-yl]carbonyl}hydrazinecarbothioamide (19) and 8-(trifluoromethyl)quinolin-3-yl]carbonyl}-hydrazinecarboxamide (20) against drug-sensitive and MDR strains of Mycobacterium. The evaluation of the activity of these compounds was carried out against different Mycobacterium strains. In comparison with INH and rifampicin, some of the synthesized compounds displayed good activity against drug-sensitive and MDR strains of Mycobacterium in comparison with INH and rifampicinCitation18. Eswaran et al. reported the synthesis and antimycobacterial activity of new quinoline derivatives and highlighted the effect of substituted hydrazones at position C-3 of the quinoline (21). Few compounds have shown good antimycobacterial activity in MIC ranging from 0.625 to 1.25 µg/mL against M. tuberculosis. The SAR indicated that 3-pyridyl, 3-(1,1,2,2-tetrafluoro ethoxy)phenyl, octyl, 2-fluoro-phenyl and 4-fluoro-phenyl at R1 position improve the activity while 5-bromo-thiophene, 5-fluoro-2-hydroxy phenyl, 2-fluoro-4-methoxy-phenyl, 3-(methylthio)propyl and 4-methoxy-phenyl decrease the activity. Also, (3R)-3-amino-N,N-dimethyl-4-(phenylthio)butanamide moiety at positions C-4 of quinoline skeleton showed profound effect of antimycobacterial activity. Compounds 21a and 21b were identified as the most active compound with a MIC of 0.625 µg/mL and found to be nontoxic up to 62.5 µg/mL when tested on mammalian Vero cell linesCitation19.
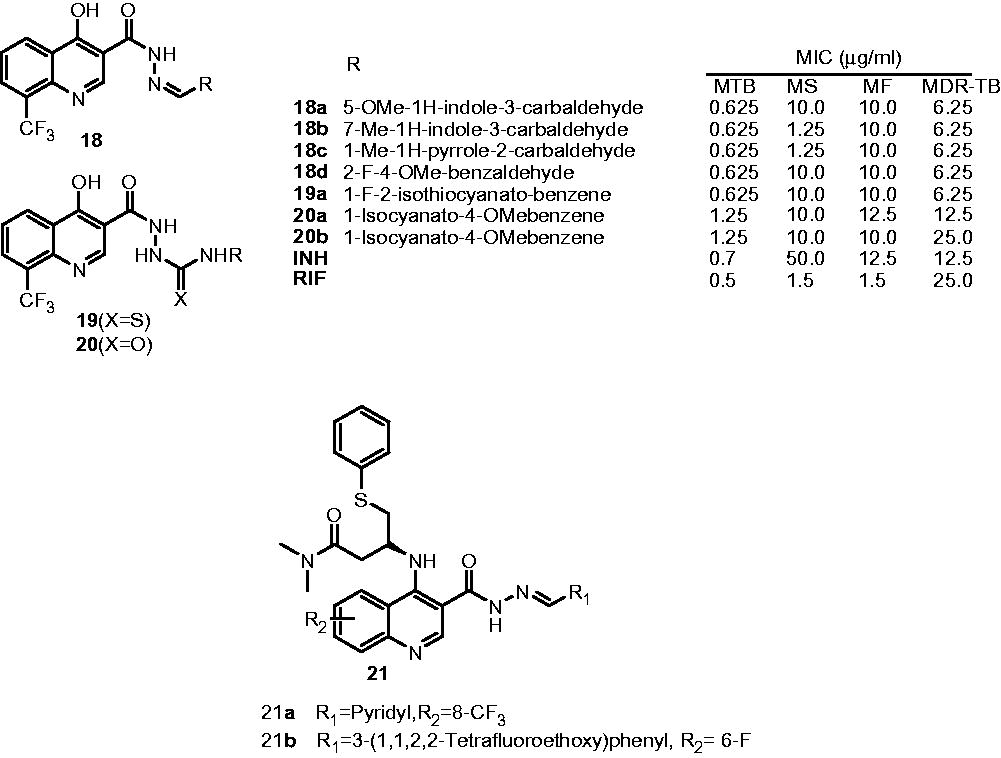
4-Substituted quinolines
Three series of quinoline-4-yl-1,2,3-triazoles possessing substituted amides (22), sulphonamides (23) and amidopiperazines (24) were developed and evaluated for their antimycobacterial activityCitation20. All three series of compounds displayed moderate-to-good activity against M. tuberculosis (MIC: 0.625–1.25 µg/mL) compared with INH and rifampicin (MIC: 0.7 and 0.5 µg/mL, respectively). The SAR indicated that acetyl, methoxy, fluoro and trifluoromethyl groups increase the activity while cyclo-butyl and 2-chloro-benzoyl decrease the activityCitation20. Thomas et al. also developed two series of hybrid quinoline-oxazolidinone molecules (25, 26) as enoyl-ACP reductase ligands and antimycobacterial agents using structure-based drug design. It was observed that in both series of molecules, smaller groups like methyl and dimethylamino at R-position are favorable for the activity (MIC: 0.625 µg/mL) while bulkier groups such as 4-ethylpiperazinyl, 4-acetylpiperazinyl and 4-isopropylpiperazinyl are detrimental for the activity (MIC: 5–10 µg/mL)Citation21.
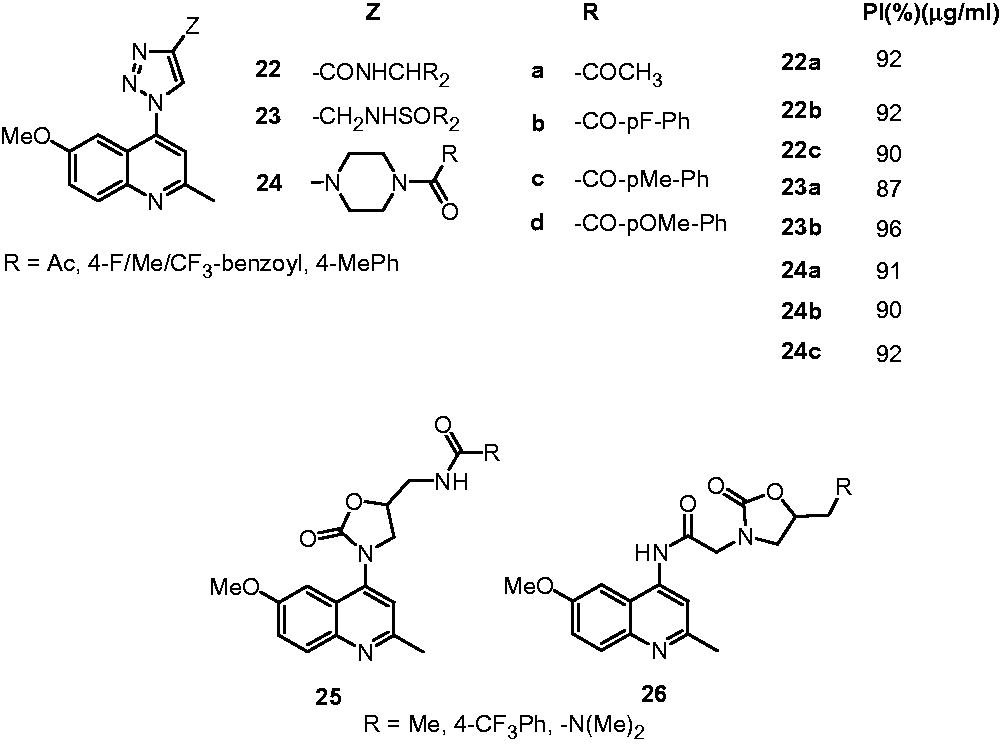
Carmo et al. reported the synthesis and antimycobacterial activity (M. tuberculosis H37Rv) of 4-amino-7-chloroquinolines (27) and their platinum complexes (27a). However, these compounds (27) showed poor activity (MIC: 12.5–250 µg/mL) compared to the standard drug INH (0.03 µg/mL). Also, platinum-complexed compounds (27a) did not award any improvement in activityCitation22. These compounds were found to be cytotoxic on mouse peritoneal macrophages (cytotoxic IC50: 6.90–13.36 µg/mL).
Candea et al. synthesized a series of 7-chloro-4-quinolinylhydrazones (28) as antimycobacterial agents. Some of the synthesized compounds displayed moderate-to-good activity (MIC: 2.5 to 6.25 µg/mL) compared with standard drug ethambutol (MIC: 3.25 µg/mL). The SAR indicated that the compounds with fluoro, chloro, hydroxy and methoxy groups possess good activity while bromo, nitro and cyano analogs showed decreased activity. These compounds were evaluated for their cytotoxicities by Mosmans’s assay on BCG-infected macrophages cell line J774. In this assay, 28a showed 100% cell viability at the dose of 100 µg/mL and thus found to be noncytotoxic to the host cells in the effective concentrations (MIC 2.5 µg/mL) to inhibit the growth M. tuberculosisCitation23. De Souza et al. studied the effect of number of carbon in intermediate chain between two nitrogen atoms (as shown in 29) of 4-amino-quinolines on the antimycobacterial activity (29). The compounds with two to four carbon length showed poor activity (MIC: 100 µg/mL). Further increment in the number of carbon, namely six, eight and ten carbons, led to the significant increase in activity as shown by MIC of 25.0, 6.25 and 3.12 µg/mL, respectively. The number of carbon atoms in intermediate chain is directly associated with the hydrophobicity of the compounds (log P), and hence contributing for higher activityCitation24.
