
Abstract
An efficient method to obtain ethyl 5-amino-1-tosyl-1H-pyrazole-4-carboxylate (3) was outlined using condensation reactions of 4-methylbenzenesulfonylhydrazide with (E)-ethyl 2-cyano-3-ethoxyacrylate. The cyclocondensation reaction of this substrate and its hydrazide derivative with urea, thiourea, formamide, formic acid, d-glucose, o-phenylenediamine, 4-dimethylaminobenzaldehyde, anthracene-9-carbaldehyde, thioglycolic acid and carbon disulphide then with hydrazine hydrate analogues furnished a series of pyrazolo[3,4-d]pyrimidine, pyrazolo[3,4-d]oxazin-4-one, pyrazole-4-glucoside, 4-benzo[d]imidazole, 1,3-thiazolidinone, 1,3,4-oxadiazol-2(3H)-thione and 1,2,4-triazol-5(4H)-thione derivatives respectively. The structure of the compound 3 was supported by X-Ray crystallographic data. Orally administrated, one of each of the series of pyrazoles showed significant effects in mouse tumor model cancer cell lines (EAC) and two human cancer cell lines of Colon cancer (HCT-29) and Breast cancer (MCF-7) with docking studies.
Introduction
The chemistry of 1H-pyrazole-containing compounds is particularly interesting because of their potential application in medicinal chemistry as analgesicCitation1,Citation2, anti-inflammatoryCitation3,Citation4, anti-tumorCitation5,Citation6, antimicrobialCitation7–11 and therapeutic agentsCitation12, as well as based on their wide applications in agriculture as potent insecticidesCitation13,Citation14 and herbicidesCitation15,Citation16, although scarcely found in natureCitation17. Due to many promising pharmacological, agrochemical, and analytical applications, a number of substituted pyrazoles are being used as inhibitors of heat-shock protein 90 (HSP90) and as therapeutics of cancer and therefore they have been the focus of many synthetic targets over the past decadesCitation18. Furthermore, it has also been found that 1H-pyrazole based heterocyclic structures have attracted synthetic interest for being an essential moieties in many chemotherapeutic agents with potential anti-parasiticCitation19,Citation20, anti-malarialCitation21 and antiviral activitiesCitation22,Citation23. As far as the anticancer activity is concerned, literature citation revealed that a wide range of pyrazole derivatives were reported to contribute to a variety of anti-neoplastic potentials against a wide range of cancer cell linesCitation24–27. Moreover, many pyrazole derivatives are associated with anti-fungal, anti-bacterialCitation28 and anti-pyreticCitation29 properties. Pyrazolopyrimidines and related fused heterocycles are of interest as potential bioactive molecules. They are known to exhibit pharmacological activities such as CNS-depressantCitation30, anticancerCitation31–33 and tuberculostaticCitation34 activities. Furthermore, some pyrazolo(3,4-d)pyrimidine derivatives demonstrated significant antiviral activityCitation35,Citation36. There is no much difference in the basic structures of pyrazolopyrimidines and purinesCitation37. The above facts and our interestCitation38 in the synthesis of new biologically active isolated and fused substituted heterocycles prompted us to synthesize new substituted pyrazoles linked to five membered heterocycles and fused pyrazolopyrimidine and oxazine ring systems in order to increase its biological activity and evaluate its activity as anticancer agents.
Experimental
Chemistry
General procedures
All melting points were measured using a Reichert Thermovar apparatus (Depew, NY) and are uncorrected. Yields listed are of isolated compounds. The IR spectra were recorded on a perkin-Elmer model 1720 FTIR spectrometer (Waltham, MA) for KBr disc. NMR spectra were recorded on a varian Gemini 300 BB NMR Spectrometer (Buffalo, NY) at 300 MHz for 1H and 75 MHz for 13C. Chemical shifts were reported in δ scale (ppm) relative to TMS as a reference standard and the coupling constants J values are given in Hz. Mass spectra were recorded on GC/MS Finnegan SSQ 7000 spectrophotometer and GC Ms-QP 1000 EX mass spectrometer at 70 eV (Waltham, MA). The progress of the reactions was monitored by TLC using aluminum silica gel plates 60 F245. Elemental analyses were performed at the Microanalytical Centre at Faculty of Science, Cairo University, Egypt.
Reaction of sulfonylhydrazide 1 with cyanoacrylate 2
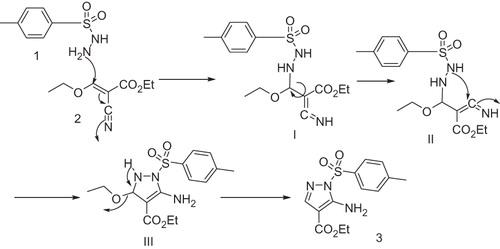
To a solution of 4-methylbenzenesulfonylhydrazide (1) (1.86 g, 0.01 mol) in absolute ethanol (20 ml) was added (E)-ethyl 2-cyano-3-ethoxyacrylate (2) (1.69 g, 0.01 mol). The reaction mixture was heated at reflux temperature for 16 h, cooled to room temperature. The solvent was evaporated under reduced pressure. The resulting solid was recrystallized from ethanol to afford ethyl 5-amino-1-tosyl-1H-pyrazole-4-carboxylate (3) as a white solid, 2.01 g, (64.97%) m.p. 135–137 °C; dec. Diffraction-quality crystal was grown by slow diffusion of ethanol solution. IR (KBr) cm−1: ν (NH2) 3480, ν (C=O) 1693, ν (C=N) 1615; 1H NMR (DMSO-d6, 300 MHz): δ = 1.29 (t, 3H, J = 7.2, O–CH2CH3), 2.34 (s, 3H, CH3), 4.18 (q, 2H, J = 2.7, O–CH2CH3), 4.22 (br.s, 2H, NH2 exchangeable), 7.46 (d, 2H, J = 8.1 Hz, Ar–H), 7.84 (d, 2H, J = 8.1 Hz, Ar–H), 7.90 (s, 1H, pyrazole N=CH) ppm; 13C NMR (DMSO-d6, 75 MHz): δ = 15.3 (CH3), 21.2 (CH3), 61.2 (CH2), 106.5 (C-4), 128–140 (Ar. C + C-3), 148.5 (C-5), 160.5 (C=O) ppm; GC/MS: m/z (%) 309 M+ (8.37), 245 (6.07), 199 (5.90), 155 (12.81), 91 (33.88), 63 (91.51), 44 (100). Anal. Calcd. for C13H15N3O4S (309.34): C, 50.47; H, 4.89; N, 13.58; Found: C, 50.39; H, 4.90; N, 13.45%.
Reaction of pyrazolocarboxylate 3 with formic acid
A suspension of 3 (3.09 g, 0.01 mol) in formic acid (85%; 20 ml) was heated at reflux temperature for 10 h. A solid product was obtained after cooling at room temperature was filtered off and recrystallized from ethanol to give 1-tosylpyrazolo(3,4-d1,3)oxazin-4(1H)-one (4) as a red powder 1.6 g (54.92%), m.p. 135–136 °C, IR (KBr) cm−1: ν (C=O) 1644; 1H NMR (DMSO-d6, 300 MHz) ppm: δ = 2.34 (s, 3H, CH3), 7.46 (d, 2H, J = 8.1 Hz, Ar–H), 7.84 (d, 2H, J = 8.1 Hz, Ar–H), 7.9 (s, 1H, pyrazole N=CH), 8.14 (s, 1H, oxazine N=CH); 13C NMR (DMSO-d6, 75 MHz): δ = 21.2 (CH3), 106.5 (C-3a), 128–140 (Ar. C + C-3), 147.0 (C-7a), 159.5 (C=O), 162 (C-6); Anal. Calcd. for C12H9N3O4S (291.28): C, 49.48, H, 3.11, N, 14.43; Found: C, 49.56, H, 3.0, N, 14.30.
