
Abstract
A new series of N-heteroarylsubstituted triazolosulfonamide compounds were synthesized and their inhibitory effects on the activity of purified human carbonic anhydrase (hCA) I and II were evaluated. Compounds (3 a–k) were prepared by propargylation of N-heteroaryl compounds. Compound 5 was obtained from sulfanilamide and sodium nitrite followed by addition of sodium azide. The products (6 a–k) were synthesized from compounds 3 and 5. The results showed that all the synthesized compounds were inhibited the CA isoenzymes activity. Figure 6a (IC50 = 0.52 µM for hCA I and 0.34 µM for hCA II) has the most inhibitory effect among the synthesized compounds.
Introduction
The carbonic anhydrases (CAs, EC.4.2.1.1) are a class of ubiquitous zinc containing enzymes involved in important physiological and pathological functions, such as pH and CO2 homeostasis, respiration and transport of between tissues and the lungs. CA enzymes present in prokaryotes and eukaryotes, which are encoded by four evolutionarily unrelated gene families (the α-, β-, γ-, and ξ-CAs)Citation1,Citation2.
Up to now, there are 16 isozymes differ in their subcellular localization, catalytic activity and susceptibility to different classes of inhibitors. Some of the isoforms are cytosolic (CA I, II, III, VII and XIII), the others are membrane bound (CAIV, IX, XII, XIV), two are mitochondrial and one is secreted in saliva. CAs have been obtained from many different tissues including human erythrocytes, fish liver, goat erythrocytes, fish gills and fish erythrocytesCitation3. Biological activities of this metalloenzyme family have several medicinal applications such as treatment of glaucoma, anti-convulsant, diuretics, several neurological disorders, including epilepsy, in the treatment of Alzheimer’s disease, whereas several agents are in clinical evaluationsCitation1,Citation4–8.
Nitrogen heterocycles have received a great deal of attention in the literature owing to their biological properties. Most of the drugs used in human medicine are heterocyclic compounds. Morphine, Lipitor, Penicillin and non-steroidal anti-inflammatory agents used as drug have at least one hetero atom in their structureCitation9.
Aryl and heteroaryl sulfonamides are therapeutically used to inhibit the catalytic activity of carbonic anhydrases (CAs). Using a “click-tail” approach a novel class of glycoconjugate benzene sulfonamides have been synthesizedCitation10. Triazole and its derivatives are very important class of heterocycles with wide ranging pharmacological activities such as anti-fungalCitation11, anti-bacterialCitation12, anti-cancerCitation13. Many azole derivatives such as triazole (Fluconazole, Itraconazole) and imidazole (Miconazole, Ketoconazole) have been extensively used as drugsCitation14. Cyclic imides such as succinimide, maleimide and phthalimide have potential biological activity and pharmaceutical use. The compounds have hydrophobic and neutral groups and therefore can cross biological membranes in vivoCitation15. Cyclic imides and their derivatives have received great attention in the treatment of psychopathologies such as anxiety, schizophrenia, epilepsy and depressionCitation16,Citation17.
There are many classes of compounds for CA inhibitors in the literature such as sulfonamides and their isoesters (sulfamates/sulfamides)Citation18, coumarinesCitation19, polyaminesCitation20, phenolsCitation21 and urea/thioureasCitation22,Citation23. Sulfonamides have an important inhibition effect on the CA enzyme due to gaining ionic structure easily and they constitute an important class of drugs used extensively as pharmaceutical and agricultural agentsCitation24. Many sulfonamide derivatives have been widely used clinically as anti-glaucoma drugs including orally administered acetazolamide, ophthalmic suspension of brinzolamide and ophthalmic solution of dorzolamideCitation25.
In this study, a newly synthesized N-heteroarylsubstituted triazole sulfonamide compounds were synthesized and their inhibitory effects on the activity of purified human carbonic anhydrase (hCA) I and II were evaluated.
Materials and methods
General procedure of N-heteroarylsubstituted triazolosulfonamide compounds
Melting points were taken on a Yanagimoto Barnstead Electrothermal (Surrey, UK) micro-melting point apparatus and were uncorrected. IR spectra were measured on a SHIMADZU Prestige-21 (200 VCE) spectrometer (Kyoto, Japan). 1H and 13C NMR spectra were measured on spectrometer at VARIAN Infinity Plus (Palo Alto, CA) 300 and 75 MHz, respectively. 1H and 13C chemical shifts were referenced to the internal deuterated solvent. The elemental analysis was carried out with a Leco CHNS-932 instrument (St. Joseph, MI). All chemicals were purchased from Merck (Darmstadt, Germany), Alfa Aesar (Ward Hill, MA) and Sigma-Aldrich (Taufkirchen, Germany). Synthesis of N-heteroarylsubstituted triazolosulfonamide compounds was prepared according to Scheme 1.