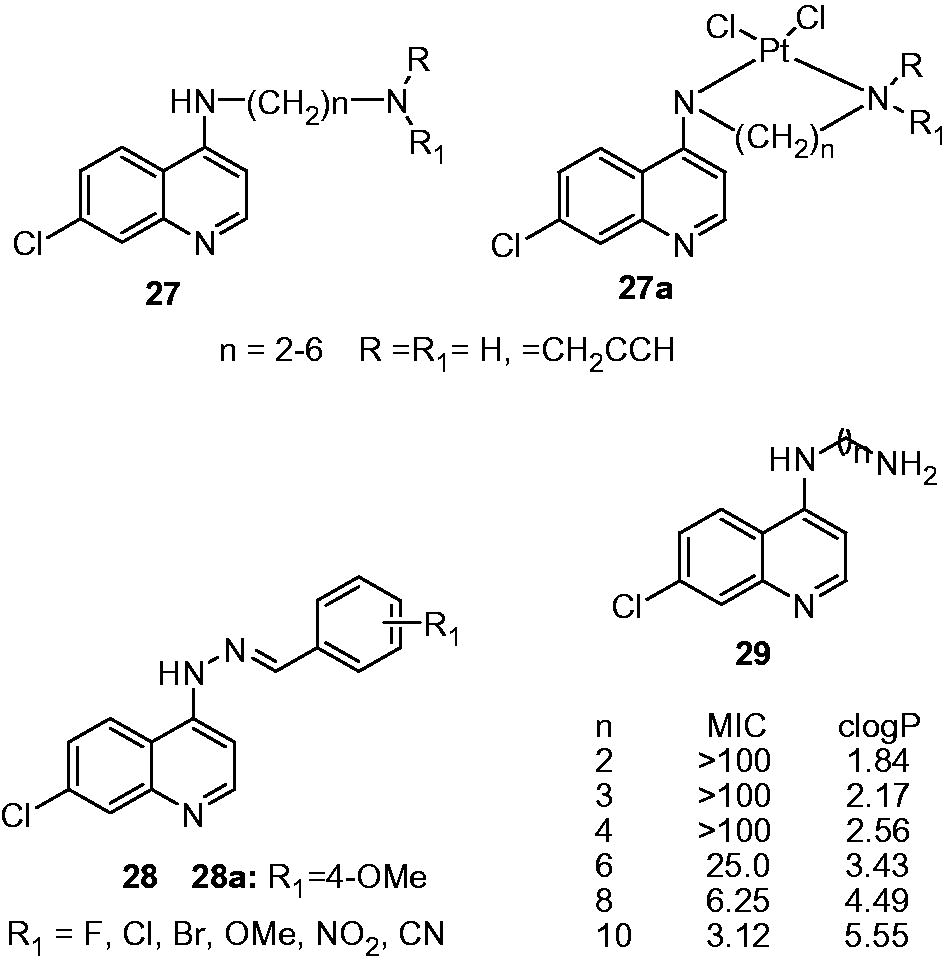
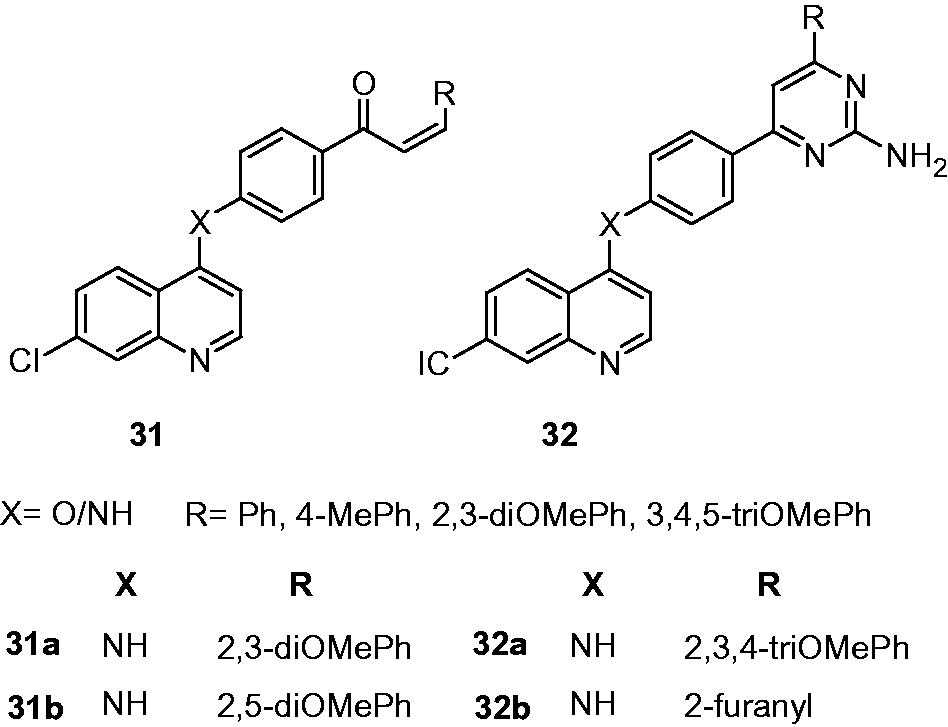
Novel quinolyl-hydrazone derivatives (30) and their antimycobacterial activity were reported by Gemma et al. These derivatives displayed moderate-to-good antimycobacterial activity (MIC: 0.6 to 10.0 µM) when compared with standard INH (MIC: 0.36 µM). Two derivatives 30a and 30b have shown 100% growth inhibition at the MIC of 0.6 µM. In vitro cytotoxicity (IC50) of 30a and 30b in the Vero cell line was found to be 0.45 µg/mL (SI: 2.27) and 0.21 µg/mL (SI: 1.05), respectively. SAR indicated that functionalities at both the quinoline (R2) and arylhydrazone moiety (R3) modulate the antimycobacterial activity. Among the quinoline substituents, the 7-OMe is favorable for antimycobacterial activity, especially when coupled to a naphthyl ring at the hydrazone moiety. Based on the structure–activity pattern, authors suggested that the electronic effects of the quinoline-pharmacophore and the overall lypophilicity of the molecule play an important role in determining optimal antimycobacterial activityCitation25. Antimycobacterial activity of substituted quinolinyl-4-chalcones (31) and quinolinyl-4-pyrimidines (32) were reported by Sharma et al. Quinolinyl-chalcones (31a and 31b) showed excellent activity as shown by MIC of 3.12 µg/mL compared to antimycobacterial drug pyrazinamide (MIC 50.0 µg/mL). Quinolinyl-chalcones also appeared to be better than quinolinyl-pyrimidines (32a and 32b, MIC: 12.5 µg/mL). Compounds 31a and 31b were found to be noncytotoxic when tested for cytotoxicity against Vero and MBMDM cell lines (IC50 > 31.2 µg/mL)Citation26.

Quinoline-based fused ring system
Parekh et al. reported the synthesis and antimycobacterial activity of phenyl pyrazolone-substituted 1H-benzo[g]pyrazolo[3,4-b]quinoline-3-yl amine derivatives (33). These derivatives showed moderate to poor activity (62.5–1000 µg/mL) against M. tuberculosis H37Rv. Two derivatives 33a and 33b displayed comparable antimycobacterial activity (MIC: 62.5 µg/mL) with the standard rifampicin (MIC: 40 µg/mL). Among various inhibitors, the chloro-substituted derivatives (mono or dichloro) exhibited better activity than methyl- or sulfonyl-substituted onceCitation27. Karthikeyan et al. reported 2-aryl-3,4-dihydro-2H-thieno[3,2-b]indoles (thienoindole, 34) as a novel antimycobacterial agentCitation28. Balamurugan et al. designed a new class of antimycobacterial agents 2,9-diaryl-2,3-dihydrothieno[3,2-b]quinolines series; 35) by incorporating the pharmacophoric features of TMC207 and thienoindole (). Among the synthesized compounds, 35a (MIC: 1.76 µg/mL) and 35b (MIC: 0.95 µg/mL) showed 6- and 12-fold higher activity against MDR-TB than standard drug INH (MIC: 11.38 µg/mL). The SAR indicated that fluoro-, chloro- and nitro-substituted inhibitors possess better antimycobacterial activity than methyl-phenyl- or 1-naphthyl-substituted analogs. Also, at C-7 position (R2), the presence of chlorine atom considerably improved the activity. It may be suggested that electron-withdrawing substituents at R1 and R2 positions are conducive for the activityCitation29.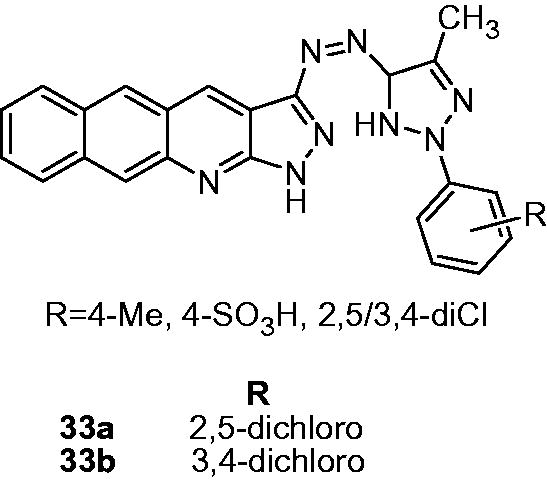
Figure 4. Design of 2,9-diaryl-2,3-dihydrothieno[3,2-b]quinolines (35).