Reaction of pyrazolocarboxylate 3 with formamide
A solution of 3 (3.09 g, 0.01 mol) in formamide (10 ml) was heated on oil bath at 180–190 °C for 4 h. The solution was then cooled to room temperature and diluted with 100 ml ice-cooled water. The precipitate that obtained was filtered off and recrystallized from ethanol to afford 1-tosyl-1H-pyrazolo(3,4-d)pyrimidin-4(5H)-one (5) as a yellow powder, 1.6 g (55.11%) m.p. 125–126 °C; IR (KBr) cm−1: ν (NH) 3165, ν (C=O) 1700, ν (C=N) 1600; 1H NMR (DMSO-d6, 300 MHz) ppm: δ = 2.34 (s, 3H, CH3), 7.46 (d, 2H, J = 8.1 Hz, Ar–H), 7.84 (d, 2H, J = 8.1 Hz, Ar–H), 7.9 (s, 1H, pyrazole N=CH), 8.0 (br.s, 1H, NH exchangeable), 9.0 (s, 1H, pyrimidine N=CH); 13C NMR (DMSO-d6,75 MHz): δ = 21.2 (CH3), 105.2 (C-3a), 128–140 (Ar. C + C-3), 145.5 (C-6), 147 (C-7a), 157.65 (C=O); Anal. Calcd. for C12H10N4O3S (290.30): C, 49.65; H, 3.47; N, 19.30; Found: C, 49.60; H, 3.35; N, 19.35%.
General procedure for preparing compounds (6a,b)
Ethyl 5-amino-1-tosyl-1H-pyrazole-4-carboxylate (3) (5 g, 1.61 mol) and urea (10 g, 16.66 mol) and/or thiourea (10 g, 13.15 mol) were heated together in an oil path at 150 °C for 30 min. The clear solution went mushy and heating was continued for ten minutes at 170 °C. The resulting solids were dissolved in dilute sodium hydroxide solution then acidified with acetic acid to obtain the crude products 6a or 6b.
1-Tosyl-1H-pyrazolo3,4-dpyrimidin-4,6(5H,7H)-dione (6a)
2.72 g (54.88%) yellow powder, m.p. 290–291 °C, (ethyl acetate); IR (KBr) cm−1: ν (NH) 3210, ν (C=O) 1690, ν (C=N) 1610; 1H NMR (DMSO-d6, 300 MHz) ppm: δ = 2.34 (s, 3H, CH3), 7.46 (d, 2H, J = 8.1 Hz, Ar–H), 7.6 (s, 1H, -NH exchangeable), 7.84 (d, 2H, J = 8.1 Hz, Ar–H), 7.90 (s, 1H, N=CH pyrazole), 8.70 (s, 1H, NH exchangeable); 13C NMR (DMSO-d6, 75 MHz): δ = 21.2 (CH3), 127.2–140.7 (Ar. C + C-3), 148.5 (C-7a), 150.6 (C-3a), 154.3, 155.5 (2C=O); Anal. calcd. for C12H10N4O4S (306.30): C, 47.06; H, 3.29; N, 18.29; Found: C, 47.05; H, 3.25; N, 18.25.
6-Thioxo-1-tosyl-6,7-dihydro-1H-pyrazolo3,4-dpyrimidin-4(5H)-one (6b)
2.63 g (52.6%) brown powder, m.p. 190–191 °C, (ethanol); IR (KBr) cm−1: ν (NH) 3216, ν (C=O) 1670, ν (C=S) 1623, ν (C=N) 1610; 1H NMR (DMSO-d6, 300 MHz) ppm: δ = 2.34 (s, 3H, CH3), 5.90 (br. s, 1H, NH exchangeable), 7.46 (d, 2H, J = 8.1 Hz, Ar–H), 7.84 (d, 2H, J = 8.1 Hz, Ar–H), 7.9 (s, 1H, N=CH pyrazole), 8.20 (br. s, 1H, NH exchangeable); 13C NMR (DMSO-d6, 75 MHz): δ = 21.2 (CH3), 127–140 (Ar. C + C-3), 148.6 (C-7a), 150.5 (C-3a), 155.23 (C=O), 172 (C=S); Anal. calcd. for C12H10N4O3S2 (322.36): C, 44.71; H, 3.13; N, 17.38; Found: C, 44.65; H, 3.17; N, 17.46%.
Formation of 5-amino-1-tosyl-1H-pyrazol-4-carboxylic acid (7)
A mixture of the pyrazolocarboxylate 3 (9.27 g, 0.03 mol) and sodium hydroxide (4 g, 0.1 mol) in 20 ml water was heated in an oil bath for 15 h. It was then cooled to 10 °C and acidified with HCl. The produced precipitate was filtered off, washed with water, and recrystallized from ethyl acetate to afford 7 as a red solid, 5.06 g (59.96%) m.p. 230–231 °C, IR (KBr) cm−1: ν (OH) 3431, (NH) 3180, ν (C=O) 1640, 1H NMR (DMSO-d6, 300 MHz) ppm: δ = 2.34 (s, 3H, CH3), 4.50 (br. s, 2H, NH2 exchangeable), 7.49 (d, 2H, J = 8.1 Hz, Ar–H), 7.84 (d, 2H, J = 8.1 Hz, Ar–H), 7.9 (s, 1H, pyrazole N=CH), 11.00 (br. s, 1H, OH exchangeable), 13C NMR (DMSO-d6,75 MHz): δ = 21.2 (CH3), 106.2 (C-4), 128–140 (Ar. C + C-3), 146.6 (C-5), 165.0 (C=O); Anal. Calcd. for C11H11N3O4S (281.29): C, 46.97, H, 3.94, N, 14.94; Found: C, 46.84, H, 3.90, N, 14.84%.
Reaction of pyrazolocarboxylic acid 7 with Ac2O
The pyrazolocarboxilic acid 7 (11.25 g, 0.04 mol) was heated with 30 ml acetic anhydride in an oil bath for 6 h. The mixture was cooled to room temperature, poured onto ice-cooled water, and stirred for 30 min. The solid obtained was filtered, washed with water, recrystallized from ethanol to afford 6-methyl-1-tosylpyrazolo(3,4-d1,3)oxazin-4(1H)-one (8) as a brown powder; 4.64 g (37.99%) m.p. 280–281 °C, IR (KBr) cm−1: ν (C=O) 1696; 1H NMR (DMSO-d6) ppm: δ = 1.70 (s, 3H, CH3), 2.34 (s, 3H, CH3), 7.46 (d, J = 8.1, 2H, Ar–H), 7.70 (d, J = 8.1, 2H, Ar–H), 7.90 (s, 1H, pyrazole CH); 13C NMR (DMSO-d6,75 MHz): δ = 21.2 (CH3), 25.2 (CH3), 106.2 (C-9), 128–140 (Ar. C + C-3), 147 (C-7a), 154.3 (C=O), 159.6 (C6); Anal. Calcd. for C13H11N3O4S (305.31): C, 51.14, H, 3.63, N, 13.76; Found: C, 51.0, H, 3.60, N, 13.80%.
