
Abstract
We synthesized a series of new naphthalene derivatives: naproxen- and 6-methoxy naphthalene acetic acid-like 1–5. In these compounds the carboxylic function, typical of the classical NSAIDs, was replaced by a methylsulfonamido (1, 2 and 6a–c) or methylsulfonyl (3–5) group present in some selective COX-2 inhibitors. We also synthesized compounds 7 and 8 in which the naphthalene portion was substituted by tetrahydronaphthalene ring. Some of the new compounds were assayed for their enzymatic inhibitory activity towards cycloxygenase enzymes. Compounds 4 and 6b, at a concentration of 10 µM exhibit percentage inhibition values of 65%, 50% and 29%, 87% towards COX-2 and COX-1, respectively. The substitution of carboxylic group with a mehylsulfonamido or a methylsulfonyl groups does not allow to direct the selectivity versus to cycloxygenase enzymes.
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are competitive inhibitors of the cycloxygenase (COX) enzymes. These enzymes exist in two isoforms: COX-1, which is constitutive, and COX-2 which is inducible. COX-1 is expressed in the most of tissues and is involved in physiological production of PGs responsible of gastric cytoprotection, COX-2 in normal conditions has lower expression levels in tissues, but its expression is increased during inflammatory responses. The up-regulation of COX-2 was also observed in premalignant and malignant conditions of various cancers such as breast, prostate and lung ones which metastasize in the bonesCitation1. Particular attention has been recently focused on the role that COX-2 enzyme could play in Alzheimer's diseases (AD)Citation2,Citation3.
After the removal from the market of some selective COX-2 inhibitors (COXIBs), due to their cardiovascular side effects, the research has focused towards the evaluation of alternative chemical structures able to maintain the COX inhibitory activity with reduced side effectsCitation4.
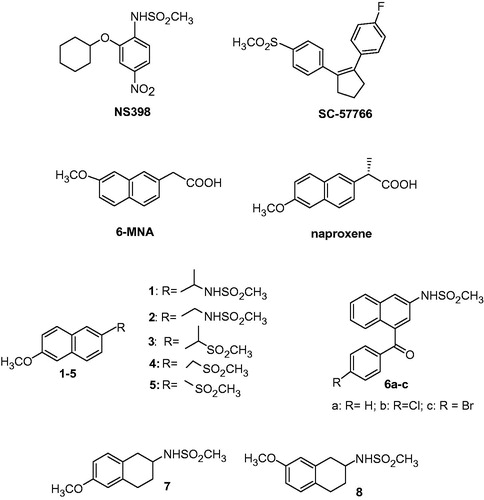
With this aim, we synthesized a series of new naphthalene derivatives naproxene and 6-methoxy naphthalene acetic acid-like (6-MNA; 1–6; ). In these compounds, the carboxylic function, typical of the classical NSAIDs, was replaced by a methylsulfonamido (1, 2, 6a–c) or methylsulfonyl (3–5) group present in some selective COX-2 inhibitors as for example NS398 or SC-57666, respectively. We also synthesized the methylsulfonamido compounds 7 and 8 in which the naphthalene portion was substituted by tetrahydronaphthalene ring.
Figure 1. General structure of NS398 and SC5766, 6-MNA, naproxene and new naphthalene 1–6 and tetrahydronaphthalene 7 and 8 compounds.
Methods
Chemistry
The synthesis of compounds 1 and 2 were carried out as depicted in Schemes 1 and 2.
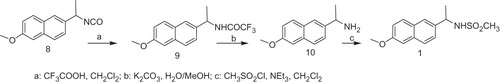
The isocyanate 8, obtained according to the synthetic method reported in literatureCitation5, treated with trifluoroacetic acid in methylene chloride gave the 2,2,2-trifluoro-N-[1-(6-methoxy-naphthalen-2-yl)-ethyl]-acetamide 9. Compound 9 by hydrolysis with potassium carbonate in aqueous methanol afforded the amine 10. By reaction of the derivative 10, with mesyl chloride and triethylamine, we obtained the desired compound 1.
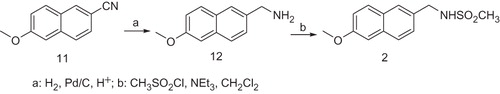
The N-(6-Methoxy-naphthalen-2-ylmethyl)-methanesulfonamide 2 was prepared as shown in Scheme 2. The catalytic hydrogenation with Pd/C in acid medium of the commercial available 6-methoxy-naphthalene-2-carbonitrile 11 afforded the amine 12, which by subsequent treatment with mesyl chloride and triethylamine furnished the compound 2.
The synthesis of the compounds 3 and 4 are shown in Schemes 3 and 4.