![Figure 4. Design of 2,9-diaryl-2,3-dihydrothieno[3,2-b]quinolines (35).](/cms/asset/f9ae67c0-1418-4cd5-8489-fc7afda4144d/ienz_a_930454_f0004_b.jpg)
Antimycobacterial activity of a series of novel isomeric hexahydro-2H-pyrano[3,2-c]quinoline analogs (36) was reported by Kantevari et al.Citation30. These analogs were designed by combining the hexahydro-2H-pyrano[3,2-c]quinoline scaffold with benzofurobenzopyran pharmacophores (). In in-vitro activity evaluation, the halogen-substituted analogs were found to be more active than unsubstituted/methyl/methoxy-substituted analogs. Among halogen-substituted analogs, the maximum activity was observed in fluoro analogs (R = F, X = N–CH3; MIC: 3.13 µg/mL; M. tuberculosis H37Rv), similar to that of ethambutol (MIC: 3.13 µg/mL). The isomeric analogs showed similar activityCitation30.
Figure 5. Design of hexahydro-2H-pyrano[3,2-c]quinoline analogs (36).
![Figure 5. Design of hexahydro-2H-pyrano[3,2-c]quinoline analogs (36).](/cms/asset/90922750-9f27-4875-a35a-a7610c530acd/ienz_a_930454_f0005_b.jpg)
Quinoline derivatives derived from drugs
Derived from mefloquine
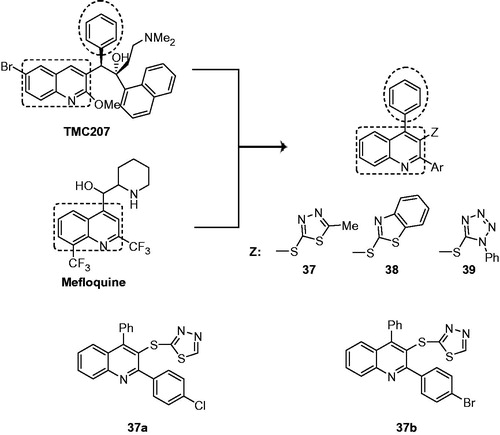
Mefloquine (antimalarial drug, ) targets ATP synthase in M. tuberculosis and possesses weak antimycobacterial activity (MIC for M. tuberculosis H37Rv: mefloquine 33 µM; ethambutol 15.9 µM)Citation31. TMC207 also acts on the proton pump of M. tuberculosis ATP synthase and possesses excellent potency against drug-sensitive and drug-resistant strains of MycobacteriumCitation3. Therefore, Chitra et al. proposed new antimycobacterial compounds by incorporating the chemical features of both TMC207 and mefloquine. The three new series of 2-aryl-3-heteroarylthioquinolines, that is, 2-aryl-4-phenyl-3-quinolyl(5-methyl-1,3,4-thiadiazol-2-yl)sulfides (37), 1,3-benzothiazol-2-yl [2-aryl-4-phenyl-3-quinolyl]sulfides (38) and 2-aryl-4-phenyl-3-[(2-phenyl-2H-1,2,3,4-tetraazol-5-yl)sulfanyl]quinoline (39) bearing pharmacophoric features of TMC207 and mefloquine (), were synthesized and evaluated for their antimycobacterial activityCitation32. The study showed that compounds of series 37 possess superior antimycobacterial activity profile (MIC: 3.2–30.4 µM) than compounds of series 38 (MIC: 5.9–55.9 µM) and 39 (MIC: 23.3–54.6 µM). The two compounds 37a and 37b exhibited an MIC of 3.2 and 3.5 µM, respectively, for M. tuberculosis H37Rv and showed better activity than ciprofloxacin (MIC: 4.7 µM) and ethambutol (MIC: 7.6 µM). The in vitro cytotoxicity (IC50) of compounds 37a and 37b on mouse embryonic fibroblasts cell line (NIH 3T3) was reported as 1263 and 2050 mM, respectively, indicating that 37a and 37b are not cytotoxic to the normal fibroblasts (NIH 3T3). In compounds of series 37, for aryl groups, the order of activity was 4-BrC6H4 > 4-ClC6H4 > 4-PhC6H4 > 2-naphthyl > 4-MeOC6H4 > 4-MeC6H4 > C6H5. In all three series, the presence of halogens in aryl ring at C-2 position augmented the activityCitation32.
Figure 6. Design of 37, 38 and 39.
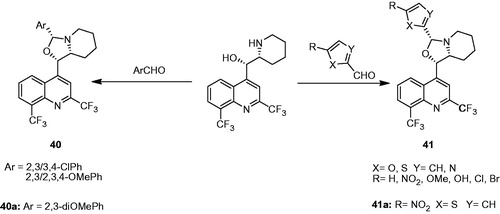
Gonçalves et al. designed two series of mefloquine–oxazolidine analogs (40 and 41) by condensing the mefloquine with aryl and heterocyclic aldehydes (). The study was designed to evaluate the effect of (a) the introduction of conformational restriction in the rotation of the piperidinyl ring of mefloquine by the construction of an oxazolidine ring and (b) influence of substituted aryl/heteroaryl functions attached to the oxazolidine ring on the antimycobacterial activity (). The compounds were evaluated for their activity against M. tuberculosis H37Rv and M. tuberculosis T113. Compounds 40a and 41a exhibited approximately 2.7 times more potency (MIC: 11.9 and 12.1 µM, respectively) than mefloquine (MIC: 33.0 µM) in both strains. They also displayed higher activity than ethambutol (MIC: 15.9 and 61.2 µM for M. tuberculosis H37Rv and T113, respectively). In Mosmans’s cytotoxicity assay on Murine Macrophages Cells, 40a and 41a showed 100% cell viability at the dose of 100 µM. The study showed that the electronic nature of substituents in the aryl ring plays a significant role on the antimycobacterial activity. Inhibitors having electron-releasing groups, such as hydroxy and methoxy groups, were more active than electron-withdrawing chloro or nitro groupsCitation31,Citation33.
Figure 7. Design of mefloquine–oxazolidine derivatives (40 and 41).
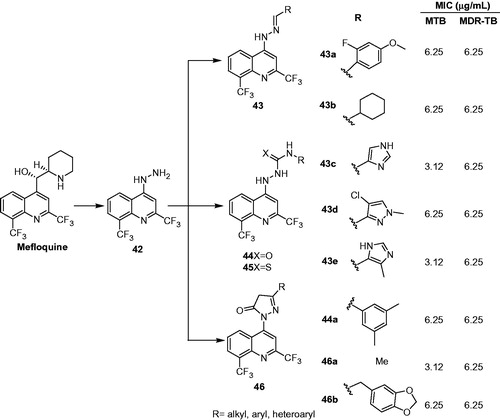
Eswaran et al. designed four new series of hydrazine-linked-quinolines based on the structure of mefloquine (). The compounds were screened against M. tuberculosis and MDR-TB. Most of the compounds possess moderate-to-good antimycobacterial activity (MIC ranging from 6.25 to 12.5 µg/mL) and were found to be more active than INH and rifampicin. Compounds 43a, 43b, 43c, 43d, 43e, 44a, 46a and 46b were found to be two- and four-fold more potent than INH and RIF (MIC: 6.25 µg/mL; INH: 12.5 µg/mL; RIF MIC: 25 µg/mL). It was suggested that the good antimycobacterial activity of these compounds is attributed to the presence of heteroaryl groups, namely pyrazole, imid-azole and indole. Compound 46a showed excellent antimycobacterial activity against both TB strains (MIC: 3.12 and 6.25 µg/mL, respectively). The authors suggested that the presence of electron-donating –CH3 group stabilize the pyrazole ring and thus make the 46a more active against M. tuberculosisCitation34.
Figure 8. Design of hydrazine-linked-quinolines based on the structure of mefloquine.