Reaction of pyrazolocarboxylic acid 7 with o-phenylenediamine
A mixture of 7 (2.81 g, 0.01 mol) and o-phenylenediamine (1.08 g, 0.01 mol) in DMF (30 ml) was heated at reflux temperature for 6 h. The reaction mixture was allowed to cool at room temperature and poured onto cold water. The solid formed was filtered off, recrystallized from ethanol to afford 4-(1H-benzodimidazol-2-yl)-1-tosyl-1H-pyrazol-5-amine (9) as a brown solid; 2 g (56.59%) m.p. 170–172 °C, IR (KBr)cm−1: ν (NH2) 3307, ν (NH) 3190, ν (C=N) 1602; 1H NMR (DMSO-d6, 300 MHz): δ = 2.34 (s, 3H, CH3), 4.22 (br.s, 2H, NH2 exchangeable), 7.19 (m, 2H, Ar–H), 7.46 (d, 2H, J = 8.1 Hz, Ar–H), 7.6 (m, 2H, Ar–H), 7.70 (br. s,1H, NH exchangeable), 7.74 (d, 2H, J = 8.1 Hz, Ar–H), 7.9 (s, 1H, pyrazole N=CH); 13C NMR (DMSO-d6,75 MHz): δ = 21.2 (CH3), 100.2 (C-4), 115.2–141.7 (Ar. C + C-3), 132.9 (C-5), 153.0 (C=N); Anal. calcd. for: C17H15N5O2S (353.40): C, 57.78; H, 4.28; N, 19.82. Found: C, 57.67; H, 4.39; N, 19.70%.
Formation of 5-amino-1-tosyl-1H-pyrazole-4-carbohydrazide (10)
A solution of the pyrazolocarboxylate 3 (3.09 g, 0.01 mol) and hydrazine hydrate (0.15 mol) in ethanol (50 ml) was stirred at room temperature for 2 h. The solid formed after filtration was recrystallized from ethanol to yield 10 as a white solid; 1.33 g (45.03%) m.p. 140–142 °C, IR (KBr) cm−1: ν (NH2) 3457, ν (NH) 3219, ν (C=O) 1630; 1H NMR (DMSO-d6, 300 MHz) ppm: δ = 2.34 (s,3H, CH3), 4.22 (br. s, 2H, NH2 exchangeable), 5.8 (br. s, 2H, NH2 exchangeable), 7.46 (d, 2H, J = 8.1 Hz, Ar–H), 7.87 (d, 2H, J = 8.1 Hz, Ar–H), 7.9 (s, 1H, pyrazole N=CH), 8.0 (br. s, 1H, NH exchangeable); 13C NMR (DMSO-d6,75 MHz): δ = 21.2 (CH3), 128–140 (Ar. C + C-3), 148.0 (C-5), 150.2 (C-4), 165.0 (C=O) ppm; Anal. Calcd. for C11H13N5O3S (295.32): C, 44.74; H, 4.44; N, 23.71; Found: C, 44.65; H, 4.36; N, 23.74%.
Reaction of carbohydrazide 10 with d-glucose
To a well stirred solution of the d-glucose (0.9 g, 5 mmol) in water (1 ml), and glacial acetic acid (1 ml); was added the carbohydrazide 10 (1.47 g, 5 mmol) in ethanol (15 ml). The mixture was heated under reflux for 3 h. and the resulting solution was concentrated and left to cool. The precipitate formed was filtered off, washed with water, then dried and recrystallized from ethanol to afford 5-amino-N′-(d-glucopyranosyl)-1-tosyl-1H-pyrazole-4-carbohydrazide (11) as a white powder, 1.37 g (59.9%) m.p. 143–144 °C; IR (KBr) cm−1: ν (OH) 3460–3449, ν (NH) 3265,ν 1695 (C=O), ν (C=N) 1616; 1H NMR (DMSO-d6, 300 MHz): δ = 2.34 (s, 3H, CH3), 3.30–3.35 (m, 2H, H-6′,6″), 3.70 (m, 1H, H-5′), 4.12 (m, 1H, H-4′), 4.27 (t, J = 7.4 Hz, 1H, H-3′), 4.22 (br. s, 2H, NH2 exchangeable), 4.34 (dd, J = 7.4 Hz, J = 7.8 Hz, 1H, H-2′), 4.44 (m, 1H, OH), 4.45 (d, J = 6.4 Hz, 1H, OH), 5.20 (m, 1H, OH), 5.60 (t, J = 4.6 Hz, 1H, OH), 5.79 (t, J = 4.6 Hz, 1H, OH), 7.0 (br. s, 1H, NH exchangeable), 7.47 (d, 2H, J = 8.1 Hz, Ph-H), 7.6 (s, 1H, N=CH), 7.87(d, 2H, J = 8.4 Hz, Ph-H), 7.89 (s, 1H, pyrazole N=CH); 13C NMR (DMSO-d6, 75 MHz): δ = 21.2 (CH3), 62.1 (C-6′), 63.0 (C-5′), 69.2 (C-4′), 74.3 (C-3′), 75.7 (C-2′), 128–142 (Ar. C + C-3), 148.0 (C-5), 150.5 (C-4), 151.5 (C-1′), 164.2 (C=O) ppm, Anal. calcd. for: C17H23N5O8S (457.46): C, 44.63; H, 5.07; N, 15.31. Found: C, 44.56; H, 5.15; N, 15.37%.
5-(5-Amino-1-tosyl-1H-pyrazol-4-yl)-1,3,4-oxadiazole-2(3H)-thione (12)
To a solution of 10 (3.37 g, 0.01 mol) in ethanol (20 ml) was added a potassium hydroxide solution (0.56 g, 0.01 mol) in water (2 ml) and carbon disulphide (4 ml). The mixture was heated under reflux for 10 h. The solvent was evaporated and the residue was dissolved in water, filtered, and the filtrate was acidified with dilute hydrochloric acid with continuous stirring. The precipitate that formed was filtered off, washed with water and recrystallized from ethanol to yield 2.7 g (80%) 12 as a yellow solid; m.p. 140–141 °C; IR (KBr) cm−1: ν (NH2) 3252, ν (NH) 3141, ν (C=S) 1670, ν (C=N) 1600; 1H NMR (DMSO-d6, 300 MHz): δ = 2.34 (s,3H, CH3), 4.20 (br. s, 2H, NH2 exchangeable), 7.1 (br. s, 1H, NH exchangeable); 7.46 (d, 2H, J = 8.1 Hz, Ar–H), 7.84 (d, 2H, J = 8.1 Hz, Ar–H), 7.90 (s, 1H, pyrazole N=CH),13C NMR (DMSO-d6, 75 MHz): δ = 21.2 (CH3), 98.2 (C-4), 128–142 (Ar. C + C-3), 148.0 (C-5), 156.5 (C=N imine), 190 (C=S) ppm; Anal. calcd. for: C12H11N5O3S2 (337.38): C, 42.72; H, 3.29; N, 20.76; Found: C, 42.65; H, 3.35; N, 20.80%.
4-Amino-3-(5-amino-1-tosyl-1H-pyrazol-4-yl)-1H-1,2,4-triazole-5(4H)-thione (13)
A solution of compound 12 (3.37 g, 001 mol) and hydrazine hydrate (0.15 mol) in ethanol (30 ml) was refluxed for 6 h. The reaction mixture was cooled at room temperature and the solvent was evaporated under reduced pressure. The solid that obtained was crystallized from ethanol to afford 13 as a white powder, 2.1 g (60%) m.p. 135–136 °C, IR (KBr) cm−1: ν (NH2) 3460, ν (NH) 3268, ν (C=S) 1696, ν (C=N) 1615; 1H NMR (DMSO-d6, 300 MHz): δ = 2.0 (br. s, 2H, NH2 exchangeable), 2.34 (s, 3H, CH3), 4.22 (br. s, 2H, NH2 exchangeable), 7.0 (br. s, 1H, NH exchangeable),7.46 (d, 2H, J = 8.1 Hz, Ar–H), 7.84 (d, 2H, J = 8.1 Hz, Ar–H), 7.90 (s, 1H, pyrazole N=CH); 13C NMR (75 MHz, DMSO): δ = 21.2 (CH3), 98.2 (C-4), 128–142 (Ar. C + C-3), 148.0 (C-5), 149.8 (C=N imine), 190.4 (C=S); Analysis Calcd. for C12H13N7O2S2 (351.41): C, 41.01, H, 3.73, N, 27.90; Found C, 41.10, H, 3.65, N, 27.80%.