Scheme 1. Synthesis of N-heteroarylsubstituted triazolosulfonamides.
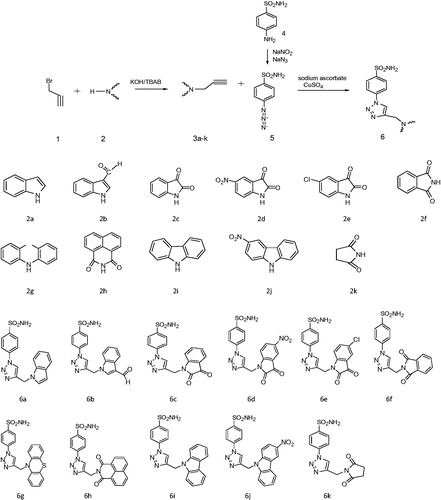
General procedure for the synthesis of Azidosulfonamide (5)
Sulfonilamide (0.5 g, 3 mmol) was dissolved in 6 M HCI (10 mL), THF (5 mL) and DMF (5 mL) and cooled to 0 °C. Sodium nitrite (248 mg, 3.6 mmol) in 15 ml of cold water was added slowly to the mixture and stirred for 25 min at 0 °C. Then, sodium azide (282 mg, 4.4 mmol) was added to the mixture and stirred overnight at room temperature. The mixture was extracted with ethyl acetate and washed with 1 M sodium hydroxide solution, saturated NaHCO3 solution and brine, respectively. The organic phase was dried over MgSO4, filtered and evaporated under vacuum. The product was purified by crystallization from CHCl3.
General procedure for the N-propargylation (3a–k)
KOH (1 mmol) and TBAB (10%) were added to dissolved compound (2) (1 mmol) in DMF and the mixture was cooled to 0 °C. Then, propargil bromide (1.2 mmol) was added and stirred overnight at room temperature. The mixture was extracted with ethyl acetate and the product was purified by crystallization from CHCl3.
General procedure for the synthesis of N-heteroarylsubstituted triazolosulfonamide compounds (6a–k)
Sodium ascorbate (0.1 mmol) and CuSO4 (0.1 mmol) in water were added to compound 5 (1.0 mmol) in DMF. Then compound 3a–k was added to the mixture and stirred overnight at room temperature. The mixture was extracted with ethyl acetate and washed with saturated NH4Cl solution. The product was purified by washing with acetone.
4-(4((1H-indol-1-yl)methyl)-1H-1,2,3-triazol-1-yl)benzenesulfonamide (6a)
Yield 75%; m.p.: 250–52 oC; IR (ν, cm−1): 3366 (NH2), 3146 (=C–H), 1312 and 1155 (S=O); 1H NMR (300 MHz; ppm; DMSO) δ 5.55 (2H, s, N–CH2), 6.45 (1H, s), 6.98–7.03 (1H, t), 7.10–7.15 (1H, t) 7.48(2H, s, –NH2), 7.51–7.62 (3H, m), 7.96–7.98 (2H, d), 8.05–8.07 (2H, d), 8.87 (1H, s, triazole ring =C–H); 13C NMR (75 MHz; ppm; DMSO) δ 145.7, 144.5, 139.1, 136.2, 129.3, 128.9, 128.1, 122.5, 121.9, 121.1, 121.0, 119.9, 110.7, 101.8, 31.37; Anal. Calcd. For C17H15N5O2S: C, 57.78; H,4.28; N, 19.82; O, 9.05; S, 9.07. Found: C, 57.99; H, 4.58; N, 19.98; O, 9.45; S, 9.46.