Derived from fluoroquinolones
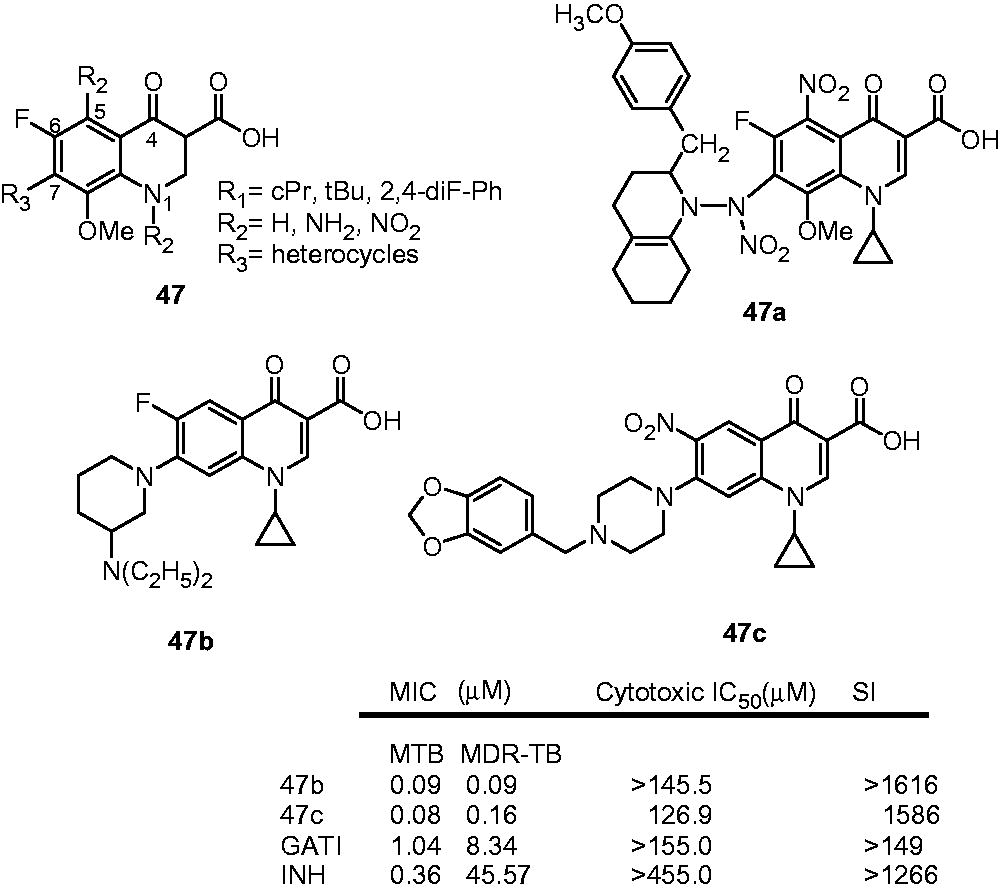
Senthilkumar et al. reported the synthesis of a new series of fluoroquinolones (47) as antimycobacterial agents. These compounds were evaluated for their antimycobacterial activities against M. tuberculosis H37Rv, MDR-TB and M. smegmatis (MCCitation2) as well as for their ability to inhibit the supercoiling activity of mycobacterial DNA gyrase. Compound 47a was found to be the most active compound in the series with MIC of 0.16, 0.33 0.68 µM against MTB, MDR-TB and MCCitation2, respectively (INH: 0.36, 45.57 and 45.57 µM, respectively). Compound 47a also inhibited the supercoiling activity of DNA gyrase with an IC50 of 30.0 µg/mL. The SAR of this series reveals that the substitution of nitro group at C-5 position increased the antimycobacterial activity up to ninefold, while the replacement of nitro group with amino group reduces the activity. Substitution pattern at C-7 demonstrated the following order of activity: fused piperazines and piperidines > (thio) morpholines > substituted piperazines > substituted piperidines. It was suggested that the introduction of bulky lipophilic secondary amines at C-7 position improved antimycobacterial activity due to more penetration of these compounds into mycobacterial cellsCitation35.
In continuation, Senthilkumar et al. also reported novel fluoroquinolones by varying the substitution at N-1 and C-7 positions (47). Some of these compounds displayed moderate-to-good activity for M. tuberculosis and MDR-TB in the MIC range 0.08–16.6 and 0.08–14.45 µM, respectively. Two compounds 47b and 47c possess excellent in vitro activity for M. tuberculosis and MDR-TB. The in vivo efficacy of both compounds in M. tuberculosis ATCC35801-infected mice was found to be comparable with gatifloxacin (GATI) and INH on a dose basis. Analysis of the substitution pattern at C-7 revealed that the order of antimycobacterial activity was substituted piperidines > substituted piperazines > fused piperazines and piperidines > (thio) morpholines. Interestingly, the study revealed that the contribution of the C-7 position to the activity was dependent on the substituent at N-1 and was in the order of substituted piperidines > substituted piperazines > fused piperazines and piperidines > (thio) morpholines when N-1 was cyclopropyl or t-butyl and substituted piperidines > substituted piperazines ≥ fused piperazines and piperidines ≥ (thio)morpholines when substitutions at N-1 was 2,4-difluorophenylCitation36,Citation37.
Dinakaran et al. studied the antimycobacterial activity profile of various 2-(substituted)-3-fluoro/nitro-5,12-dihydro-5-oxobenzothiazolo[3,2-a]quinoline-6-carboxylic acid derivatives (48) on M. tuberculosis H37Rv and MDT-TB and their efficacy to inhibit the supercoiling activity of DNA gyrase. The compounds displayed moderate to promising activity against both strains. Among the synthesized compounds, 48a showed best activity with MIC of 0.18 and 0.08 µM against MTB and MDR-TB, respectively. Compound 48a was found to be 2 and 570 times more potent than standard INH against MTB and MDR-TB, respectively (INH, MIC: 0.36 and 45.57 for MTB and MDR-TB, respectively). The structure–MTB activity relationship for the substitution pattern at C-7 showed that the order of activity was substituted piperidines > fused piperazines, pyrrolidine and piperidines > substituted piperazines, (thio) morpholines. With respect to the substitutions at R1, fluoro-substituted inhibitors were found to be more active than nitro-substitutedCitation38. In another work, Dinakaran et al. synthesized and evaluated various ofloxacin derivatives for their antimycobacterial activity on M. tuberculosis and MDR-TB (49). The most active compound 49a displayed excellent activity against M. tuberculosis and MDR-TB at the MIC of 0.19 and 0.09 µM, respectively, when compared to the ofloxacin and INH. SAR showed that, for R1, the introduction of nitro group led to the 1.1- to 8.8-fold higher activity while the introduction of amino group reduced the activity. The SAR for the substituents at R2 position follows activity order as: fused piperazines and piperidines > substituted piperidines ≥ (thio) morpholines > substituted piperazinesCitation39.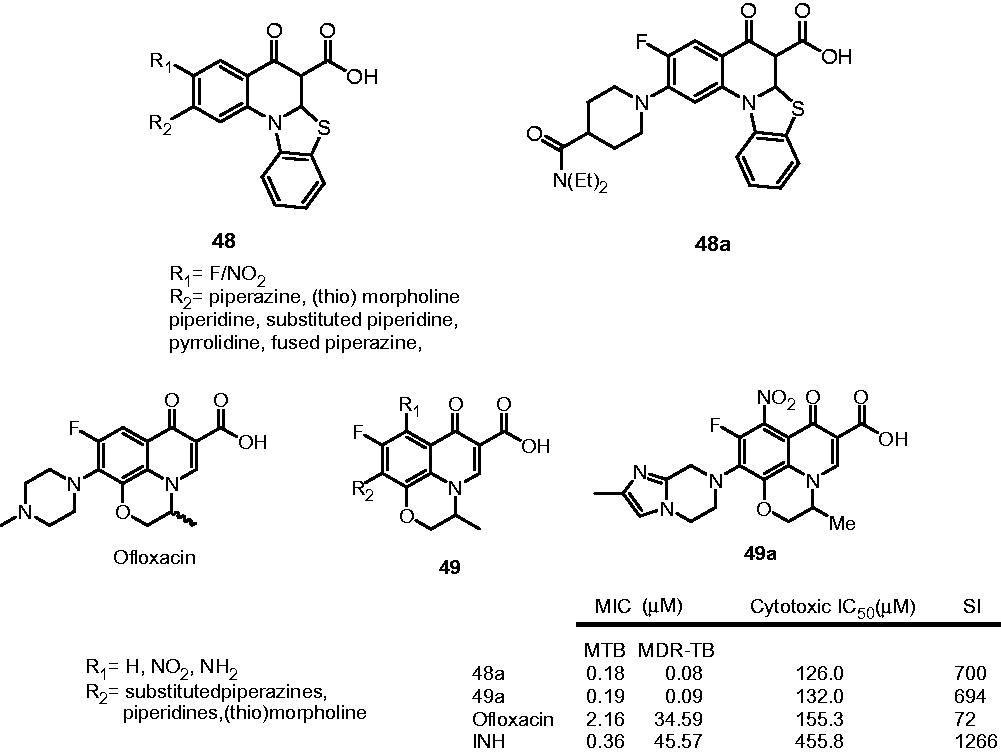
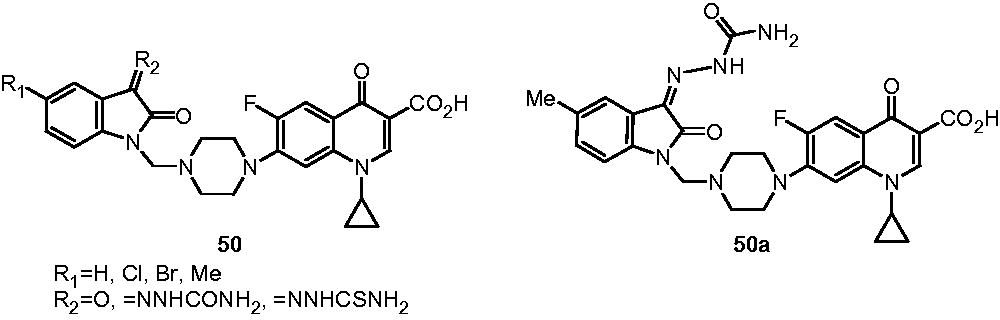
Sriram et al. reported the antimycobacterial activity profile of 7-substituted ciprofloxacin (CP) derivatives (50). The CP derivatives showed moderate-to-excellent activity, with the MIC ranging from 1.21 to 10.82 nM when compared with ciprofloxacin and moxifloxacin (MIC: 6.04 and 1.94 nM, respectively). Some derivatives exhibited 100% growth inhibition of M. tuberculosis H37Rv at the concentration of 6.25 µg/mL. However in DNA gyrase inhibitory activity assay, none of the compound showed better activity than moxifloxacin. In in vivo study on C57BL/6 female mice, 50a reduced the bacterial load in spleen tissue with 0.76-log10 protections and was suggested to be moderately active in decreasing bacterial count in spleenCitation40.