Generals procedure for preparation of the shiff’s bases (15, 18)
A solution of 10 (2.95 g, 0.01 mol) and the appropriate aldehydes, (4-(dimethylamino)-benzaldehyde 14 or anthracene-9-carbaldehyde 17 (0.01 mol) in ethanol (30 ml) and a few drops of acetic acid was heated at reflux temperature for 6 h. The reaction mixture was cooled at room temperature and the solvent was evaporated under reduced pressure. The resulting solid was crystallized from ethanol to obtain the shiff’s bases 15 and/or 18.
5-Amino-N′-(4-(dimethylamino)benzylidene)-1-tosyl-1H-pyrazole-4-carbohydrazide (15)
This compound was obtained as a brown solid (benzene), 2.34 g (54.86%) m.p. 100–101 °C, IR (KBr) cm−1: ν (NH2) 3455, ν (NH) 3266, ν (C=O) 1666, ν (C=N) 1600; 1H NMR (DMSO-d6, 300 MHz) ppm: δ = 2.34 (s, 3H, CH3), 3.0 (s, 6H, 2CH3), 4.22 (br. s, 2H, NH2 exchangeable), 6.77 (d, 2H, J = 8.4 Hz, Ar–H), 7.0 (br. s, 1H, NH exchangeable), 7.50 (d, 2H, J = 8.2 Hz, Ar–H), 7.70 (d, 2H, J = 8.4 Hz, Ar–H), 7.86 (d, J = 8.2, 2H, Ar–H), 7.90 (s,1H, pyrazole N=CH), 9.67 (s,1H, N=CH); 13C NMR (DMSO-d6, 75 MHz): δ = 21.2 (CH3), 41.0 (2CH3), 128–142 (Ar. C + C-3), 148.0 (C-5), 149.3 (N=CH), 151 (C-4), 164.2 (C=O) ppm, Anal. Calcd. for C20H22N6O3S (426.49): C, 56.32, H, 5.20, N, 19.70; Found C, 56.36, H, 5. 30, N, 19.73%.
5-Amino-N′-(anthracen-9-ylmethylene)-1-tosyl-1H-pyrazole-4-carbohydrazide (18)
This compound was obtained as a brown solid (benzene), 2.56 g (52.98%) m.p. 97–98 °C, IR (KBr) cm−1: ν (NH2) 3458, ν (NH) 3265, ν (C=O) 1666, ν (C=N) 1610; 1H NMR (DMSO-d6, 300 MHz) ppm: δ = 2.38 (s, 3H, CH3), 4.22 (br. s, 2H, NH2 exchangeable), 7.0 (br. s, 1H, NH exchangeable), 7.46 (d, J = 8.4, 2H, Ar–H), 7.6 (m, 2H, Ar–H), 7.70 (m, 2H, Ar–H), 7.84 (d, J = 8.2, 2H, Ar–H), 7.90 (s, 1H, N=CH pyrazole), 8.20 (d, J = 8.4, 2H, Ar–H), 9.0 (d, J = 9.3, 2H, Ar–H), 9.05 (s, 1H, Ar–H), 11.48 (s, 1H, -N=CH); 13C NMR (DMSO-d6,75 MHz): δ = 21.2 (CH3), 125.5–142.8 (Ar. C + C-3), 149.3 (CH=N), 148.0 (C-5), 151.6 (C-4), 164.2 (C=O), Anal. Calcd. for C26H21N5O3S (483.14): C, 64.58, H, 4.38, N, 14.48; Found C, 64.47, H, 4.25, N, 14.55%.
General procedure for preparing compounds (16, 19)
A mixture of compounds 15 or 18 (0.01 mol) and thioglycolic acid (0.01 mol) in benzene (30 ml) was stirred at room temperature for 30 min, then refluxed for 10 h, cooled to room temperature. The solvent was removed under reduced pressure and the solid that obtained was recrystallized from the proper solvent to yield 16 and/or 19.
5-Amino-N-(2-(4-(dimethylamino)phenyl)-4-oxothiazolidin-3-yl)-1-tosyl-1H-pyrazole-4-carboxamide (16)
This compound was obtained as a yellow powder (ethyl acetate) 1.9 g (37.95%) m.p. 209–210 °C, IR (KBr) cm−1: ν (NH2) 3458, ν (NH) 3266, ν (C=O) 1690, ν (C=N) 1610; 1H NMR (DMSO-d6, 300 MHz) ppm: δ = 2.39 (s, 3H, CH3), 3.0 (s, 6H, 2CH3), 3.6 (s, 2H, CH2), 4.22 (br. s, 2H, NH2 exchangeable), 5.20 (s, 1H, thiazolidine CH), 6.90 (d, 2H, J = 8.4 Hz, Ar–H), 7.0 (br. s, 1H, NH exchangeable), 7.2 (d, 2H, J = 8.2 Hz, Ar–H), 7.47 (d, 2H, J = 8.4 Hz, Ar–H), 7.84 (d, J = 8.2, 2H, Ar–H), 7.9 (s,1H, pyrazole N=CH); 13C NMR (DMSO-d6,75 MHz): δ = 21.2 (CH3), 39.4 (CH2), 41.0 (2CH3), 64.2 (CH), 128.0–145.0 (Ar. C + C-3), 148.0 (C-5), 151.5 (C-4), 165.0 (C=O), 169 (C=O); Anal. Calcd. for C22H24N6O4S2 (500.59): C, 52.78, H, 4.83, N, 16.79; Found C, 52.69, H, 4.79, N, 16.85%.
5-Amino-N-(2-(4-(anthracen-9-yl)-4-oxothiazolidin-3-yl)-1-tosyl-1H-pyrazole-4-carboxamide (19)
This compound was obtained as a yellow powder (ethanol) 2.5 g (44.83%) m.p. 200–201 °C, IR (KBr) cm−1: ν (NH2) 3456, ν (NH) 3266, ν (C=O) 1670, (C=N) 1600; 1H NMR (DMSO-d6, 300 MHz) ppm: δ = 2.34 (s, 3H, CH3), 3.6 (s, 2H, CH2), 4.22 (br. s, 2H, NH2 exchangeable), 5.13 (s,1H, CH), 7.0 (br. s, 1H, NH exchangeable), 7.13 (d, J = 7.8, 2H, Ar–H), 7.33 (d, J = 7.8, 2H, Ar–H), 7.46 (d, 2H, J = 8.4 Hz, Ar–H), 7.9 (s,1H, pyrazole N=CH), 8.20 (m, 2H, Ar–H), 8.40 (d, J = 8.1, 2H, Ar–H), 8.60 (s, 1H, Ar–H), 8.74 (d, J = 8.2, 2H, Ar–H); 13C NMR (DMSO-d6,75 MHz): δ = 21.2 (CH3), 36.4 (CH2), 64.2 (CH), 125.0–142.0 (Ar. C + C-3), 148.0 (C-5), 151.0 (C-4), 164.2 (C=O), 168.0 (C=O); Anal. Calcd. for C28H23N5O4S2 (557.64): C, 60.31, H, 4.16, N, 12.56; Found C, 60.30, H, 4.10, N, 12.55.