1-((1-(4-Sulfamoylphenyl)-1H-1,2,3-triazol-4-yl)methyl)-1H-indole-3-carbaldehyde (6b)
Yield 70%; m.p.: 316–17 °C; IR (ν, cm−1): 3398 (NH2), 3118 (=C–H), 2803 (C–H, aldehyde), 1320 and 1159 (S=O); 1H NMR (300 MHz; ppm; DMSO) δ 5.73 (2H, s, N–CH2), 7.24–7.40 (3H, m), 7.51 (2H, s, –NH2), 7.75–7.77 (1H, d), 7.98–8.01 (2H, d), 8.08–8.13 (2H, d), 8.46 (1H, s, olefinic=C–H), 9.00 (1H, s, triazole ring =C–H), 9.95 (1H, s, –CHO); 13C NMR (75 MHz; ppm; DMSO) δ 185.5, 144.6, 144.4, 141.5, 139.1, 137.5, 128.2, 125.4, 124.4, 123.3, 123.1, 121.7, 121.0, 118.2, 112.0, 42.1; Anal. Calcd. For C18H15N5O3S: C, 56.68; H, 3.96; N, 18.36; O, 12.58; S, 8.41. Found: C, 56.48; H, 3.78; N, 18.56; O, 12.79; S, 8.71.
4-(4-((2,3-Dioxoindolin-1-yl)methyl)-1H-1,2,3-triazol-1-yl)benzenesulfonamide (6c)
Yield 78%; m.p.: 282–84 °C; IR (ν, cm−1): 3367 (NH2), 3146 (=C–H), 1743 (C=O) 1322 and 1159 (S=O); 1H NMR (300 MHz; ppm; DMSO) δ 5.08 (2H, s, N–CH2), 7.12–7.23 (2H, m), 7.51 (2H, s, –NH2), 7.58–7.67 (2H, m), 7.97–8.00 (2H, d), 8.05–8.08 (2H, d), 8.97 (1H, s, triazole ring =C–H); 13C NMR (75 MHz; ppm; DMSO) δ 183.6, 158.5, 150.6, 144.5, 143.8, 139.0, 138.8, 128.2, 125.2, 124.1, 122.7, 120.7, 118.3, 111.7, 35.6; Anal. Calcd. For C17H13N5O4S: C, 53.26; H, 3.42; N, 18.27; O, 16.69; S, 8.36. Found: C, 53.56; H, 3.73; N, 18.49; O, 16.92; S, 8.16.
4-(4-((5-Nitro-2,3-dioxoindolin-1-yl)methyl)-1H-1,2,3-triazol-1-yl)benzenesulfonamide (6d)
Yield 80%; m.p.: 266–68 °C; IR (ν, cm−1): 3400 (NH2), 3095 (=C–H), 1615 (C=O); 1336 (NO2), 1316 and 1157 (S=O); 1H NMR (300 MHz; ppm; DMSO) δ 5.17 (2H, s, N–CH2), 7.43–7.46 (1H, d), 7.51 (2H, s, –NH2), 7.96–7.98 (2H, d), 8.01–8.04 (2H, d), 8.27 (1H, s), 8.49–8.53 (1H, d), 8.94 (1H, s, triazole ring =C–H); 13C NMR (75 MHz; ppm; DMSO) δ 181.5, 159.0, 154.8, 144.5, 143.8, 143.4, 139.0, 133.6, 128.2, 122.8, 120.8, 119.8, 118.8, 112.2, 31.3; Anal. Calcd. For C17H12N6O6S: C, 47.66; H, 2.82; N, 19.62; O, 22,41; S, 7.49. Found: C, 47.86; H, 2.99; N, 19.88; O, 22,61; S, 7.74.
4-(4-((5-Chloro-2,3-dioxoindolin-1-yl)methyl)-1H-1,2,3-triazol-1-yl)benzenesulfonamide (6e)
Yield 82%; m.p.: 287–89 °C; IR (ν, cm−1): 3388 (NH2), 3091 (=C–H), 1607 (C=O), 829 (Cl); 1318 and 1156 (S=O); 1H NMR (300 MHz; ppm; DMSO) δ 5.10 (2H, s, N–CH2), 7.24–7.26 (1H, d), 7.53 (2H, s, –NH2), 7.66–7.72 (2H, d), 7.98–8.01 (2H, d), 8.05–8.08 (2H, d), 8.95 (1H, s, triazole ring =C–H); 13C NMR (75 MHz; ppm; DMSO) δ 182.5, 158.3, 149.1, 144.5, 143.7, 139.0, 137.6, 128.3, 128.2, 124.7, 122.7, 120.8, 119.8, 113.5, 35.7; Anal. Calcd. For C17H12ClN5O4S: C, 48.87; H, 2.89; Cl, 8.49; N, 16.76; O, 15.32; S, 7.67. Found: C, 49.05; H, 3.05; Cl, 8.65; N, 16.96; O, 15.74; S, 7.96.