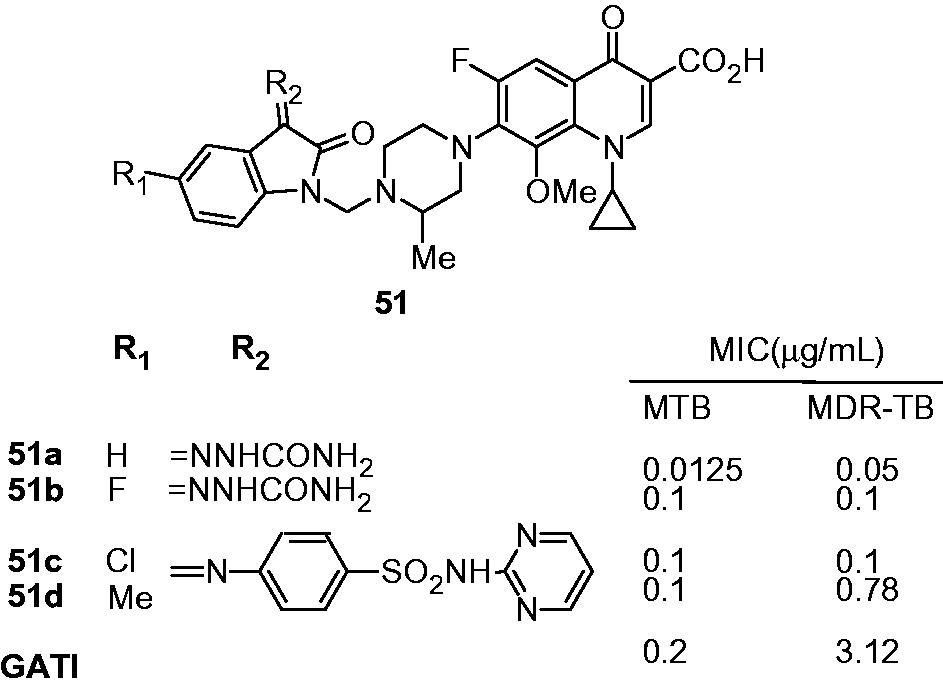
In continuation of previous work, Sriram et al. also reported the antimycobacterial activity profile of 7-substituted gatifloxacin derivatives (51). The gatifloxacin derivatives exhibited 4–62 times more activity than gatifloxacin against MDR-TB (51, MIC: 0.05 to 0.78; gatifloxacin, 3.12 µg/mL). For M. tuberculosis, only four compounds (51a, 51b, 51c and 51d) displayed better activity than gatifloxacin while others were found to be equal or lesser active derivatives. Among the active derivatives, 51d appeared as the most active one, with the MIC of 0.0125 and 0.05 µg/mL for M. tuberculosis and MDR-TB, respectively. 51d also showed wild-type M. tuberculosis DNA gyrase inhibitory activity, with an IC50 of 3.0 µg/mL, comparable with gatifloxacin. In the in vivo animal model (female CD-1 mice) 51d decreased the bacterial load in lung and spleen tissues with 3.62 and 3.76-log10 protections, respectively. Furthermore, all the derivatives were found to be nontoxic up to 62.5 µg/mL, and 51d showed selectivity index (IC50/MIC) of more than 1250. It was suggested that increasing the lipophilicity of side chain at C-7 position improved the antimycobacterial activityCitation41.
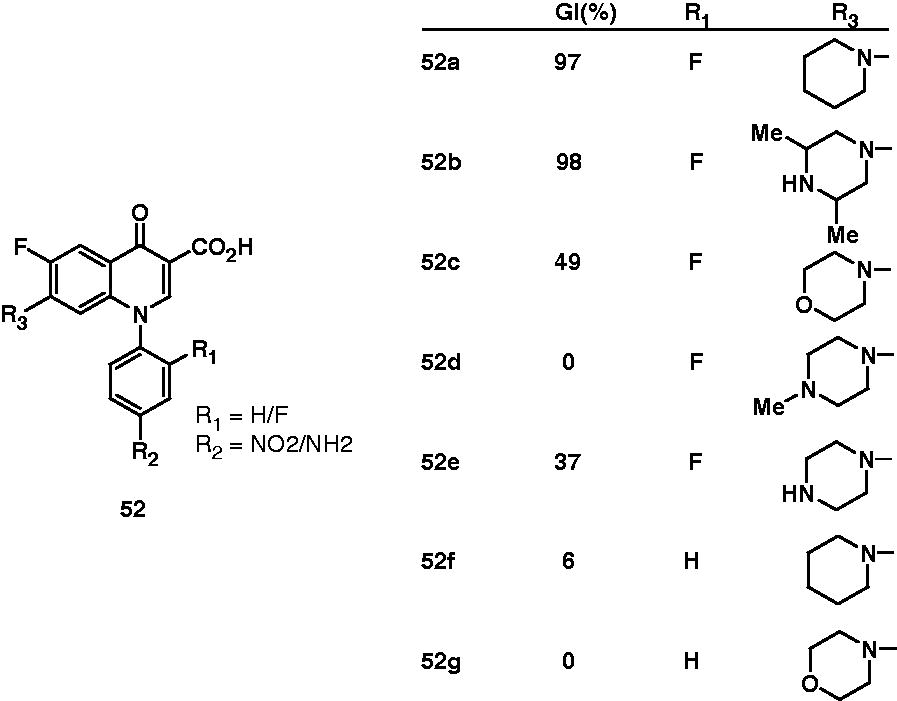
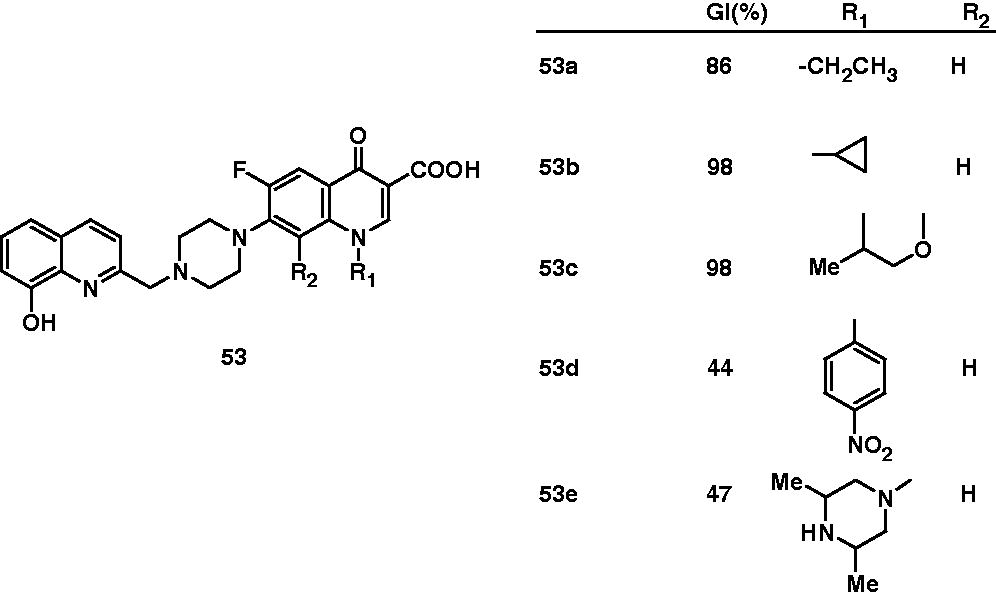
Zhao et al. found out interesting observations on the antimycobacterial activity of two types of fluoroquinolone derivatives. In the first type, C-7 position of fluoroquinolone was substituted with various six-membered heterocyclic rings like piperidine, morpholine and piperazine (52). In other type, various bifunctional fluoroquinolone–hydroxyquinoline complexes (53) were prepared, which include 8-hydroxyquinoline derivatives of norfloxacin (53a), ciprofloxacin (53b) and ofloxacin (53c). Their antimycobacterial activity against M. tuberculosis H37Rv was reported as the percentage growth inhibition (GI%) at the concentration of 6.25 µg/mL. Closure look on the activity of compounds 52f and 52g and their fluoro-substituted counterparts 52a and 52b suggested that fluoro group at R1 position is important for the activity. For R3 substituents, 7-piperidinyl derivative (52a) and 7-(3,5-dimethylpiperazinyl) derivative (52b) displayed 97% and 98% inhibition, respectively, and appeared to be more active than 7-morpholinyl (52c), 7-(4-methylpiperazinyl) (52d) and 7-piperazinyl (52e) derivatives. Interestingly, 44% growth inhibition of M. tuberculosis by (7-[4-(8-hydroxyquinolin-2-ylmethyl)piperazin-1-yl] derivative) (53d) over 7-piperadinyl (52f) and 7-morphonyl (52g) suggested that metal-chelating property of 8-hydroxyquinoline moiety is conducive antimycobacterial activity. For the bifunctional fluoroquinolone–hydroxyquinoline complexes, norfloxacin (53a, 84% inhibition), ciprofloxacin (53b, 98%) and ofloxacine (53c, 98%) derivatives were found to be more potent than 1-aryl congeners 53d and 53e, which showed only 44% and 47% M. tuberculosis growth inhibition, respectivelyCitation42. Antimycobacterial potential of fluoroquinolones (54) was also studied by Talath et al. However, these compounds showed only moderate antimycobacterial activity (MIC: 10 μg/mL)Citation43.