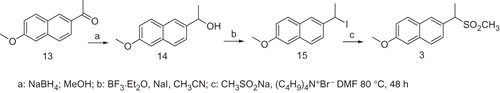
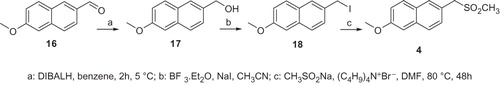
The reduction of the commercial available 1-(6-methoxynaphthalen-2-yl) ethanone 13 with NaBH4 afforded the alcoholCitation6 14 which was transformed in the corresponding iodine derivative 15. The treatment of compound 15 with the sodium salt of methane sulfinic acid in presence of tetrabutylammoniumbromide afforded the methylsulfone 3.
Commercially available 6-Methoxy-naphthalene-2-aldehyde 16 was reduced with diisobutylaluminum hydride to afford the corresponding alcohol 17. Compound 17 by treatment with NaI and BF3ċEt2O afforded the corresponding iodine derivative 18. The desidered methylsulfone 4 was obtained by treatment with the sodium salt of methane sulfinic acid of the iodine derivative 18.
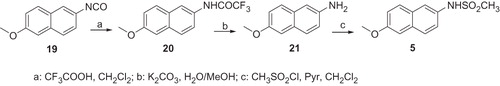
Compound 5 was obtained as reported in Scheme 5. The 6-Methoxynaphthylisocyanate 19Citation7, prepared from commercially available 6-methoxy-2-naphthoic acid by Curtius rearrangement of the corresponding acyl azide, in the presence of trifluoroacetic acid and methylene chloride, gave the 2,2,2-Trifluoro-N-(6-methoxy-naphthalen-2-yl)-acetamide 20, which by hydrolysis with potassium carbonate in aqueous methanol afforded the desidered amine 21. Compound 21 treated with mesyl chloride and pyridine, furnished the desired compound 5.
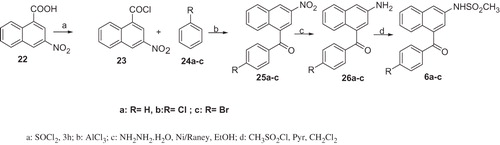
The general synthetic approach to compounds 6a–c is outlined in Scheme 6. Compound 22 obtained according to the synthetic method reported in literatureCitation8 was treated by thionyl chloride to obtain the corresponding acid chloride 23Citation8. Compound 23 treated with the appropriate benzenic derivatives 24a–c in AlCl3 afforded the 2-nitro-4-benzoyl-naphthalene derivatives 25a–c. Compounds 25a–c have been reduced by hydrazine hydrate and Ni/Raney to the corresponding amines 26a–c, and then treated with mesyl chloride and pyridine to give the desired derivatives 6a–c.
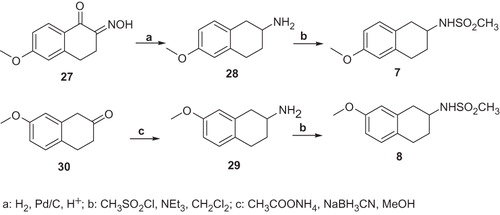
The synthesis of methylsulfonamides 7 and 8 were reported in Scheme 7. The catalytic hydrogenation of 6-methoxy-3,4-dihydro-(1,2)-naphtoquinone-2-oxime 27 with Pd/C in acid medium and the reduction of 7-methoxy-3,4-dihydro-1H-naphthalen-2-one 30 provided the amines 28, 29. The amines 28 and 29 treated with mesyl chloride and triethylamine furnished the desired compunds 7 and 8.
Biological results and conclusion
For the new compounds 1–4, 6a–b and 7 the inhibitory activity towards COX-1 and COX-2 was evaluated in vitro by measuring the PGE2 production on activated J774.2 macrophagesCitation9. The results are reported in together with those obtained in the same type of test with NS398 as reference drug.
Table 1. Biological data of compounds 1–4, 6a,b and 7.
Compounds 4 and 6b at a concentration of 10 µM exhibit percentage inhibition values of 65% and 50% toward COX-2 and 29% and 87% toward COX-1, while the other compounds resulted practically inactive. However, it can be seen how similar structures have completely different activity values as shown in for compounds naproxene-like 2 (0, 13%) and 4 (29%, 65%). These data seem to indicate that in the class of the naproxene analogues 1–4 only the methylsulfonyl group, in substitution of the carboxylic function, is suitable for the interaction towards COX-2.
Regarding the compounds 6a–b the introduction of a chlorine substituent on the benzoyl group (6b) confers a good inhibitory activity even if, no COX-1 and COX-2 selectivity, with respect to the unsubstituted analogue 6a completely devoid of any cycloxygenase inhibitory activity. In this class of compounds would seem confirmed the observation that had already been highlighted for other class of antiinflammatory drugs, namely that the presence of a halogen on the benzoyl group is able to have a positive influence on the activity.