4-(4-((1,3-Dioxoisoindolin-2-yl)methyl)-1H-1,2,3-triazol-1-yl)benzenesulfonamide (6f)
Yield 78%; m.p.: 273–75 °C; IR (ν, cm−1): 3467 (NH2), 3252 (=C–H), 1716 (C=O), 1314 and 1155 (S=O); 1H NMR (300 MHz; ppm; DMSO) δ 4.92 (2H, s, N–CH2), 7.50 (2H, s, –NH2), 7.80–7.95 (4H, m), 7.94–7.97 (2H, d) 8.05–8.08 (2H, d), 8.89 (1H, s, triazole ring =C–H); 13C NMR (75 MHz; ppm; DMSO) δ 168.05, 144.7, 144.4, 135.4, 135.2, 132.3, 128.0, 124.0, 123.9, 120.8, 33.6; Anal. Calcd. For: C17H13N5O4S: C, 53.26; H, 3.42; N, 18.27; O, 16.69; S, 8.36. Found: C, 53.63; H, 3.72; N, 18.63; O, 16.99; S, 8.62.
4-(4-((10H-phenothiazin-10-yl)methyl)-1H-1,2,3-triazol-1-yl)benzenesulfonamide (6g)
Yield 85%; m.p.: 230–32 °C; IR (ν, cm−1): 3361 (NH2), 3062 (=C–H), 1320 and 1156 (S=O); 1H NMR (300 MHz; ppm; DMSO) δ 5.24 (2H, s, N–CH2), 6.91–7.01 (4H, m), 7.11–7.16 (4H, m), 7.53 (2H, s, –NH2), 7.96–7.99 (2H, d), 8.10–8.13 (2H, d), 8.84 (1H, s, triazole ring =C–H); 13C NMR (75 MHz; ppm; DMSO) δ 145.7, 144.6, 144.4, 139.1, 128.2, 128.1, 127.4, 123.4, 123.2, 122.5, 120.8, 116.4, 44.4; Anal. Calcd. For: C21H17N5O2S2: C, 57.91; H, 3.93; N, 16.08; O, 7.35; S, 14.72. Found: C, 58.18; H, 3.83; N, 16.35; O, 7.62; S, 14.92.
4-(4-((1,3-Dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)methyl)-1H-1,2,3-triazol-1-yl)benzenesulfonamide(6h)
Yield 72%; m.p.: 306–08 °C; IR (ν, cm−1): 3364 (NH2), 3101 (=C–H), 1707 (C=O), 1326 and 1158 (S=O); 1H NMR (300 MHz; ppm; DMSO) δ 5.42 (2H, s, N–CH2), 7.49 (2H, s, –NH2), 7.89–7.97 (4H, m), 8.06–8.09 (2H, d), 8.50–8.56 (4H, t), 8.86 (1H, s, triazole ring =C–H);13C NMR (75 MHz; ppm; DMSO) δ 163.9, 146.2, 144.3, 135.2, 131.6, 129.5, 128.1, 127.9, 124.5, 122.7, 122.0, 120.7, 118.6, 36.8; Anal. Calcd. For: C21H15N5O4S: C, 58.19; H, 3.49; N, 16.16; O, 14.77; S, 7.40 Found: C, 58.65; H, 3.72; N, 16.36; O, 14.97; S, 7.62.
4-(4-((9H-Carbazol-9-yl)methyl)-1H-1,2,3-triazol-1-yl)benzenesulfonamide (6i)
Yield 86%; m.p.: 286–87 °C; IR (ν, cm−1): 3317 (NH2), 3099 (=C–H), 1315 and 1155 (S=O); 1H NMR (300 MHz; ppm; DMSO) δ 5.76 (2H, s, N–CH2), 7.18–7.23 (2H, t), 7.43–7.78 (2H, t), 7.48 (2H, s, –NH2), 7.78–7.80 (2H, d), 7.92–7.95 (2H, d), 8.02–8.05 (2H, d), 8.12–8.15 (2H, d), 8.91 (1H, s, triazole ring =C–H); 13C NMR (75 MHz; ppm; DMSO) δ 145.3, 144.4, 140.4, 139.1, 128.1, 126.4, 122.9, 122.3, 120.9, 119.8, 110.3, 38.1, 31.3; Anal. Calcd. For: C21H17N5O2S: C, 62.52; H, 4.25; N,17.36; O, 7.93; S, 7.95. Found: C, 62.82; H, 4.66; N, 17.16; O, 7.73; S, 8.25.