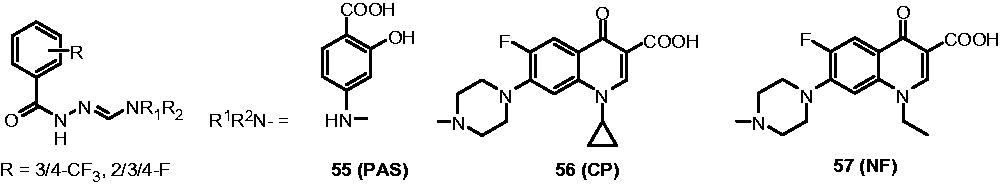
Vavrikova et al. synthesized hydrazine-linked derivatives of p-aminosalicylic acid (PAS, 55), ciprofloxacin (CP, 56) and norfloxacin (NF, 57) and evaluated their antimycobacterial activity against drug-sensitive (H37Rv) and MDR (MDR A8 241) strains of M. tuberculosis. For MDR-TB, PAS-hydrazones and CP-hydrazones exhibited better activity (MIC: 0.5 µg/mL) than INH, CP and NF alone (MIC: INH 1.0 µg/mL; PAS 0.25 µg/mL; CP: 1.5 µg/mL; NF: 10 µg/mL). For M. tuberculosis H37Rv, the CP-hydrazones appeared to be better (MIC: 1 µg/mL) than PAS-hydrazones and NF-hydrazones (MIC: 2–6 µg/mL). All hydrazones were found to be noncytotoxic for human hepatocytes, PBMC cells and human SH-SY5Y neuroblastoma cells (cytotoxic IC50: 0.03 to 1.28 µM)Citation44.
Derived from ferroquine
Ferroquine (FQ, SSR97193), an antimalarial compound, also possesses moderate antimycobacterial activity (MIC: 10–15 µg/mL; M. tuberculosis MCCitation2 7000)Citation45,Citation46. It is a hybrid of 4-amino-7-chloro-quinoline and ferrocene. Few ferrocenyl derivatives quinoline–ferrocene hybrid (58) and ferrocene-based hydrazones (59–61) were developed by Mahajan et al. and evaluated in vitro for their efficacy against Mycobacterium (M. tuberculosis MCCitation2 7000). It was found that only quinoline–ferrocene hybrid (58) possesses good antimycobacterial activity (MIC: 2.5–5 µg/mL; M. tuberculosis MCCitation2 7000) comparable to that of ethambutol (MIC: 1–2.5 µg/mL), while all ferrocene-based hydrazones (59–61) failed to show antimycobacterial activity (MIC > 100 µg/mL). It was suggested that the good antimycobacterial activity of 58 may be attributed to the presence of the quinoline ring in the ferrocenyl derivativesCitation46.
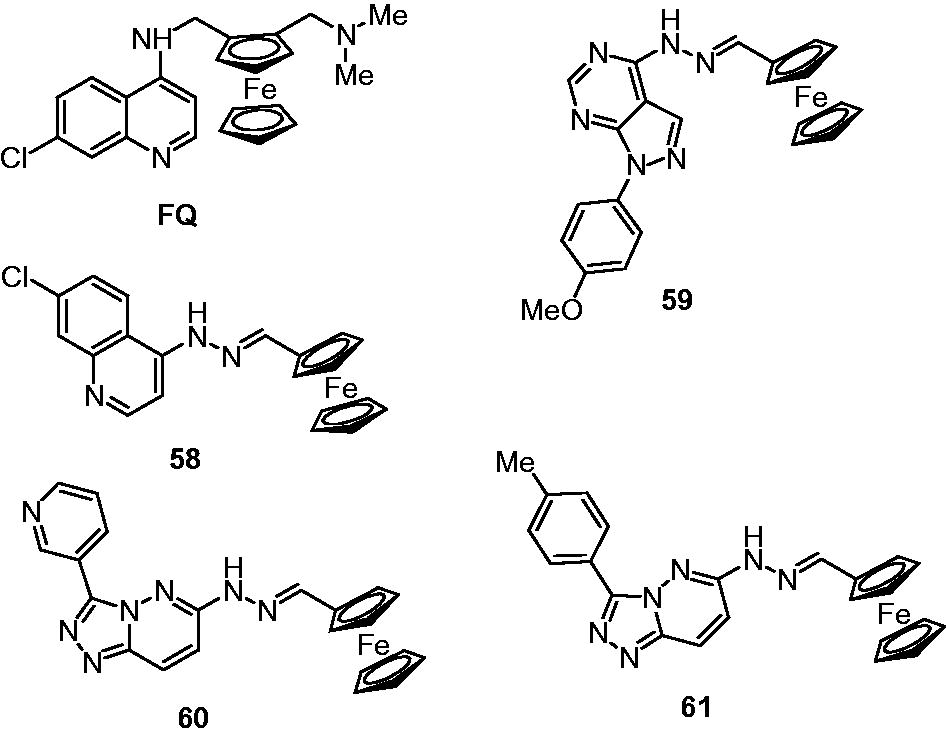
Miscellaneous
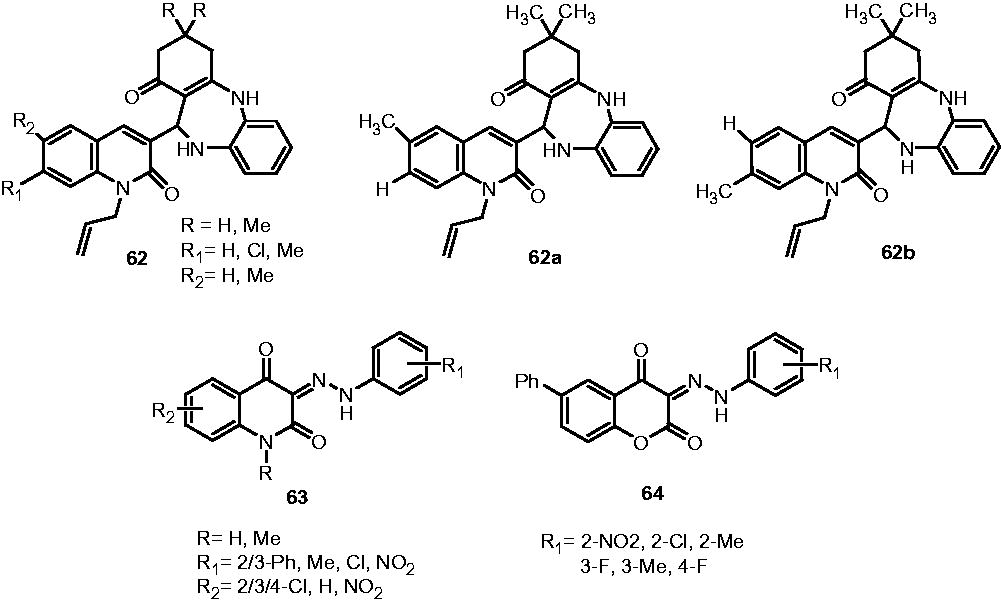
Parmar et al. studied the antimycobacterial effect of quinolones linked with diazepinones (62). They synthesized a novel series of quinolyldibenzo[b,e][1,4]diazepinones, and among the evaluated compounds, 62a and 62b showed 96% and 91% growth inhibition activity, respectively, for M. tuberculosis H37Rv at the concentration of 250 µg/mLCitation47. Manvar et al. studied the antimycobacterial potential of various hydrazides of substituted quinolones (63) and coumarins (64). The quinolone analogs displayed three- to four-fold better activity profile (GI: 98–100%) compared to the coumarin analogs (GI: 17–25% at MIC < 6.25 µg/mL). The quinolone analogs with bromo, phenyl and methyl at R1 position impart excellent growth inhibitory activity (GI: 98–100%), while nitro, fluoro and dichloro groups compromised the activity of quinolone analogs (GI: 60–89%). This may be suggested that hydrophobic substituents at R1 are favorable for the growth inhibitory activity while electron-withdrawing substituents are detrimental for the activityCitation48.