Experimental procedures
Chemistry
Analytical grade reagents and solvent were purchased from Sigma-Aldrich (St. Louis, MO), and were used as supplied. Solvents were dried according to standard methods. All chemical reactions were monitored by thin layer chromatography (TLC) using alumina plates coated with silica gel 60 F254 (Merck, Darmstadt, Germany) containing a fluorescent indicator; spots were detected under UV light (254 nm). Column flash chromatography separations were performed on silica gel Merck 230–400 mesh ASTM Evaporations were made in vacuo (rotating evaporator); Na2SO4 was always used as the drying agent. Melting points were determined by a Kofler apparatus (A) and are uncorrected. IR spectra for comparison of compounds were taken as paraffin oil mulls or as liquid films on a Unicam Mattson 1000 FT-IR spectrometer (Cambridge, UK). 1H-NMR spectra were obtained with a Varian Gemini CTF 20 spectrometer (Mountain View, CA) operating at 80 MHz, at 25 °C in ca. 5% solution of CDCl3. Chemical shifts (δ) are reported in ppm, coupling constants J are reported in Hertz. The following abbreviations are used: singlet (s), doublet (d), triplet (t), broad singlet (bs) and multiplet (m). Elemental analyses were performed in our analytic laboratory and agree with the theoretical values to within ± 0.4%. Mass spectrometry data were collected by spectrophotometer Hewelett Packard 5988A (Palo Alto, CA) by direct introduction at a nominal electron energy of 70 eV and a source temperature at 350 °C.
2,2,2-Trifluoro-N-[1-(6-methoxynaphthalen-2-yl)-ethyl]-acetamide (9)
To a solution of 2-(1-Isocyanato-ethyl)-6-methoxynaphthalene 8 (384 mg, 1.69 mmol) in CH2Cl2 (15 ml) was added dropwise trifluoroacetic acid (0.175 ml, 2.28 mmol). After 10 h at reflux temperature, the mixture was cooled at room temperature and then washed with a solution of NaHCO3, dried and filtered to give 9. Yield 84%; m.p.: 175–180 °C; IR (ν, cm−1): 3335 (NH), 1700 (C=O); 1H-NMR δ: 7.82–7.11 (m, 6H, aromatic-H), 4.85–4.78 (m, 1H, CH), 3.9 (s, 3H, CH3O), 1.85 (bs, 1H, NH), 1.43 (d, 3H, J = 6.4 Hz, CH3); MS: 297 (M+, 30), 185 (100); Anal. Calcd. for C15H14 F3NO2 (297.27): C, 60.60; H, 4.75; N, 4.71%. Found: C, 61.00; H, 5.07; N, 4.32%.
1-(6-Methoxynaphthalen-2-yl)-ethylamine (10)
To a solution of 2,2,2-trifluoro-N-[1-(6-methoxynaphthalen-2-yl)-ethyl]-acetamide (9) (331 mg, 1.11 mmol) in MeOH (20 ml) was added a solution of K2CO3 (255 mg, 1.85 mmol) in H2O/MeOH (1:1, 5 ml). The resulting mixture was stirred at room temperature for 48 h, then the solvent was evaporated and the crude residue was dissolved in CH2Cl2, extracted with HCl 5%, alkalized, and finally extracted with AcOEt. The organic phase was then dried, filtered and evaporated, to give an oily residue, which was purified by transformation in the corresponding hydrochloride salt, and crystallised from Et2O/HCl. The hydrochloride salt of 10 was converted into a free base by treating an aqueous solution of the salt with solid KOH and extracting the free base with CHCl3. The organic layer was washed (H2O) filtered and evaporated to give 10 as a solid. 10: Yield 64%; m.p.: 77–78 °C; 1H-NMR δ: 7.72–7.06 (m, 6H, Ar), 4.3 (m, 1H, CH), 3.89 (s, 3H, CH3O), 2.5 (bs, 1H, NH), 1.54 (d, 3H, J = 6.4 Hz, CH3); MS: 201 (M+, 18), 186 (100); Anal. Calcd. for C13H15NO (201.26): C, 77.58; H, 7.51; N, 6.96%. Found: C, 77.13; H, 7.34; N, 6.98%.
(6-Methoxynaphthalen-2-yl)-methylamine (12)
A solution of 6-methoxynaphthalene-2-carbonitrile 11 (1 g, 5.46 mmol) in CHCl3 (10 ml), absolute EtOH (20 ml) and 37%HCl (1 ml) was hydrogenated for 24 h in presence of Pd/C (400 mg). The catalyst was removed by filtration. The evaporation of the ethanolic solution provided 12 as hydrochloride which was crystallized from MeOH/Et2O. 12: Yield 81%, m.p.: 236–240 °C; IR (ν, cm−1): 2654 (NH2); 1H-NMR (D2O) δ: 7.90–7.23 (m, 6H, Ar), 4.05 (m, 2H, CH2), 3.96 (s, 3H, CH3O); MS: 187 (M+, 100), 171 (38); Anal. Calcd. for C12H13NO√HCl (223.63) C, 64.43; H, 6.31; N, 6.26%. Found: C, 64.05; H, 5.96; N, 6.10%.