4-(4-((3-Nitro-9H-carbazol-9-yl)methyl)-1H-1,2,3-triazol-1-yl)benzenesulfonamide (6j)
Yield 82%; m.p.: 310–12 °C; IR (ν, cm−1): 3326 (NH2), 3143 (=C–H), 1333 (NO2), 1330 and 1162 (S=O); 1H NMR (300 MHz; ppm; DMSO) δ 5.89 (2H, s, N–CH2), 7.30–7.35 (1H, t), 7.50 (2H, s, –NH2), 7.55–7.60 (1H, t), 7.87–8.04 (6H, m), 8.34–8.41 (2H, t), 8.94 (1H, s, triazole ring =C–H), 9.17–9.19 (1H, m); 13C NMR (75 MHz; ppm; DMSO) δ 144.6, 144.5, 143.7, 141.7, 140.9, 139.0, 128.1, 123.0, 122.8, 122.6, 122.0, 121.5, 120.9, 117.9, 111.3, 110.6, 38.6; Anal. Calcd. For C21H16N6O4S: C, 56.24; H, 3.60; N, 18.74; O, 14.27; S, 7.15. Found: C, 56.54; H, 3.39; N, 18.94; O, 14.50; S, 7.55.
4-(4-((2,5-Dioxopyrrolidin-1-yl)methyl)-1H-1,2,3-triazol-1-yl)benzenesulfonamide (6k)
Yield 72%; m.p.: 227–28 °C; IR (ν, cm−1): 3333 (NH2), 3112 (=C–H), 1688 (C=O), 1329 and 1163 (S=O); 1H NMR (300 MHz; ppm; DMSO) δ 2.64 (4H, s), 4.69 (2H, s, N–CH2), 7.52 (2H, s, –NH2), 8.00–8.06 (2H, d), 8.08–8.09 (2H, d), 8.77 (1H, s, triazole ring =C–H); 13C NMR (75 MHz; ppm; DMSO) δ 177.8, 144.5, 144.3, 139.1, 128.2, 122.3, 120.9, 33.95, 28.84; Anal. Calcd. For C13H13N5O4S: C, 46.56; H, 3.91; N, 20.88; O, 19.08; S, 9.56. Found: C, 46.06; H, 4.21; N, 20.68; O, 19.28; S, 9.76.
CA enzyme assay
Preparation of hemolysate and purification from blood red cells
Blood samples (25 mL) were taken from healthy human volunteers. They were anti-coagulated with ACD (acid-citrate-dextrose), centrifuged at 2000 × g for 20 min at 4 °C and the supernatant was removed. The packed erythrocytes were washed three times with 0.9% NaCI and then hemolysed in cold water. The ghosts and any intact cells were removed by centrifugation at 2000 × g for 25 min at 4 °C, and the pH of the hemolysate was adjusted to pH 8.5 with solid Tris-base. The 25 mL hemolysate was applied to an affinity column containing l-tyrosine-sulfonamide-Sepharose-4BCitation26 equilibrated with 25 mM Tris–HCl/0.1 M Na2SO4 (pH 8.5). The affinity gel was washed with 50 mL of 25 mM Tris–HCl/22 mM Na2SO4 (pH 8.5). The hCA isozymes were then eluted with 0.1 M NaCl/25 mM Na2HPO4 (pH 6.3) and 0.1 M CH3COONa/0.5 M NaClO4 (pH 5.6), which recovered hCA I and II, respectively. Fractions of 3 mL were collected and their absorbance measured at 280 nm.
In vitro inhibition studies
CA activity was measured by the Maren method based on the determination of the time required for the pH to decrease from 10.0 to 7.4 due to CO2 hydrationCitation27. The assay solution was 0.5 M Na2CO3/0.1 M NaHCO3 (pH 10.0) and Phenol Red was added as the pH indicator. CO2-hydratase activity (enzyme units (EU)) was calculated by using the formula t0−tc/tc, where t0 and tc are the times for pH change of the non-enzymatic and the enzymatic reactions, respectively.