Biology
Materials used in the study
EAC cell lines were supplied from National Cancer Institute, Cairo, Egypt. Colon cancer (HCT-29) and Breast cancer (MCF-7), cell lines were obtained from Centre of Mycology and Biotechnology, Al-Azhar University, DMSO, crystal violet and trypan blue dye were purchased from Sigma (St. Louis, MO). DMEM, RPMI-1640, Fatal bovine serum (FBS), HEPES buffer solution, l-glutamine, gentamycin and 0.25% trypsin-EDTA were purchased from (BioWhittaker@ Lonza, Belgium). Anticancer activity was performed at Centre of Mycology and Biotechnology, Al-Azhar University, Nasr city, Cairo, Egypt.
Evaluation of cytotoxic effects of the chemical compounds
Method of trypan blue exclusion cytotoxicity assay
The in vitro cytotoxicity of the newly synthesized pyrazole derivatives were subjected to a preliminary screening for their anti-tumor activity in breast mouse tumor (EAC) using trypan blue exclusion methodCitation39. Stock solution of each of the tested samples was prepared of 100 µg of the tested material dissolved in 100 µL DMSO and 900 µL saline. From the stock solution, various dilutions of the tested materials were prepared. EAC cells were obtained by needle aspiration of Ascite under aseptic conditions. 10% of the tumor cell suspension (2.5 × 106) was prepared in saline (0.1 ml Ascite + 0.9 ml saline). From the stock solution, five concentrations of the tested compound (200, 100, 50, 30 and 10) µg/ml were prepared in five sterile test tubes, then 100 µL of 10% Ascite suspension was added, the volume was then completed to 1 ml by adding saline solution. The test tubes were incubated at 37 °C for 1 h. The supernatant was discarded from each tube, and one drop of trypan blue solution was added to the pellet. The number of viable cells were counted for trypan blue dye exclusion in a hemocytometer and examined by light microscopy. The viability assay for each compound was performed in triplicate.
Cell viability assay for human cell lines using crystal violet
Crystal violet stain (1%) is composed of 5% (w/v) crystal violet and 50% methanol then made up to volume with DD water and filtered through Whatmann filter paper. The Colon cancer cells (HCT) and Breast cancer cells (MCF-7) were propagated in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fatal bovine serum, 1% glutamine, HEPES buffer and 50 µg/ml gentamycine. All cells were maintained at 37 °C in a humidified atmosphere with 5% CO2 and were subtracted two times a week.
X-ray crystallography
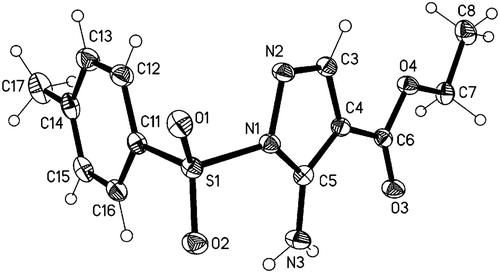
A single crystal of compound 3 was obtained by slow evaporation from a solution of ethanol. The crystal structure was solved and refined using data collection. The chemical structure and ring-labeling system is shown in . Crystallographic data (excluding structure factors) for the structure in this paper have been publishedCitation40.
Results and discussion
Chemistry
Reaction of p-toluenesulfonylhydrazide (1) with (E)-ethyl 2-cyano-3-ethoxyacrylate (2) in absolute ethanol under reflux afforded the pyrazole derivative 3.
The structure of compound 3 was evidenced from its spectral and analytical data. Its IR spectrum showed bands at 3483, 1693 and 1615 cm−1 corresponding to NH2, C=O and C=N groups respectively, its 1H NMR spectrum showed signals at δ 1.29 ppm as triplet (3H) and a quartet at δ 4.18 ppm (2H) indicating the presence of the ethyl ester group and also showed a singlet at δ 2.34 ppm (3H) for the tosylmethyl group, in addition to an exchangeable singlet for NH2 at 4.22 ppm. The CH of the pyrazole at position 3 was appeared as a singlet at δ 7.90 ppm and the phenyl ring protons were appeared as two doublets at 7.46 and 7.84 ppm with J coupling 8.1 Hz. 13C NMR of compound 3 supported its structure. The mass spectrum of 3 revealed the presence of the molecular ion peak at m/z 309 (M+, 8.37%). Moreover, the X-ray diffractionCitation40 of 3 () added a clear evidence for its proposed structure following the cyclization pathway of the intermediate outlined in Scheme 1.
Figure 1. ORTEP Diagram of compound 3.
Scheme 1. A proposed mechanism for the reaction of 4-methylbenzenesulfonylhydrazide (1) with (E)-ethyl 2-cyano-3-ethoxyacrylate (2) at refluxing temperature.
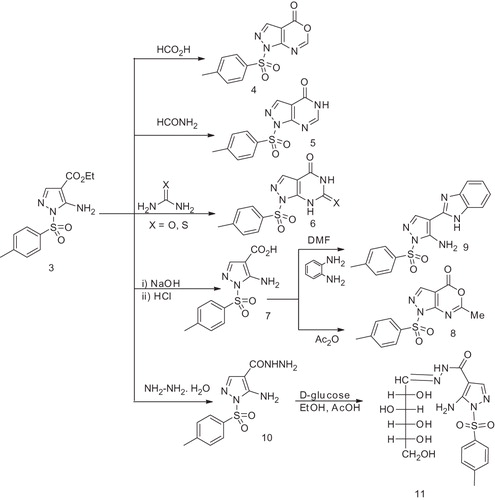
When compound 3 was refluxed with formic acid, the pyrazolo(3,4-d1,3)oxazinone derivative 4 was obtained. IR spectrum of 4 showed the band of C=O of the oxazinone ring at 1644 cm−1. Its 1H NMR spectrum displayed a singlet at 8.13 ppm of the CH of the oxazinone ring and 13C NMR spectrum of 4 showed a signal at δ 154.0 ppm of the C=O group. However, refluxing compound 3 with formamide afforded the pyrazolo(3,4-d)pyrimidin-4(5H)-one derivative 5. Its IR spectrum revealed bands at 3164, 1700 and 1590 cm−1 characteristic for NH, C=O and C=N groups respectively, while its 1H NMR spectrum showed an exchangeable singlet signal of the NH proton at δ 8.0 ppm in addition to a singlet at 9.0 ppm attributed to the CH of the pyrimidine ring. 13C NMR spectrum of 5 showed a signal of the C=O group at 157.6 ppm in addition to the other signals for carbons of the fused rings.
On the other hand, fusion of 3 with urea and/or thiourea at 150 °C yielded the pyrazolo(3,4-d)pyrimidine-4,6-dione derivative 6a and/or 6-thioxo-pyrazolo(3,4-d)pyrimidine-4(5H)-one derivative 6b respectively. The structures of compounds 6a, b were confirmed by studying their IR, 1H NMR, 13C NMR spectra and elemental analyses which are in accord with the assigned structures cf. experimental. While refluxing 3 with NaOH (10% solution) followed by acidification with HCl afforded the acid derivative 7, which upon heating with Ac2O gave the pyrazolo(3,4-d1,3)oxazin-4(1H)-one derivative 8. Furthermore, reaction of 7 with o-phenylenediamine in DMF at reflux temperature afforded the benzo(d)imidazolopyrazole derivative 9. The structures of compounds 7, 8 and 9 were confirmed by IR, 1H NMR, 13C NMR spectra and elemental analyses cf. experimental.
Reaction of 3 with hydrazine hydrate in absolute ethanol at reflux temperature yielded the acid hydrazide derivative 10. Its IR spectrum showed absorption bands of NH2, NH and C=O groups respectively at 3457, 3219 and 1630 cm−1. The 1H NMR spectrum of 10 showed a singlet at δ 8.0 ppm corresponding to the NH group and a singlet at δ 5.8 ppm of the –NH2 group of the hydrazide. The 13C NMR spectrum of 10 showed a signals of the C=O group at 165.0 ppm.