N-[1-(6-Methoxynaphthalen-2-yl)-ethyl]-methanesulfonamide (1) and N-(6Methoxy- naphthalen-2-yl)methyl)-methanesulfonamide (2)
To a solution of appropriate hydrochloride salts of 10, 12 (0.72 mmol) and NEt3 (0.2 ml, 1.44 mmol) in anhydrous CH2Cl2 (8 ml) at 0 °C, was added dropwise CH3SO2Cl (0.056 ml, 0.72 mmol). The mixture was stirred for 3 h at room temperature and after evaporated. The residue was dissolved in CH2Cl2, washed with 5% HCl and H2O. The organic phase was dried over anhydrous Na2SO4, and the solvent evaporated to afford the methansulfonamides 1 and 2 which were crystallized by CHCl3/hexane. 1: Yield 69%; m.p.: 154–155 °C; IR (ν, cm−1): 3227 (NH); 1H-NMR δ: 7.77–7.09 (m, 6H, Ar), 4.73–4.65 (m, 1H, CH), 3.90 (s, 3H, CH3SO2), 1.57 (s, 1H, NH), 1.54 (d, 3H, J = 6.4 Hz, CH3); MS: 281 (M+2, 1), 280 (M+1, 2), 279 (M+, 20), 185 (100); Anal. Calcd. for C14H17NO3S (279.35) C, 60.19; H, 6.13; N, 5.01%. Found: C, 59.90; H, 6.06; N, 4.99%. 2: yield 75%; m.p.: 174–175 °C; IR (ν, cm−1): 3246 (NH); 1H-NMR δ: 7.85–7.05 (m, 6H, Ar), 4.32–4.24 (d, 2H, J = 6.4 Hz, CH2), 3.88 (s, 3H, CH3O), 2.95 (s, 3H, CH3SO2); MS: 267 (M+2, 1), 266 (M+1, 3), 265 (M+, 28), 185 (100); Anal. Calcd. for C13H15NO3S (265.33) C, 58.85; H, 5.70; N, 5.28%. Found: C, 59.11; H, 5.93; N, 5.14%.
1-(6-Methoxynaphthalen-2-yl)-ethanol (14)
To a solution of 1-(6-methoxynaphthalen-2-yl)-ethanone 13 (500 mg, 2.5 mmol) in MeOH (20 ml) was added NaBH4 (277 mg, 7.5 mmol). The reaction mixture was stirred for 8 h, at r.t then evaporated at reduced pressure. The residue, dissolved in Et2O/CH2Cl2 (1:1), was washed with H2O, 10% HCl and with a saturated solution of NaCl. The organic phase was dried over anhydrous Na2SO4 and after evaporated. The residue obtained, crystallized from hexane afforded the pure compound 14. 14: Yield 55%; m.p.: 113–114 °CCitation6; IR (ν, cm−1): 3338 (OH); 1H-NMR δ: 7.74–7.07 (m, 6H, Ar), 4.97–5.04 (m, 1H, CH), 3.90 (s, 3H, CH3O), 1.87 (s, 1H, OH), 1.56 (d, 3H, J = 6.4 Hz, CH3); MS: 202 (M+, 54), 144 (100); Anal. Calcd. for C13H14O2 (202.10) C, 77.20; H, 6.98%. Found: C, 77.05; H, 6.83%.
(6-Methoxynaphthalen-2-yl)-methanol (17)
A solution of 1-(6-Methoxynaphthalen-2-yl)-ethanone 16 (800 mg, 4.3 mmol) in benzene anhydrous, cooled at 0–5 °C, was added slowly dropwise under N2 atmosphere DIBALH (6.45 mmol). The reaction mixture was stirred at 5 °C for 2 h, than was quenched with MeOH excess. Aluminum salts were removed by filtration and washed with warm MeOH; the filtrate was evaporated under reduced pressure. The crude product crystallized by CH2Cl2/hexane, gave pure 17. 17: Yield 81%; m.p.: 116–118 °C; IR (ν, cm−1): 3261 (OH); 1H-NMR δ: 7.74–7.04 (m, 6H, Ar), 4.78 (s, 2H, CH2), 3.89 (s, 3H, CH3O); MS: 188 (M+, 100), 115 (80); Anal. Calcd. for C12H12O2 (188.08) C, 76.57; H, 6.43%. Found: C, 76.19; H, 6.81%.
2-(1-Iodo-ethyl)-6-methoxynaphthalene (15) and 2-Iodomethyl-6-methoxynaphthalene (18)
To a solution of appropriate alcohol 14, 17 (3.19 mmol) and NaI (957 mg, 6.38 mmol) in anhydrous CH3CN (25 ml) was added dropwise, in 15 minutes, BF3 Et2O freshly distilled (9.57 mmol). The reaction mixture was stirred for 5 h (for compound 15) and for 45 minutes (for compound 18), then the organic phase was treated with brine (30 ml), a solution of 15% Na2S2O3 and finally extracted with Et2O. Ether phase washed with H2O and with saturated solution of NaCl, dried and evaporated afforded the iodine derivatives 15 and 18 which were crystallized by hexane and AcOEt/hexane, respectively. 15: Yield 46%; 1H-NMR δ: 7.71–7.01 (m, 6H, Ar), 4.73–4.65 (m, 1H, CH), 3.91 (s, 3H, CH3O), 1.63 (d, 3H, J = 6.4 Hz, CH3); MS: 186 (M+-I); Anal. Calcd. for C13H13IO (312.15) C, 52.02; H, 4.20%. Found: C, 52.34; H, 4.36%. 18: Yield 32%; 1H-NMR δ: 7.69–7.05 (m, 6H, Ar), 4.60 (s, 2H, CH2), 3.89 (s, 3H, CH3O); MS: 171 (M+-I, 100), 128(80); Anal. Calcd. for C12H11IO (298.12) C, 48.35; H, 3.72%. Found: C, 48.60; H, 4.01%.