For the inhibition studies of sulfonamide, different concentrations of these compounds were added to the enzyme. Activity % values of CA for different concentrations of each compound were determined by regression analysis using Microsoft Office 2000 Excel. CA enzyme activity without a sulfonamide solution was accepted as 100% activity. For the sulfonamide having an inhibition affect, the inhibitor concentration causing up to 50% inhibition (IC50 values) was determined from the graphs.
Results and discussion
To evaluate the physiologically relevant human CA isozymes hCAI and II inhibitory activity, several N-heteroarylsubstituted triazolosulfonamide compounds were subjected to CA inhibition assay with CO2 as substrate.
Compounds 3a–k were prepared by propargylation of N-heteroaryl compounds in the presence of KOH-TBAB in DMF at room temperature. Compound 5 was obtained from sulfonilamide and sodium nitrite with HCl in THF-DMF mixture and followed by addition of sodium azide to the reaction mixture. The products (6a–k) were synthesized by reacting compounds 3a–k and 5 with sodium ascorbate and CuSO4 in DMF.
The prepared compounds were characterized by 1H NMR, 13C NMR, IR and elemental analysis. The 1H NMR results depict the successful synthesis of the compounds. The resonances between 7.00 ppm and 8.50 ppm can be assigned to aromatic protons and about 7.50 ppm for –SO2NH2 protons and 4.50–6.00 ppm CH2 next to the triazole ring and around 9.00 ppm for =C–H in triazole ring. In addition to the analysis of 1H NMR, 13C NMR resonance assignments were also studied. The peaks of carbonyl carbons were observed at around 190 and 163 ppm, respectively. In the infrared spectra of compounds 6a–k, it was possible to observe the absorptions between 3300 and 3500 cm−1 relating to NH2 stretch, absorptions around 1700 cm−1 from carbonyl moiety stretching. As seen in the literatureCitation28, there are two peaks assigned to S=O as symmetric and asymmetric stretching. The peak of asymmetric and symmetric stretch appeared around 1300 and 1100 cm−1, respectively.
Sulfonamides are coordinated to the zinc (II) ion within the hCAs active site, whereas its organic scaffold fills the entire enzyme cavity, making an extensive series of van der Waals and polar interactions with amino acid residues both at the bottom, middle and entrance of the active site cavityCitation29. Inhibitors of carbonic anhydrase play an important role in ophthalmology where they are used to reduce elevated intraocular pressure (IOC). Acetazolamide that is less active against CAI than against CAII in erythrocytes is the most widely used inhibitorsCitation30. For evaluation the physiologically relevant human CA isozymes hCA I and II inhibitory activity, newly synthesized sulfonamide compounds were subjected to CA inhibition assay with CO2 as substrate. The obtained results showed that all the compounds inhibited the enzyme activity and the inhibition constants against CAs are given in the . It is determined that the inhibition values are between 0.52 and 1.36 µM for hCA I and 0.34 and 1.84 µM for hCA II. The following should be noted regarding the CA inhibitory data of :
The N-heteroarylsubstituted triazole sulfonamide derivatives investigated here showed good inhibitory properties against the slow cytosolic isoform hCA I. Compound 6a exhibited the lowest inhibition of this isoform, with IC50 value of 0.52 µM. Compound 6k (IC50 value of 1.36) is the least active molecule of the compounds due to the small pendent group.
A better inhibitory activity has been observed with compound 6a for the inhibition of the rapid cytosolic isozyme hCA II. However, 6k is the least active molecule with the value of 1.80 µM. One of the best hCA II inhibitor in the compounds was the bulky 6h (IC50 value of 0.35 µM).
Table 1. The IC50 values of N-heteroarylsubstituted triazolosulfonamides.
Although sulfonamide compounds are mentioned as very potent inhibitor of the cytosolic isoform hCAsCitation31, all the synthesized substituted sulfonamides have good inhibitory activity for the hCAs. Despite the similarities of the structures of CAs I, II, VII and XIV, there are several differences in the active sites of amino acids. Particularly, hydrophobic residues (Phe131, Val135 and Leu204) on the surface of CAII have an important stabilizing role when interacting with the hydrophobic groups of sulfonamidesCitation2. Sulfonamides inhibit hCAI and II by coordination of the sulfonamide moiety to the Zinc metal from the enzyme active site, and participation in hydrophobic and hydrophilic interactions of the organic scaffold of the inhibitor with amino acids residuesCitation32. It is assumed that the effects are the results of hydrophobic and van der Waals interactions between large pendant part of the inhibitor molecule (indole, isatin, carbazole, etc.) whereas less hydrophobic and van der Waals interactions occur between small pendant group such as succinimide and active site of amino acid residues.