The hydrazide 10 is used as a key starting material for the preparation of novel interesting heterocyles and sugar hydrazide. Thus, when compound 10 was allowed to react with d-glucose in an aqueous solution of ethanol and a catalytic amount of glacial acetic acid, the corresponding hydrazone 11 was obtained. The IR spectrum of 11 showed the bands of the OH groups of the glucose at 3460–3449 cm−1. Its 1H NMR spectrum displayed the presence of the CH2, CH and OH groups of the sugar at their specific regions and so the 13C NMR spectrum showed the presence of the sugar hydrazone carbons as outlined in the experimental section (Scheme 2).
Scheme 2. Preparation pathways for compounds 4–11.
On the other hand, refluxing the acid hydrazide 10 with carbon disulfide in alcoholic KOH afforded 1,3,4-oxadiazole-2(3H)-thione derivative 12. Reaction of 12 with hydrazine hydrate in ethanol gave the pyrazolothione derivative 13 in a good yield. The IR spectra of 12 and 13 revealed bands attributable to NH2, NH and C=N groups, respectively. Further support for their assigned structures is gained from their 1H NMR that revealed the presence of signals of NH2, NH groups in 12 as well as one additional singlet signal for NH2 group at 2.0 ppm for compound 13. Their 13C NMR spectra correspond very well with their assigned structures. Heating of the hydrazide derivative 10 with 4-dimethylaminobenzaldehyde 14 and/or anthracene-9-carbaldehyde 17 in ethanol and in the presence of a catalytic amount of acetic acid, the Shiff’s bases 15, 18 were afforded in good yields, which upon reaction with thioglycolic acid in benzene at reflux temperature afforded the thiazolidinone derivatives 16, 19 in good yields (Scheme 3).
Scheme 3. Preparation pathways for compounds 12–19.
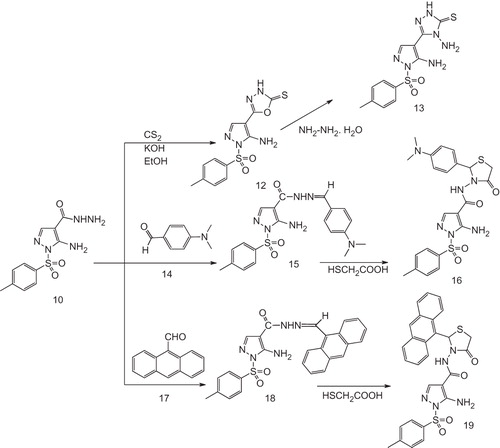
The structures of compounds 15–19 were evidenced from their IR spectra that revealed bands characteristic for NH2, NH and CO groups. The 1H NMR and 13C NMR spectra correspond very well with their assigned structures.
Anticancer activity
The anticancer activity of the newly synthesized compounds were assessed in mouse tumor model cancer cell line (EAC) and two human cancer cell lines, Colon cancer (HCT-29), and Breast cancer (MCF-7) cell lines, using trypan blue exclusion assay and crystal violet for cytotoxicity assay. These assays are based on the alteration in the membrane integrity as determined by the uptake of the dye by dead cells, thereby giving a direct indication on cell viability. Cytotoxicity was considered as anticancer activity. Treatment of cell lines by chemical materials caused inhibition of the growth of cells.
Cytotoxicity evaluation
The cells were seeded in 96-well plate at a cell concentration of 1 × 104 cells per well in 100 µL of growth medium. Fresh medium containing different concentrations of the tested samples was added after 24 h of seeding. Serial two-folds dilutions of the tested chemical compounds were added to confluent cell monolayer dispensed into 96-well, flat-bottomed microtiter plates were incubated at 37 °C in a humidified incubator with 5% CO2 for 48 h. Three wells were used for each concentration of the test sample. Control cells were incubated without test sample and/or without DMSO. (NB: The low DMSO percent used in the assay (maximal 0.1%) was found to be not effective). After incubation of cells for 24 h at 37 °C, various concentrations of samples (1.56, 3.125, 6.25, 12.5, 25, and 50) µg were added. Then the incubation continued for 48 h and the viable cells yield were determined by using trypan blue exclusion assay. All experiments were carried out in triplicate. The cytotoxicity of each compound was calculated, and the percentage of viable cells was plotted against the concentration. The preliminary investigation of the anticancer activity of the newly synthesized pyrazole derivatives was screened against EAC. The IC50 values of these compounds were determined compared to Doxorubicin.
Results and discussion for biology
The data expressed in ( and ) represents the cytotoxicity effect of the two compounds 6a and 18 in colon cancer cells (HCT), the data showed that compound 18 exhibit the most potent antitumor activity (IC50 = 10.24 µ/ml) as compared to compound 6a (IC50 = 29.58 µ/ml). The anticancer activity of the two compounds were also screened against breast cancer cell lines (MCF-7), in which both compounds exhibited anticancer activity with IC50 values (18 = 10.24 µ/ml and 6a = 24.34 µ/ml) respectively (, ).
Figure 2. In vitro cytotoxicity of the compounds 6a and 18 on human cancer cell line (HCT).
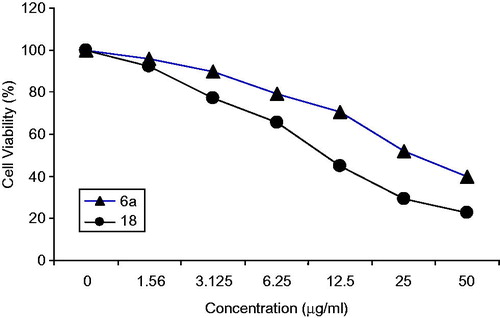
Figure 3. In vitro cytotoxicity of the compounds 6a and 18 on breast cancer cell line (MCF-7).
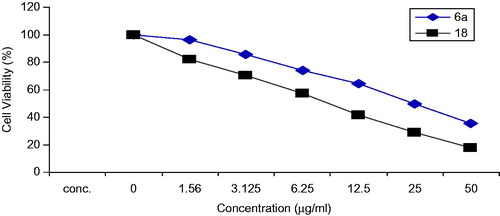
Table 1. Docking score, binding energy and IC50 of the new compounds against mouse tumor model (EAC), HCT (colon) and MCF-7 (breast cancer cell lines).
From the data we concluded that the higher anticancer activity of compound 18 is attributed to the presence of the tosyl, C=N, and C=O groups in addition to NH2 group that facilitates the H-Bonding with the active sites which increase its reactivity.
Molecular modeling
Ligand preparation
The structures of the newly synthesized compounds were drawn by using ChemDraw and converted to 3D structure with the help of 3D optimization tool. By using the LigPrep, the drawn ligand was geometry optimized by using the Optimized Potentials for Liquid Simulations-2005(OPLS-2005) force field with the Steepest Descent followed by truncated Newton Conjugate gradient protocol. Partial atomic charges were computed using the OPLS-2005force field. The LigPrep is a utility in Schrodinger software suite that combines tools for generating3D structures from 1D (Smiles) and 2D (SDF) representation, searching for tautomers and steric isomers and geometry minimization of ligand. Finally, different poses had been prepared with different tautomeric and steric features for docking studies.
Protein preparation
The X-ray crystal structure of Cyclin Dependent Kinase2 (CDK2) was obtained from the RCSB protein data bankCitation41 (pdb: 3pj8). After evaluating numbers of entries, the best protein was selected by analyzing the protein with Ramachandran plot and ProCheck using SAVS server based on ligand and number of disallowed regionsCitation42,Citation43. After selection, protein preparation wizard of Schrodinger suiteCitation44 has been used to prepare protein. The protein was preprocessed separately by deleting the substrate cofactor as well as the crystallographic ally observed water molecules (water without H bonds), correcting the mistakes in PDB file, optimizing hydrogen bonds. After assigning charge and protonation state finally energy minimization with root mean square deviation (RMSD) value of 0.30 Å was done using OPLS2005 force field.