2-(1-Methanesulfonyl-ethyl)-6-methoxynaphthalene (3) and 2-Methanesulfonylmethyl-6-methoxynaphthalene (4)
A solution of the appropriate iodine derivatives 15, 18 (1.04 mmol), CH3SO2Na (111 mg, 1.09 mmol), tetrabutylammoniumbromide (351 mg, 1.09 mmol) in anhydrous DMF (8 ml) was stirred at 80 °C for 48 h. After, the reaction mixture was treated with H2O/ice and extracted with CHCl3. The organic phase, washed with H2O, dried, evaporated under reduced pressure, afforded the derivatives 3 and 4 which were crystallized by CHCl3/hexane. 3: Yield 42%; 1H-NMR δ: 7.71–7.01 (m, 6H, Ar), 4.85–4.75 (m, 1H, CH), 3.9 (s, 3H, CH3O), 2.74 (s, 3H, CH3SO2), 1.43 (d, 3H, J = 6.4 Hz, CH3); MS: 265 (M+1, 0.67), 264 (M+, 3), 185 (100); Anal. Calcd. for C14H16O3S (264.34) C, 63.61; H, 6.10%. Found: C, 63.26; H, 6.45%. 4: Yield 60%; m.p.: 148 °C, 1H-NMR δ: 7.76-7.10 (m, 6H, Ar), 4.35 (s, 2H, CH2), 3.91 (s, 3H, CH3O), 2.74 (s, 3H, CH3SO2); MS: 251 (M+1, 1), 250 (M+, 2), 171 (100); Anal. Calcd. for C13H14O3S (250.31) C, 62.38; H, 5.64%. Found: C, 61.99; H, 5.82%.
2,2,2-Trifluoro-N-(6-methoxynaphthalen-2-yl)-acetamide (20)
To a solution of 2-Isocyanato-6-methoxynaphthalene 19 (337 mg, 1.69 mmol) in CH2Cl2 (15 ml) was added dropwise trifluoroacetic acid (0.175 ml, 2.28 mmol). The reaction mixture was refluxed at 40 °C under stirring for 48 h. The organic solution cooled, washed with NaHCO3, dried and evaporated under reduced pressure gave pure 20. 20: Yield 70%; m.p.: 132–140 °C, IR (ν, cm−1): 3275 (NH), 1703 (C=O); 1H-NMR δ: 7.96–7.45 (m, 6H, Ar), 3.90 (s, 3H, CH3O); MS: 269 (M+, 100); Anal. Calcd. for C13H10F3NO2 (269.22) C, 58.00; H, 3.74; N, 5.20%. Found: C, 58.33; H, 4.03; N, 4.85%.
6-Methoxynaphthalen-2-ylamine (21)
To a solution of 2,2,2-Trifluoro-N-(6-methoxynaphthalen-2-yl)-acetamide 20 (300 mg, 1.85 mmol) in MeOH (20 ml) was added a solution of K2CO3 (225 mg, 1.85 mmol) in H2O/MeOH (1:1, 5 ml). The mixture was stirred at 40 °C for 48 h, then evaporated under reduced pressure. The residue was dissolved in CH2Cl2 extracted with HCl 5%, alkalized, and finally extracted with AcOEt. The organic phase, dried and evaporated under reduced pressure, gave 21 which in presence of Et2O/HCl was transformed into the corresponding hydrochloride. The hydrochloride salt of 20 was converted into a free base by treating an aqueous solution of the salt with solid KOH and extracting the free base with CHCl3 the organic layer was washed (H2O) filtered and evaporated to give 21 as a solid. 21:Yield 80%; m.p.: 241–245 °C, IR (ν, cm−1): 3384 (NH2), 1703 (C=O); 1H-NMR δ: 7.81–7.33 (m, 6H, Ar), 3.90 (s, 3H, CH3O), 2.45 (bs, 1H, NH2); MS: 173 (M+, 72), 158 (82), 130 (100); Anal. Calcd. for C11H11NO (173.31) C, 76.28; H, 6.40; N, 8.09%. Found: C, 75.90; H, 6.70; N, 8.10%.