In conclusion, we have reported the synthesis, characterization and biological activity evaluation for the inhibition of the physiologically relevant CA isozymes of a series of N-heteroarylsubstituted triazole sulfonamide derivatives. They inhibit the CAs with the inhibition constants of 0.52–1.36 µM for hCA I and 0.34–1.80 µM for hCA II. Compound 6a behaved as a good inhibitory activity on hCA I and II with an inhibition constant of 0.52 µM and 0.34 µM, respectively, because of large pendant groups. Compound 6k has a weak inhibitory activity 1.36 µM for hCA I and 1.80 µM for hCA II due to small pendant group. Enzyme inhibition is more important issue for drug design and biochemical applicationsCitation33–36. The results put forward that new sulfonamide derivatives inhibited the hCA I and II enzyme activity. Therefore, our results suggested that the sulfonamide derivatives may be used for some medical applications and they may be taken for further evaluation in in vivo studies.
Declaration of interest
This work was supported by Research Fund of the Sakarya University. Project Number: 2012-02-04-033.
References
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81
- Hen N, Bialer M, Yagen B, et al. Anticonvulsant 4-aminobenzenesulfonamide derivatives with branched-alkylamide moieties: X-ray crystallography and inhibition studies of human carbonic anhydrase isoforms i, II, VII, and XIV. J Med Chem 2011;54:3977–81
- Demirdag R, Comakli V, Senturk M, et al. Purification and characterization of carbonic anhydrase from sheep kidney and effects of sulfonamides on enzyme activity. Bioorg Med Chem 2013;21:1522–5
- Innocenti A, Durdagi S, Doostdar N, et al. Nanoscale enzyme inhibitors: fullerenes inhibit carbonic anhydrase by occluding the active site entrance. Bioorg Med Chem 2010;18:2822–8
- Ekinci D, Cavdar H, Durdagi S, et al. Structure-activity relationships for the interaction of 5,10-dihydroindeno[1,2-b] indole derivatives with human and bovine carbonic anhydrase isoforms I, II, III, IV and VI. Eur J Med Chem 2012;49:68–73
- Alp C, Maresca A, Alp NA, Gültekin MS. Secondary/tertiary benzenesulfonamides with inhibitory action against the cytosolic human carbonic anhydrase isoforms I and II. J Enzyme Inhib Med Chem 2013;28:294–8
- Maresca A, Supuran CT. (R)-/(S)-10-Camphorsulfonil-substituted aromatic/heterocyclic sulfonamides selectively inhibit mitochondrial over cytosolic carbonic anhydrases. Bioorg Med Chem Lett 2011;21:1334–7
- Riafrecha LE, Rodriguez OM, Vullo D, et al. Synthesis of C-cinnamoyl glycosides and their inhibitory activity against mammalian carbonic anhydrases. Bioorg Med Chem 2013;21:1489–94
- Joule JA, Mills K. Heterocyclic chemistry at a glance. Chichester, UK: John Wiley & Sons, 2012
- Wilkinson BL, Bornaghi LF, Houston TA, et al. A novel class of carbonic anhydrase Inhibitors: glyco-conjugate benzene sulfonamides prepared by click tailing. J Med Chem 2006;49:6539–48
- Collin X, Sauleau A, Coulon J. 1,2,4-Triazolo mercapto and aminonitriles as potent antifungal agents. Bioorg Med Chem Lett 2003;13:2601–5
- Bayrak H, Demirbas A, Bektas H, et al. Synthesis and antimicrobial activities of some new 1,2,4-triazole derivatives. Turkish J Chem 2010;34:835–46
- Mavrova AT, Wesselinova D, Tsenov YA, Denkova P. Synthesis, cytotoxicity and effects of some 1,2,4-triazole and 1,3,4-thiadiazole derivatives on immunocompetent cells. Eur J Med Chem 2009;44:63–9
- Zhang YY, Zhou CH. Synthesis and activities of naphthalimide azoles as a new type of antibacterial and antifungal agents. Bioorg Med Chem Lett 2011;21:4349–52