Docking studies
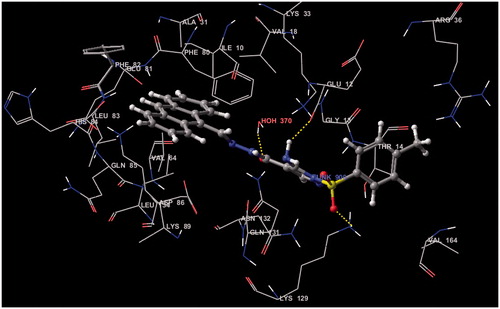
The docking studies on compounds prepared through LigPrep were carried out in the active site of the protein. Receptor Vander Waals scaling for the non-polar atoms was set to 0.9 which makes the protein site “roomier” by moving back the surface of non-Polar Regions of the protein and ligand. This kind of adjustments emulate to some extent the effect of breathing motion to the protein site, it is a kind of giving breathing to the receptor, this approach softens the active site region of the receptor making it flexibleCitation45. The prepared protein and the ligand were employed to build energy grids using the default value of protein atom scaling (1.0 Å) within a cubic box, centered on the centroid of the X-ray ligand pose. After Grid generation, the ligand was docked with the protein by using Glide 5.5 module in extra precision mode (XP) which uses MCSA (Monte Carlo Based Simulated Algorithm) based minimization. The best docked pose (with lowest Glide Score value) obtained from Glide was analyzedCitation46,Citation47. The binding energy was calculated by Prim module. All the data are shown in . represents the Electrostatic and hydrogen bond interactions of compound 18 with active site of CDK2 and the interaction with amino acids in the active pocket of CDK2.
Figure 4. Electrostatic and hydrogen bond interactions of compound 18 with active site of CDK2.
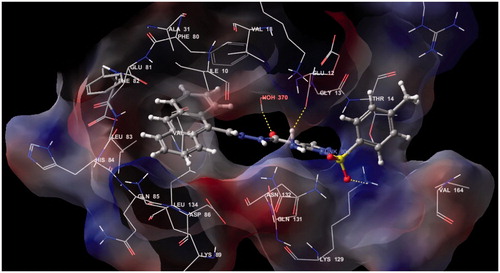
Figure 5. Schematic interaction of compound 18 with amino acids in the active pocket of CDK2.
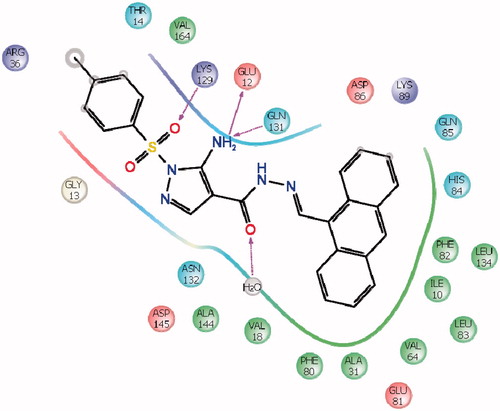
Figure 6. Hydrogen bond interactions of compound 18 with active site of CDK2.
Conclusion
Ethyl 5-amino-1-tosyl-1H-pyrazole-4-carboxylate (3) was synthesized. The structure of the compound 3 was supported by X-Ray crystallographic data. Reactions of this substrate with urea, thiourea, formamide and formic acid furnished the bicyclic pyrazolooxazine and pyrazolo-pyrimidine ring systems. The hydrazide derivative was used as a key compound for synthesizing 1,3,4-oxadiazole, 1,2,4-triazole, thiazole and glucoside derivatives. Among the compounds screened, compound 18 showed the most potent anticancer activity and compound 6a exhibited moderate anticancer activity towards (EAC) cancer cell line, while the rest of the other tested compounds exerted week anticancer activity. The two compounds 6a and 18 were subjected to further evaluation of their antitumor activity in the two human cell lines (HCT-29 and MCF-7) using Crystal violet cytotoxicity assay and showed good to high activities. The results from docking study also represent that compound 18 had the more Electrostatic and hydrogen bond interactions with active site of CDK2.
Acknowledgements
The authors thank Prof. Dr. Peter G. Jones Institut fur Anorganische und Analytische Chemie, Technische Universitat, Braunschweig, Postfach 3329, 38023 Braunschweig, Germany for solving the crystal structure for Ethyl 5-amino-1-(4-methylbenzenesulfonyl)- pyrazole-4-carboxylate (3).
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Wise LD, Butler DE, Dewald HA, et al. 1,3-Dialkyl-4-(iminoarylmethyl)-1H-pyrazol-5-ols. A series of novel potential antipsychotic agents. J Med Chem 1987;30:1807–12
- Gursoy SA, Demirayak S, Capan G, Vural K. Synthesis and preliminary evaluation of new 5-pyrazolinone derivatives as analgesic agents. Eur J Med Chem 2000;35:359–64
- Tsuji K, Nakamurana K, Konishi N, et al. Studies on anti-inflammatory agents. IV. Synthesis and pharmacological properties of 1,5-diarylpyrazoles and related derivatives. Chem Pharm Bull 1997;45:987–95
- Badawey AM, El-Ashmawey IM. Non-steroidal anti-inflammatory agents – Part 1: Anti-inflammatory, analgesic and antipyretic activity of some new 1-(pyrimidin-2-yl)-3-pyrazolin-5-ones and 2-(pyrimidin-2-yl)-1,2,4,5,6,7-hexahydro-3H-indazol-3-ones. Eur J Med Chem 1998;33:349–61
- Daidone G, Maggio B, Plescia S, et al. Antimicrobial and antineoplastic activities of new 4-diazopyrazole derivatives. Eur J Med Chem 1998;33:375–82
- (a) Hilgard P, Thornes RD. Anticoagulants in the treatment of cancer. Eur J Cancer 1976;12:755–62. (b) Taylor C, Patel H, Kumar H. Synthesis of pyrazolo 3,4-dpyrimidine analogues of the potent agent N-4-2-2-amino-4 3H-oxo-7H-pyrrolo 2,3-dpyrimidin-5-ylethylbenzoyl-l-glutamic acid (LY231514). Tetrahedron 1992;48:8089–100
- Thumar NJ, Patel MP. Synthesis, characterization, and antimicrobial evaluation of carbostyril derivatives of 1H-pyrazole. Saudi Pharm J 2011;19:75–83
- Nauduri D, Reddy GP. Antibacterials and antimycotics. Chem Pharm Bull 1998;46:1254–60
- Foks H, Pancechowska-Ksepko D, Kedzia A, et al. Synthesis and antibacterial activity of 1H-pyrazolo[3,4-b]pyrazine and pyridine derivatives. Farmaco 2005;60:513–17
- Dardari Z, Lemrani M, Sebban A, et al. Antileishmanial and antibacterial activity of a new pyrazole derivative designated 4-[2-(1-(ethylamino)-2-methyl-propyl)- phenyl]-3-(4-methyphenyl)-1-phenylpyrazole. Arch Pharm Chem Life Sci 2006;339:291–8
- Gilbert AM, Failli A, Shumsky J, et al. Pyrazolidine-3,5-diones and 5-hydroxy-1H-pyrazol-3(2H)-ones, inhibitors of UDP-N-acetylenolpyruvyl glucosamine reductasen. J Med Chem 2006;49:6027–36
- Jiang JB, Hesson DP, Dusak BA, et al. Synthesis and biological evaluation of 2-styrylquinazolin-4(3H)-ones, a new class of antimitotic anticancer agents which inhibit tubulin polymerization. J Med Chem 1990;33:1721–8
- Parlok JJ. Synthesis of pyrazolecarbonylaminopyridinecarboxamides as herbicides. J Heterocycl Chem 1998;35:1493–9
- Saad HA, Osman NA, Moustafa AH. Synthesis and analgesic activity of some new pyrazoles and triazoles bearing a 6,8-dibromo-2-methylquinazoline moiety. Molecules 2011;16:10187–201