N-(6-Methoxynaphthalen-2-yl)-methanesulfonamide (5)
To a solution of 6-methoxynaphthalen-2-ylamine 21 (123 mg, 0.71 mmol), pyridine (0.114 ml, 1.42 mmol) in anhydrous CH2Cl2 (8 ml), cooled at 0 °C, CH3SO2Cl (0.055 ml, 0.71 mmol) was added dropwise. The mixture was stirred for 2 h at room temperature, evaporated under reduced pressure. The residue obtained was dissolved in CHCl3, washed with H2O and 5% HCl. Organic phase, dried and evaporated under reduced pressure, afforded 5 as a crude oil that was purified by flash chromatography on silica gel (EtOAc/n-hexane 1:1). 5: Yield 48%; m.p.: 177–178 °C, IR (ν, cm−1): 3324 (NH); 1H-NMR δ: 7.75–7.08 (m, 6H, Ar), 6.7 (s, 1H, NH), 3.9 (s, 3H, CH3O), 2.77 (s, 3H, CH3SO2); MS: 253 (M+2,1), 252 (M+1, 2), 251 (M+, 18), 172 (100); Anal. Calcd. for C12H13NO3S (251.30) C, 57.35; H, 5.21; N, 5.57%. Found: C, 57.00; H, 5.43; N, 5.28%.
3-Nitro-naphthalene-1-carbonyl chloride (23)
A solution of 3-Nitro-naphthalene-1-carboxylic acid 22Citation8 (3 g, 0.014 mol) in SOCl2 (18 ml) was refluxed for 3 h. The thionyl chloride was evaporated under reduced pressure, and the solid obtained, triturated with hexane, gave 23 that was purified by crystallization from CCl4. 23Citation8: Yield 76%; m.p.: 139–140 °C, IR (ν, cm−1): 1751 (C=O); 1H-NMR δ: 8.88–7.69 (m, 6H, Ar); MS: 235 (M+, 0.65), 126 (100).
General procedure for the synthesis of 2 nitro-4-benzoyl-naphthalenes (25a–c)
To a suspension of 3-nitro-naphthalene-1-carbonyl chloride 23 (1 g, 4.25 mmol) with appropriate benzene derivative 24a–c was added, at 0 °C, in 15 minutes, AlCl3 (851 mg, 6.38 mmol). The reaction mixture was stirred at room temperature for 15 h, then triturated in ice, acidified with 37% HCl to pH = 2 and extracted with Et2O. The ether phase, washed with 1 N NaOH, H2O, dried and evaporated under reduced pressure, gave the respective 2 nitro-4-benzoyl-naphthalenes 25a–c, which was purified by crystallization from CH3COCH3 for compounds 25a and 25b and isopropanol for compound 25c. 25a: Yield 46%; m.p.: 140-143 °C, IR (ν, cm−1): 1665 (C=O); 1H-NMR δ: 9.10–7.46 (m, 11H, Ar); MS: 277 (M+, 20), 105 (100), 77 (70); Anal. Calcd. for C17H11NO3 (277.27) C, 73.64; H, 4.00; N, 5.05%. Found: C, 73.39; H, 3.81; N, 5.05%. 25b: Yield 54%; m.p.: 146-148 °C, IR (ν, cm−1): 1672 (C=O); 1H-NMR δ: 8.89–7.37 (m, 10H, Ar); MS: 311 (M+, 20), 139 (100); Anal. Calcd. for C17H10ClNO3 (311.72) C, 65.50; H, 3.23; N, 4.49%. Found: C, 65.55; H, 3.61; N, 4.75%. 25c: Yield 30%; m.p.: 155–156 °C, IR (ν, cm−1): 1672 (C=O); 1H-NMR δ: 8.90–7.22 (m, 10H, Ar); MS: 356 (M+, 34), 185 (98), 126 (100); Anal. Calcd. for C17H10BrNO3 (356.17) C, 57.33; H, 2.83; N, 3.93%. Found: C, 57.65; H, 2.73; N, 3.66%.
General procedure for the synthesis of 4-benzoyl-2-naphthylamines (26a–c)
To a solution of the opportune 2 nitro-4-benzoyl-naphthalenes 25a–c (1.80 mmol) in absolute EtOH (15 ml) was added a catalytic portion of Ni-Raney. The suspension was refluxed and NH2NH2 H2O (0.393 ml, 8.10 mmol) in absolute EtOH (2 ml) was added dropwise. The mixture was refluxed for 1 h compound 26a, 12 h compound 26b, 8–10 h compound 26c. The suspension was filtered through celite and evaporated under reduced pressure. The residue obtained was dissolved in H2O, washed with Et2O and treated with saturated solution of Et2O/HCl to obtained the opportune amines 26a–c as hydrochloride, which were purified by crystallization from MeOH/Et2O. 26a: Yield 57%; m.p.: 117 °C, IR (ν, cm−1): 2613 (NH2), 1665 (C=O); 1H-NMR δ: 7.69–7.13 (m, 11H, Ar); MS: 247 (M+-HCl, 92), 115 (100), 105 (58), 77 (78); Anal. Calcd. for C17H13NO√HCl (283.75) C, 71.96; H, 4.97; N, 4.94%. Found: C, 71.80; H, 5.25; N, 4.57%. 26b: Yield 45%; m.p.: 144–149 °C, IR (ν, cm−1): 2620 (NH2), 1672 (C=O); 1H-NMR δ: 7.95–7.15 (m, 10H, Ar); MS: 281 (M+-HCl, 60), 115 (100); Anal. Calcd. for C17H12ClNO√HCl (318.20) C, 64.17; H, 4.12; N, 4.40%. Found: C, 54.55; H, 4.46; N, 4.10%. 26c: Yield 40%; m.p.: 119-125 °C, IR (ν, cm−1): 2590 (NH2), 1675 (C=O); 1H-NMR δ: 8.10–7.35 (m, 10H, Ar); MS: 325 (M+-HCl, 38), 115 (100); Anal. Calcd. for C17H12BrNO√HCl (362.65) C, 56.30; H, 3.61; N, 3.86%. Found: C, 56.58; H, 3.75; N, 3.51%.