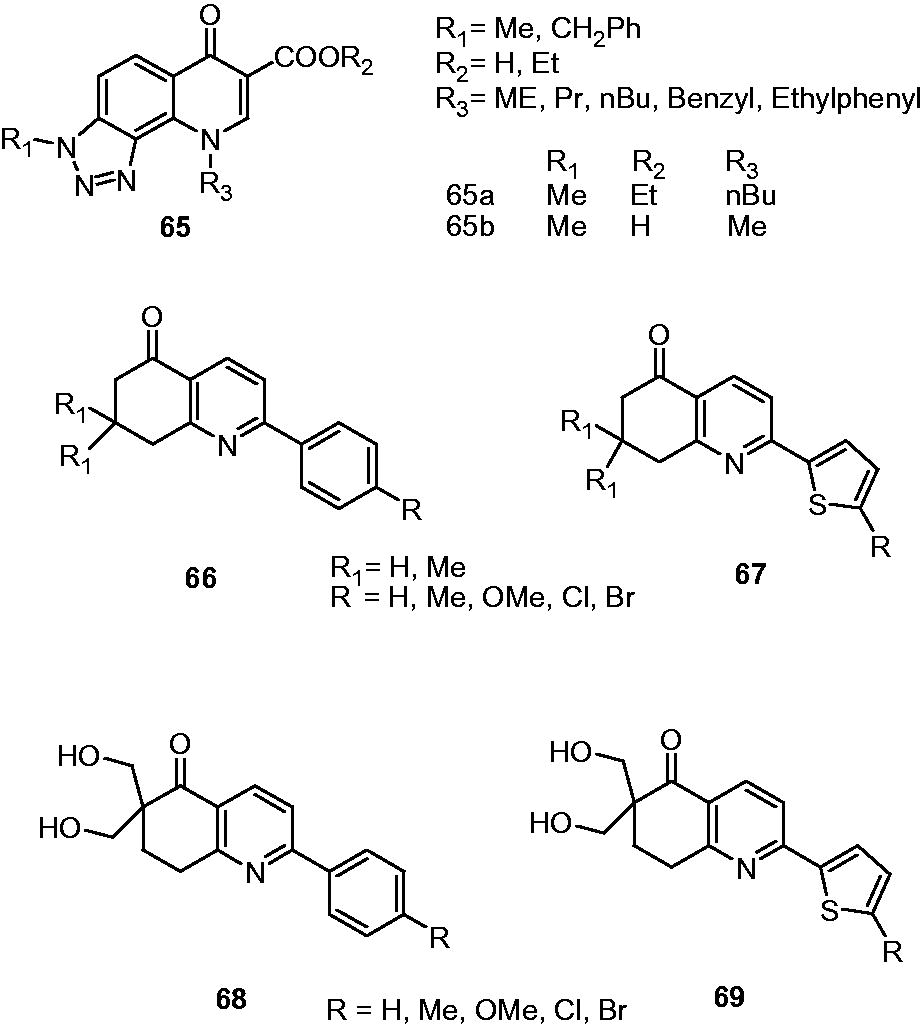
Carta et al. reported 3-methyl-9-substituted-6-oxo-6,9-dihydro-3H-[1,2,3]-triazolo[4,5-h]quinolone-carboxylic acids and their esters (65) as a new class of antimycobacterial agents against MDR-TB. Screening of these derivatives against M. tuberculosis H37Rv and 11 other clinically isolated strains of M. tuberculosis displayed MIC90 in the range of 0.5–3.2 µg/mL. 65a was identified as the most effective derivative (MIC90: 0.5 µg/mL) in comparison with rifampin (MIC90: 0.7–4.0 g/mL) against all M. tuberculosis strains. Furthermore, 65a showed no cytotoxicity for both human macrophages and Hep-2 cells up to the concentration of 50 µg/mL. The presence of alkyl substituents at N-1 position was found to be more favorable for the activity than propenyl, benzyl or phenylethyl groups. Also, free carboxylic acid derivatives appeared to be superior compared to ester analogsCitation49,Citation50.
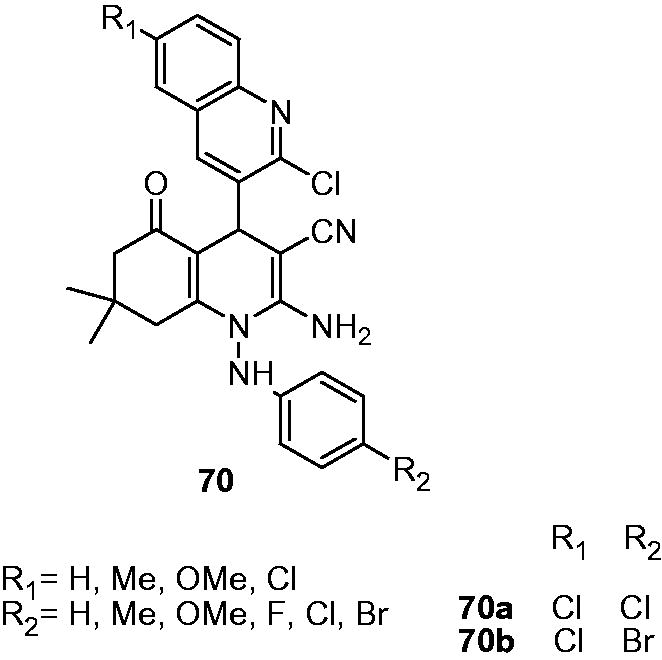
Kantevari et al. assessed the antimycobacterial activity of substituted aryl-tethered dihydro-6H-quinolin-5-ones (66), thiophenyl-tethered dihydro-6H-quinolin-5-ones (67) and their methyl hydroxyl analogs (68 and 69). In comparison with aryl-tethered dihydro-6H-quinolin-5-ones (66, MIC: 12.5 µg/mL), thiophenyl-tethered dihydro-6H-quinolin-5-ones displayed better antimycobacterial activity (67, MIC: 3.13–6.25 µg/mL). The substitution with chloro and bromo enhance the activity while methyl and methoxy groups proved to be detrimental for the activity. The hydroxyl methyl analogs (68 and 69) showed poor activity (MIC: >25 µg/mL). The study indicated that the antimycobacterial activity mainly depends upon the lipophilicity of these compounds. The thiophenyl-tethered dihydro-6H-quinolin-5-ones (67; log P ∼ 2.91 to 4.32) are relatively more lipophilic than aryl-tethered dihydro-6H-quinolin-5-ones (66; log P ∼ 3.39 to 4.53). The hydroxyl methyl analogs possess least lipophilicity and poor activity (68 and 69; log P ∼ 2.06 to 2.88)Citation51. A new series of N-arylaminobiquinoline derivatives (70) as antimycobacterial agents was reported by Shah et al. Among the tested compounds, 70a and 70b displayed 87% and 85% growth inhibitory activity, respectively, at the concentration of 6.25 µg/mL compared with 98% activity of rifampicinCitation52.
Concluding remarks
Exploration of chemical space around quinoline scaffold led to the identification of several new series of compounds with notable antimycobacterial activity against drug-sensitive and MDR-TB. For instance, 2,9-diaryl-2,3-dihydrothieno[3,2-b]quinoline 35a (MIC: 1.76 µg/mL) and 35b (MIC: 0.95 µg/mL) showed 6- and 12-fold higher activity against MDR-TB than standard drug INH (MIC: 11.38 µg/mL). Also, 4-hydroxy-8-(trifluoromethyl)quinoline-3-carbohydrazone (18a–d) and 2-{[4-hydroxy-8-(trifluoromethyl)quinolin-3-yl]carbonyl}hydrazinecarbothioamide (19a) appeared as 2–4 times more active against MDR-TB than INH and rifampicin, respectively. Among the compounds derived from existing drugs, 47a (a fluoroquinolone; MIC: 0.33 µM) appeared to be 138 times more active than INH against MDR-TB and also inhibited the supercoiling activity of DNA gyrase with an IC50 of 30.0 µg/mL. Several of quinoline derivatives appear to be safe when tested for their cytotoxicity. These studies also emphasize the substitution patterns in quinoline and quinolone scaffolds to improve their antimycobacterial activity against drug-sensitive and MDR strains. It can be clearly substantiated that quinoline and quinolone are potential scaffolds to develop future antimycobacterial drugs. This systematic review will provide a comprehendible information to researchers to design and develop potent quinoline/quinolone-based antimycobacterial drugs.
Acknowledgements
Authors thank Chairman, ISF College of Pharmacy, Moga (PB), India, for his valuable support and encouragement.
Declaration of interest
The authors have declared no conflict of interest.