- Han X, Xu-hong H, Xiao-mao Z, et al. Synthesis and herbicidal activity of 5-heterocycloxy-3-substituted-1-(3-trifluoromethyl)phenyl-1H-pyrazole. Chem Res Chinese Universities 2012;28:824–7
- Schallner O, Heinz KH, Karl K. Ger. Offen DE19615259, 1997. Chem Abstr 1997;127:346387
- Eicher T, Hauptmann S. The Chemistry of heterocycles: structure, reactivity, syntheses and applications. New York: Thieme-Verlag; 1995
- (a) Jolly C, Morimoto RI. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J Natl Cancer Inst 2000;92:1564–1572. (b) Maloney A, Workman P. HSP90 as a new therapeutic target for cancer therapy. Expert Opin Biol Ther 2002;2:3–24. (c) Workman P. Overview: translating Hsp90 biology into Hsp90 drugs. Curr Cancer Drug Targets 2003;3:297–300. (d) Drysdale MJ, Dymock BW, Barril-Alonso X, et al. Preparation of 3,4-diarylpyrazoles as inhibitors of heat shock protein 90 (HSP90) and their use in the therapy of cancer. PCT Int. Appl. WO 2003/055860; 2003
- Rathelot P, Azas N, El-Kashef H, et al. 1,3-Diphenylpyrazoles: synthesis and antiparasitic activities of azomethine derivatives. Eur J Med Chem 2002;37:671–9
- Bernardino AMR, Gomes AO, Charret KS, et al. Synthesis and leishmanicidal activities of 1-(4-X-phenyl)-N′-[(4-Y-phenyl)methylene]-1H-pyrazole-4-carbohydrazides. Eur J Med Chem 2006;41:80–7
- Katiyar SB, Srivastava K, Purib SK, Chauhana PMS. Synthesis of 2-[3,5-substituted pyrazol-1-yl]-4,6-trisubstituted triazine derivatives as antimalarial agents. Bioorg Med Chem Lett 2005;15:4957–60
- Moukha-Chafiq O, Taha ML, Lazrek HB, et al. Synthesis and biological activity of some 4-substituted 1-[1-(2,3-dihydroxy-1-propoxy)methyl-1,2,3-triazol-(4&5)-yl methyl]-1H-pyrazolo[3,4-d]pyrimidines. Farmaco 2002;57:27–32
- Allen SH, Johns BA, Gudmundsson KS, et al. Synthesis of C-6 substituted pyrazolo[1,5-a]pyridines with potent activity against herpesviruses. Bioorg Med Chem 2006;14:944–54
- Baraldi PG, Beria I, Cozzi P, et al. Cinnamoyl nitrogen mustard derivatives of pyrazole analogues of tallimustine modified at the amidino moiety: design, synthesis, molecular modeling and antitumor activity studies. Bioorg Med Chem 2004;12:3911–21
- Daidone G, Raffa D, Maggio B, et al. Synthesis and antiproliferative activity of triazenoindazoles and triazenopyrazoles: a comparative study. Eur J Med Chem 2004;39:219–24
- Gopalsamy A, Yang H, Ellingboe JW, et al. Pyrazolo[1,5-a]pyrimidin-7-yl phenyl amides as novel anti-proliferative agents: parallel synthesis for lead optimization of amide region. Bioorg Med Chem Lett 2005;15:1591–4
- Cocco MT, Congiu C, Lilliu V, Onnis V. Amidrazones as precursors of biologically active compounds-Synthesis of Diaminopyrazoles for Evaluation of Anticancer Activity. Arch Pharm Chem Life Sci 2006;339:7–13
- Tanitame A, Oyamada Y, Ofugi K, et al. Synthesis and antibacterial activity of a novel series of potent DNA gyrase inhibitors. Pyrazole Derivatives. J Med Chem 2004;47:3693–96
- Tsurumi K, Abe A, Fujimura H, et al. General pharmacological actions of l-(m-chlorophenyl)-3-N,N-dimethylcarbamoyl-5-methoxypyrazole (PZ-177). Folia Pharmacol Jpn 1976;72:41–52
- Bakavoli M, Bagherzadeh G, Vaseghifar M, et al. Molecular iodine promoted synthesis of new pyrazolo[3,4-d]pyrimidine derivatives as potential antibacterial agents. Eur J Med Chem 2010;45:647–50
- Capdeville R, Buchdunger E, Zimmermann J, Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat Rev Drug Disc 2002;1:493–502
- Taylor EC, Patel H, Kumar H. Synthesis of pyrazolo 3,4-dpyrimidine analogues of the potent agent N-4-2-2-amino-4 3 H-oxo-7 H-pyrrolo 2,3-dpyrimidin-5-yl ethylbenzoyl-l-glutamic acid (LY231514). Tetrahedron 1992;48:8089–100
- Al-Saadi MS, Rostom SAF, Faidallah HM. 4-{2-[(E)-Cyclo-pentyl-idene]hydrazin-1-yl}benzene-sulfonamide. Arch Pharm Chem Life Sci 2008;341:181–90
- Ghorab MM, Ismail ZH, Abdel-Gawad SM, Abdel Aziem A. Antimicrobial activity of amino acid, imidazole, and sulfonamide derivatives of pyrazolo[3,4-d]pyrimidine. Heteroatom Chem 2004;15:57–62
- Anderson JD, Cottam HB, Larson SB, et al. Synthesis of certain pyrazolo[3,4-d]pyrimidin-3-one nucleosides. J Heterocycl Chem 1990;27:439–53
- Rashad AE, Hegab MI, Abdel-Megeid R, et al. Synthesis and antiviral evaluation of some new pyrazole and fused pyrazolopyrimidine derivatives. Bioorg Med Chem 2008;16:7102–6
- Holla BS, Mahalinga M, Karthikeyan MS, et al. Synthesis of some novel pyrazolo[3,4-d]pyrimidine derivatives as potential antimicrobial agents. Bioorg Med Chem 2006;14:2040–7
- Nassar IF, Assaly SA-E. Synthesis, reactivity and antitumor activity of some new pyrazolo[3,4-d] pyrimidine and their triazole derivatives. Der Pharma Chemica 2011;3:229–38
- Ribeiro DA, Marques ME, Salvadori BDM. In vitro cytotoxic and non-genotoxic effects of gutta-percha solvents on mouse lymphoma cells by single cell gel (comet) assay. Braz Dent J 2006;17:228–32
- Elgazwy A-SSH, Nassar IF, Jones PG. Ethyl 5-amino-1-[(4-methylphenyl)sulfonyl]-1H-pyrazole-4-carboxylate. Acta Cryst 2013;E69:o1376
- An information portal to biological macromolecular structures. Available from: http://www.rcsb.org/pdb
- Laskowaski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst 1993;26:283–91
- Ramachandran GN, Ramakrishnan C, Sasisekharan V. Stereochemistry of polypeptide chain configurations. J Mol Biol 1963;7:95–9
- Schrodinger LLC. New York, NY: Schrodinger Suite; 2010
- Taverna DM, Goldstein RA. Why are proteins marginally stable? Proteins 2002;46:105–9
- Hamilton-Miller JMT. Antimicrobial properties of tea (Camellia sinensis L). Antimicrobial Agents Chemother 1995;39:2375–7
- Halgren TA, Murphy RB, Friesner RA, et al. Glide: a new approach for rapid, accurate docking and scoring 2. Enrichment factors in database screening. J Med Chem 2004;47:1750–9