N-(4-benzoyl-2-naphthyl)-methanesulfonamides (6a–c)
The sulfonamides 6a–c were synthesized from 4-benzoyl-2-naphthylamines 26a–c as previously reported for compound 5 and were purified by crystallization from Et2O/hexane, CHCl3/Et2O and AcOEt/hexane respectively. 6a: Yield 51%; m.p.: 48 °C, IR (ν, cm−1): 3270 (NH), 1657 (C=O); 1H-NMR δ: 7.86–7.34 (m, 11H, Ar), 7.11 (s, 1H, NH), 3.04 (s, 3H, CH3SO2); MS: 327 (M+2, 8), 326 (M+1, 15), 325 (M+, 50), 77 (100); Anal. Calcd. for C18H15NO3S (325.38) C, 66.44; H, 4.65; N, 4.30%. Found: C, 66.54; H, 4.87; N, 4.21%. 6b: Yield 40%; m.p.: 135–136 °C, IR (ν, cm−1): 3269 (NH), 1657 (C=O); 1H-NMR δ ppm 7.81–7.23 (m, 10H, Ar), 7.11 (s, 1H, NH), 3.06 (s, 3H, CH3SO2); MS: 361 (M+2, 22), 360 (M+1, 10), 359 (M+, 58), 111 (100), 75 (65); Anal. Calcd. for C18H14ClNO3S (359.04) C, 60.08; H, 3.92; N, 3.89%. Found: C, 59.78; H, 3.49; N, 3.47%. 6c: Yield 35%; m.p.: 105–115 °C, IR (ν, cm−1): 3278 (NH), 1659 (C=O); 1H-NMR δ: 7.84–7.22 (m, 10H, Ar), 3.03 (s, 3H, CH3SO2); MS: 406 (M+2, 5), 405 (M+1, 25), 404 (M+, 40), 183 (100); Anal. Calcd. for C18H14BrNO3S (404.28) C, 53.48; H, 3.49; N, 3.46%. Found: C, 53.37; H, 3.36; N, 3.22%.
6-Methoxy-1,2,3,4-tetrahydro-naphthalen-2-ylamine (28)
A solution of 6-Methoxy-3,4-dihydro-[1,2]-naphthoquinone-2-oxime 27 (1 g, 4.9 mmol) in MeOH (50 ml) and HCl 10% (9 ml) was hydrogenated at 50 °C at 1 atm in the presence of Pd/C (500 mg). After 20 h the catalyst was reactivated and the hydrogenation continued for another 20 h. The reaction mixture was filtrated and evaporated under reduced pressure. The crude residue crystallized from EtOH furnished the desired hydrochloride derivative 28. The hydrochloride salt of 28 was converted into the free base by treating an aqueous solution of the salt with solid KOH, then extracting the free bases with CHCl3. The organic layer washed (H2O) filtered and evaporated gave 28 as a solid. 28: Yield 60%; m.p.: 230–240 °C, IR (ν, cm−1): 3146, 2621 (NH2); 1H-NMR δ: 7.22–6.59 (m, 3H, Ar), 3.75 (s, 3H, CH3O), 2.91–2.76 (m, 3H, aliphatic), 1.56–1.26 (m, 4H, aliphatic); MS: 177 (M+, 0.89), 174 (82); Anal. Calcd. for C11H15NO (177.24) C, 74.54; H, 8.53; N, 7.90%. Found: C, 74.25; H, 8.86; N, 7.61%.
7-Methoxy-1,2,3,4-tetrahydro-naphthalen-2-ylamine (29)
To a solution of 7-Methoxy-3,4-dihydro-1H-naphthalen-2-one 30 (500 mg, 2.84 mmol) in anhydrous MeOH (15 ml) was added CH3COONH4 (2.19 g, 28.4 mmol) and NaBH3CN (125 mg, 199 mmol). The mixture was stirred for 48 h, then acidified with HCl (37%) to pH < 2, evaporated under reduced pressure. The residue was dissolved in H2O, washed with Et2O, alkalized and extracted with Et2O. The ether phase, dried and evaporated under reduce pressure, gave 7-Methoxy-1,2,3,4-tetrahydro-naphthalen-2-ylamine 29 which, in the presence of Et2O/HCl was transformed into the corresponding hydrochloride. The hydrochloride salt of 29 was converted into the free base by treating an aqueous solution of the salt with solid KOH then extracting the free base with CHCl3. The organic layer was washed (H2O) filtered and evaporated to give 29 as a solid. 29: Yield 52%; m.p.: 220-225 °C, IR (ν, cm−1): 3490, 2930 (NH2); 1H-NMR (δ ppm): 7.22–6.58 (m, 3H, Ar), 3.75 (s, 3H, CH3O), 2.91–2.76 (m, 3H, aliphatic), 1.57–1.25 (m, 4H, aliphatic); MS: 177 (M+, 0.79), 174 (82); Anal. Calcd. for C11H15NO (177.24) C, 74.54; H, 8.53; N, 7.90%. Found: C, 74.98; H, 8.90; N, 7.55%.