References
- World Health Organization (WHO). Available from: http://www.who.int/mediacentre/factsheets/fs104/en/index.html [last accessed May 6 2013]
- Engohang-Ndong J. Antimycobacterial drugs currently in Phase II clinical trials and preclinical phase for tuberculosis treatment. Expert Opin Investig Drugs 2012;12:1789–800
- Andries K, Verhasselt P, Guillemont J, et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 2005;5707:223–7
- Matteelli A, Carvalho AC, Dooley KE, Kritski A. TMC207: the first compound of a new class of potent anti-tuberculosis drugs. Future Microbiol 2010;5:849–58
- Haagsma AC, Abdillahi-Ibrahim R, Wagner MJ, et al. Selectivity of TMC207 towards mycobacterial ATP synthase compared with that towards the eukaryotic homologue. Antimicrob Agents Chemother 2009;3:1290–2
- Ibrahim M, Andries K, Lounis N, et al. Synergistic activity of R207910 combined with pyrazinamide against murine tuberculosis. Antimicrob Agents Chemother 2007;51:1011–15
- Diacon AH, Pym A, Grobusch M, et al. The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N Engl J Med 2009;360:2397–405
- Avorn J. Approval of a tuberculosis drug based on a paradoxical surrogate measure. JAMA 2013;309:1349–50
- Adhvaryu M, Vakharia B. Drug-resistant tuberculosis: emerging treatment options. Clin Pharmacol 2011;3:51–67
- Mistry BM, Jauhari, S. Synthesis and in vitro antimicrobial and anti-tubercular evaluation of some quinoline-based azitidinone and thiazolidinone analogues. Med Chem Res 2013;22:635–46
- Jain AK, Vaidya A, Ravichandran V, Kashaw SK. Recent developments and biological activities of thiazolidinone derivatives: a review. Bioorg Med Chem 2012;20:3378–95
- Paul N, Murugavel M, Muthusubramanian S, Sriram D. Camphorsulfonic acid catalysed facile tandem double Friedlander annulation protocol for the synthesis of phenoxy linked bisquinoline derivatives and discovery of antitubercular agents. Bioorg Med Chem Lett 2012;22:1643–8
- Mungra DC, Patel MP, Rajani DP, Patel RG. Synthesis and identification of β-aryloxyquinolines and their pyrano[32-c] chromene derivatives as a new class of antimicrobial and antituberculosis agents. Eur J Med Chem 2011;46:4192–200
- Upadhayaya RS, Vandavasi JK, Vasireddy NR, et al. Design synthesis biological evaluate on and molecular modeling studies of novel quinoline derivatives against Mycobacterium tuberculosis. Bioorg Med Chem 2009;17:2830–41
- Eswaran S, Adhikari AV, Kumar RA. New 13-oxazolo[45-c]quinoline derivatives: synthesis and evaluation of antibacterial and antituberculosis properties. Eur J Med Chem 2010;45:3957–66
- Nayyar A, Monga V, Malde A, et al. Synthesis anti-tuberculosis activity and 3D-QSAR study of 4-adamantan-1-yl-2-substituted quinolines. Bioorg Med Chem 2007;15:626–40
- Nayyar A, Patel SR, Shaikh M, et al. Synthesis anti-tuberculosis activity and 3D-QSAR study of amino acid conjugates of 4-adamantan-1-yl group containing quinolines. Eur J Med Chem 2009;44:2017–29
- Thomas KD, Adhikari AV, Telkar S, et al. Design synthesis and docking studies of new quinoline-3-carbohydrazide derivatives as antitubercular agents. Eur J Med Chem 2011;46:5283–92
- Eswaran S, Adhikari AV, Pal NK, Chowdhury IH. Design and synthesis of some new quinoline-3-carbohydrazone derivatives as potential antimycobacterial agents. Bioorg Med Chem Lett 2010;20:1040–4
- Thomas KD, Adhikari AV, Chowdhury IH, Sumesh E. New quinolin-4-yl-1 2 3-triazoles carrying amides sulphonamides and amidopiperazines as potential antitubercular agents. Eur J Med Chem 2011;46:2503–12
- Thomas KD, Adhikari AV, Imran H, et al. Design synthesis and docking studies of quinoline-oxazolidinone hybrid molecules and their antitubercular properties. Eur J Med Chem 2011;46:4834–45
- Carmo AML, Silva FMC, Machado PA, Fontes APS. Synthesis of 4-aminoquinoline analogues and their platinum: II complexes as new antileishmanial and antitubercular agents. Biomed Pharmacother 2011;65:204–9
- Candéa ALP, Ferreira MDL, Pais KC, et al. Synthesis and antitubercular activity of 7-chloro-4-quinolinylhydrazones derivatives. Bioorg Med Chem Lett 2009;19:6272–4
- De Souza MVN, Pais KC, Kaiser CR, Peralta MA. Synthesis and in vitro antitubercular activity of a series of quinoline derivatives. Bioorg Med Chem 2009;17:1474–80
- Gemma S, Savini L, Altarelli M, et al. Development of antitubercular compounds based on a 4-quinolylhydrazone scaffold. Further structure–activity relationship studies. Bioorg Med Chem 2009;17:6063–72
- Sharma M, Chaturvedi V, Manju YK, et al. Substituted quinolinylchalcones and quinolinylpyrimidines as a new class of anti-infective agents. Eur J Med Chem 2009;44:2081–91
- Parekh NM, Maheria KC. Antituberculosis and antibacterial evaluations of some novel phenyl pyrazolone-substituted 1h-benzo[g]pyrazolo[34-b]quinoline-3-ylamine derivatives. Med Chem Res 2012;21:4168–76
- Karthikeyan SV, Perumal S, Shetty KA, et al. A microwave-assisted facile regioselective Fischer indole synthesis and antitubercular evaluation of novel 2-aryl-34-dihydro-2H-thieno[32-b]indoles. Bioorg Med Chem Lett 2009;19:3006–9
- Balamurugan K, Jeyachandran V, Perumal S, Manjashetty TH. A microwave-assisted facile regioselective Friedlander synthesis and antitubercular evaluation of 2,9-diaryl-2 3-dihydrothieno-[3 2-b]quinolines. Eur J Med Chem 2010;45:682–8
- Kantevari S, Yempala T, Muthusubramanian S, Manisankar P. Synthesis and antitubercular evaluation of novel dibenzo[bd]furan and 9-methyl-9H-carbazole derived hexahydro-2H-pyrano[32-c]quinolines via Povarov reaction. Eur J Med Chem 2011;46:4827–33
- Gonçalves RSB, Kaiser CR, Lourenço MCS, De Souza MVN. Synthesis and antitubercular activity of new mefloquine-oxazolidine derivatives. Eur J Med Chem 2010;45:6095–100
- Chitra S, Paul N, Muthusubramanian S, et al. Synthesis of 3-heteroarylthioquinoline derivatives and their in vitro antituberculosis and cytotoxicity studies. Eur J Med Chem 2011;46:4897–903
- Gonçalves RSB, Kaiser CR, Lourenço MCS, et al. Mefloquine–oxazolidine derivatives derived from mefloquine and arenecarbaldehydes: in vitro activity including against the multidrug-resistant tuberculosis strain T113. Bioorg Med Chem 2012;20:243–8
- Eswaran S, Adhikari AV, Chowdhury I, et al. New quinoline derivatives: synthesis and investigation of antibacterial and antituberculosis properties. Eur J Med Chem 2010;45:3374–83
- Senthilkumar P, Dinakaran M, Banerjee D, et al. Synthesis and antimycobacterial evaluation of newer 1-cyclopropyl-14-dihydro-6-fluoro-7-:substituted secondary amino-8-methoxy-5-:sub-4-oxoquinoline-3-carboxylic acids. Bioorg Med Chem 2008;16:2558–69
- Senthilkumar P, Dinakaran M, Yogeeswari P, et al. Antimycobacterial activities of novel fluoroquinolones. Biomed Pharmacother 2009;63:27–35
- Senthilkumar P, Dinakaran M, Yogeeswari P, et al. Synthesis and antimycobacterial activities of novel 6-nitroquinolone-3-carboxylic acids. Eur J Med Chem 2009;44:345–58
- Dinakaran M, Senthilkumar P, Yogeeswari P, et al. Antimycobacterial activities of novel 2-sub-3 fluoro/nitro-5 12-dihydro-5-oxobenzothiazolo [3-2a]quinoline-6-carboxylic acid. Bioorg Med Chem 2008;16:3408–18
- Dinakaran M, Senthilkumar P, Yogeeswari P, et al. Novel ofloxacin derivatives: synthesis, antimycobacterial and toxicological evaluation. Bioorg Med Chem Lett 2008;18:1229–36
- Sriram D, Yogeeswari P, Basha JS, et al. Synthesis and antimycobacterial evaluation of various 7-substituted ciprofloxacin derivatives. Bioorg Med Chem 2005;13:5774–8
- Sriram D, Aubry A, Yogeeswari P, Fisher LM. Gatifloxacin derivatives: synthesis, antimycobacterial activities and inhibition of Mycobacterium tuberculosis DNA gyrase. Bioorg Med Chem Lett 2006;16:2982–5
- Zhao YL, Chen YL, Sheu JY, et al. Synthesis and antimycobacterial evaluation of certain fluoroquinolone derivatives. Bioorg Med Chem 2005;13:3921–6
- Talath S, Gadad AK. Synthesis antibacterial and antitubercular activities of some7-[4-5-amino-[1, 3, 4] thiadiazole-2-sulfonyl-piperazin-1-yl] fluoroquinolonic derivatives. Eur J Med Chem 2006;41:918–24
- Rikova EV, Polanc S, Kocevar M, et al. New fluorine-containing hydrazones active against MDR-tuberculosis. Eur J Med Chem 2011;46:4937–45
- Barends M, Jaidee A, Khaohirun N, et al. In vitro activity of ferroquine:SSR 97193 against Plasmodium falciparum isolates from the Thai-Burmese border. Malar J 2007;27:81–5
- Mahajan A, Kremer L, Louw S, et al. Synthesis and in vitro antitubercular activity of ferrocene-based hydrazons. Bioorg Med Chem Lett 2011;21:2866–8
- Parmar NJ, Barad HA, Pansuriya BR, Teraiya SB. An efficient one-pot synthesis structure antimicrobial and antioxidant investigations of some novel quinolyldibenzo[b,e][1,4]diazepinones. Bioorg Med Chem Lett 2012;22:3816–21
- Manvar A, Bavishi A, Radadiya S, et al. Diversity oriented design of various hydrazides and their in vitro evaluation against Mycobacterium tuberculosis H37Rv strains. Bioorg Med Chem Lett 2011;21:4728–31
- Carta A, Palomba M, Paglietti G, et al. [1,2,3]Triazolo[4,5-h]quinolones: a new class of potent antitubercular agents against multidrug resistant Mycobacterium tuberculosis strains. Bioorg Med Chem Lett 2007;17:4791–4
- Carta A, Palomba M, Briguglio I, et al. Synthesis and anti-mycobacterial activities of triazoloquinolones. Eur J Med Chem 2011;46:320–6
- Kantevari S, Patpi SR, Sridhar B, et al. Synthesis and antitubercular evaluation of novel substituted aryl and thiophenyl tethered dihydro-6H-quinolin-5-ones. Bioorg Med Chem Lett 2011;21:1214–17
- Shah NM, Patel MP, Patel RG. New N-arylaminobiquinoline derivatives: synthesis, antimicrobial, antituberculosis and antimalarial evaluation. Eur J Med Chem 2012;54:239–47