- Al-Suavidan IA, Alanazi AM, El-Azab AS, et al. Molecular design, synthesis and biological evaluation of cyclic imides bearing benzenesulfonamide fragment as potential COX-2 inhibitors. Part 2. Bioorg Med Chem Lett 2013;23:2601–5
- Goehring RR, Greenwood TD, Pisipati JS, Wolfe JF. Synthesis and anticonvulsant evaluation of some new 2-benzylsuccinimides. J Pharm Sci 1991;80:790–2
- de Oliveira KN, Costa P, Santin JR, et al. Synthesis and antidepressant-like activity evaluation of sulfonamides and sulfonil-hydrazones. Bioorg Med Chem 2011;19:4295–306
- Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2010;20:3467–74
- Maresca A, Temperini C, Vu H, et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–62
- Carta F, Temperini C, Innocenti A, et al. Polyamines inhibit carbonic anhydrases by anchoring to the zinc-coordinated water molecule. J Med Chem 2010;53:5511–17
- Sarikaya SBO, Topal F, Sentürk M, et al. In vitro inhibition of alpha-carbonic anhydrase isozymes by some phenolic compounds. Bioorg Med Chem Lett 2011;21:4259–68
- Celik F, Arslan M, Yavuz E, et al. Synthesis and carbonic anhydrase inhibitory properties of novel 1,4-dihydropyrimidinone substituted diarylureas. J Enzyme Inhib Med Chem 2014;29:18–22
- Pacchiano F, Aggarwal M, Avvaru BS, et al. Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureido-benzensulfonamides and correlate to inhibitor potency. Chem Comm 2010;46:8371–3
- Jain P, Saravanan C, Singh SK. Sulfonamides: deserving class as MMP inhibitors? Eur J Med Chem 2013;60:89–100
- Supuran CT, Maresca A, Gregan F, Remko M. Three new aromatic sulfonamide inhibitors of carbonic anhydrases I, II, IV and XII. J Enzyme Inhib Med Chem 2013;28:289–93
- Arslan O, Nalbantoglu B, Demir N, et al. A new method for the purification of carbonic anhydrase isozymes by affinity chromatography. Turk J Med Sci 1996;26:163–6
- Maren TH. A simplified micromethod for the determination of carbonic anhydrase and its inhibitors. J Pharm Exp Ther 1960;130:2629–34
- Şen E, Alım Z, Duran H, et al. Inhibitory effect of novel pyrazole carboxamide derivatives on human carbonic anhydrase enzyme. J Enzyme Inhib Med Chem 2013;28:328–36
- Maresca A, Temperini C, Pochet L, et al. Decipheringthe mechanism of carbonic anhydrase ınhibition with coumarins and thiocoumarins. J Med Chem 2010;53:335–44
- Arslan O, Cakır U, Ugras HI. Synthesis of new sulfonamide inhibitors of carbonic anhydrase. Biochemistry (Moscow) 2002;67:1055–7
- Winum JY, Vullo D, Casini A, et al. Carbonic anhydrase inhibitors. Inhibition of cytosolic isozymes I and II and transmembrane, tumor-associated isozyme IX with sulfamates including EMATE also acting as steroid sulfatase inhibitors. J Med Chem 2003;46:2197–204
- Wagner J, Avvaru BS, Robbins AH, et al. Coumarinyl-substituted sulfonamides strongly inhibit several human carbonic anhydrase isoforms: solution and crystallographic investigations. Bioorg Med Chem 2010;18:4873–8
- Gencer N, Ergün A, Demir D. In vitro effects of some anabolic compounds on erythrocyte carbonic anhydrase I and II. J Enzy Inhib Med Chem 2012;27:208–10
- Sinan S, Gencer N, Turan Y, Arslan O. In vitro inhibition of the carbonic anhydrase from saanen goat (capra hircus) with pesticides. Pest Biochem Phys 2007;88:307–11
- Demir D, Gencer N, Er A. Purification and characterization of prophenoloxidase from Galleria mellonella L. Artif Cells Blood Subs Biotec 2012;40:391–5
- Gencer N, Ergun A, Demir D. In vitro effects of some herbicides and fungicides on human erythrocyte carbonic anhydrase activity. Fresenius Environ Bull 2012;21:549–52