N-(6-Methoxy-1,2,3,4-tetrahydro-naphthalen-2-yl)-methanesulfonamide (7) N-(7-Methoxy-1,2,3,4-tetrahydro-naphthalen-2-yl)-methanesulfonamide (8)
Sulfonamides compounds 7 and 8 were synthesized from compounds 28 and 29 as previously reported for 1 and 2. Compounds 7, 8 were purified by crystallization from CH2Cl2/hexane and CH3COCH3/hexane, respectively. 7: Yield 20%; m.p.: 132–134 °C, IR (ν, cm−1): 3268 (NH); 1H-NMR δ: 6.36–6.71 (m, 3H, Ar), 4.43 (bs, 1H, NH), 3.71 (s, 3H, CH3O), 2.97 (s, 3H, CH3SO2), 3.04–2.55 (m, 4H, aliphatic), 2.56–2.12 (m, 3H, aliphatic); MS: 257 (M+2, 0.87), 256 (M+1, 1.76), 255 (M+, 12), 160 (100); Anal. Calcd. C12H17NO3S (255.33) C, 56.45; H, 6.71; N, 5.49%. Found: C, 56.76; H, 6.78; N, 5.21%. 8: Yield 51%; m.p.: 101–108 °C, IR (ν, cm−1): 3261 (NH); 1H-NMR δ: 6.91–6.56 (m, 3H, Ar), 4.5 (bs, 1H, NH), 3.74 (s, 3H, CH3O), 2.98 (s, 3H, CH3SO2), 3.06–2.75 (m, 4H, aliphatic), 2.02 (m, 3H, aliphatic); MS: 257 (M+2, 1), 256 (M+1, 2), 255 (M+, 10), 160 (100); Anal. Calcd. C12H17NO3S (255.33) C, 56.45; H, 6.71; N, 5.49%. Found: C, 56.13; H, 6.92; N, 5.28%
Enzyme assays
Compounds 1–4, 6a,b and 7 were tested following the procedure previously describedCitation9 in intact cell assays to verify their capacity to inhibit PGE2 production, considered as an index of activity on COX-1 and COX-2.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Suraneni MV, Reddy GV, Pallu R. NSAIDs, COXIBs, CLOXIBs and cancer pain. Horizons Cancer Res 2012;47:253–70
- Townsed KP, Patricò D. Novel therapeutic opportunities for Alheimer’s disease: focus on nonsteroidal anti-inflammatory drugs. FASEB J 2005;19:1592–601
- Nivsarkar M, Banerjee A, Padh H. Cyclooxygenase inhibitor: a novel direction for Alzheimer’s management. Pharmacol Rep 2008;60:692–8
- Narsinghani T, Sharma R. Lead optimization on conventional non-steroidal anti-inflammatory drugs: an approach to reduce gastrointestinal toxicity. Chem Biol Drug Des 2014. [Epub ahead of print]. doi: 10.1111/cbdd.12292:1-23
- Spahn H, Langguth P. chiral amines derived from 2-arylpropionic acids: novel reagents for the liquid chromatography (LC) fluorescence assay of optically active carboxylic xenobiotics. Pharm Res 1990;7:1262–8
- Matsunaga N, Kaku T, Akio O, et al. C17, 20-lyase inhibitors. Part 2: Design, synthesis and structure – activity relationships of (2-naphthylmethyl)-1H-imidazoles as novel C17,20-lyase inhibitors. Bioorg Med Chem 2004;12:4313–36
- MacMillan JB, Xiong-Zhou G, Skepper CK, Molinski TF. Phorbasides A-E, cytotoxic chlorocyclopropane macrolide glycosides from the marine sponge Phorbas sp. CD determination of c-methyl sugar configurations. J Org Chem 2008;73:3699–706
- Lee J.-J, Noll BC, Smith BD. Fluorescent chemosensor for chloroalkanes. Org Lett 2008;10:1735–8
- Balsamo A, Coletta I, Domiano P, et al. Synthesis and COX-2 inhibitiory properties on N-Phenyl-and N-benzyl-substituted amides of 2-(4-methylsulfonylphenyl)cyclopent-1-ene-1-carboxylic acid and their pyrazole, thiophene and isoxazole analogs. Eur J Med Chem 2002;37:391–8