
Abstract
New benzenesulfonamides incorporating water solubilizing moieties were synthesized using N-α-acetyl-l-lysine or γ-aminobutyric acid as scaffolds followed by the conversion of their terminal amino group to the guanidine one. Their inhibition activity was assessed by determining their KIs values against the human (h) carbonic anhydrase (CA, EC 4.2.1.1) isoforms hCA I, II, IX and XII. Some of these compounds were medium potency inhibitors of the cytosolic (CA I, II) and transmembrane (CA IX) isoforms and highly effective, nanomolar inhibitors of the second transmembrane isoform hCA XII. Some of these sulfonamides possessing good selectivity inhibition for the tumor-associated CA XII isoform over the cytosolic and physiologically dominant isoforms CA I and II may be used as tools to develop new anticancer agents.
Introduction
Carbonic anhydrases (CAs, EC 4.2.1.1) are metalloenzymes that catalyze a simple and physiologically relevant reaction in all life kingdoms: the carbon dioxide hydration to bicarbonate and protonsCitation1–7. In humans, 15 CA isoforms belonging to the α-CA family are known to be involved in many physiologic processes. Due to the fact that different isoforms are involved in diverse physiologic or pathologic processesCitation1,Citation2, this class of enzyme constitutes drug targets for diuretics, antiglaucoma agents, antiepileptics, antiobesity and antitumor drugsCitation1–7.
It has been found that isoforms CA IX and XII are transmembrane proteins characterized by an N-terminal extracellular domain, a putative transmembrane α-helix and an intramolecular C-terminal domainCitation1,Citation8. Both of them are overexpressed in a large number of human cancers, such as glioblastomaCitation9, colorectalCitation10 and breast cancerCitation11 as markers of hypoxia and thus used in clinical settingsCitation1,Citation2,Citation8.
Although it has been demonstrated that the overexpression of CA IX can be initiated by the activation of the hypoxia-inducible factor-1 (HIF-1) cascadeCitation6, the molecular mechanisms at the basis of the induction of CA XII are not so well understood yetCitation4. However, CA XII is known to be involved in breast cancer and recently it has been shown to be regulated by the estrogen receptor α (ERα), a hormone-regulated transcription factor belonging to the superfamily of nuclear receptors, which is expressed in about 70% of breast cancersCitation12. CA XII has also been identified in brain tumors, such as astrocytoma and glioblastomaCitation13–16.
The main consequence of an overexpression of hypoxia-induced CA IX and CA XII isoforms is a pH imbalance, with most of hypoxic tumors having lower pH value (about 6–6.5) compared with that characteristic of normal tissue equal to 7.4Citation17–19.
Therefore, hypoxia-induced CA IX and CA XII overexpression can be considered as the major tumor pH-regulating enzymes, and their inhibition may represent a promising target for antitumor therapyCitation1,Citation8,Citation13–16.
Although all α-CAs, including CA IX and XII, are inhibited by several main classes of inhibitors, such as the inorganic anionsCitation20, the phenolsCitation21 and the coumarinsCitation22 the sulfonamides and their bioisosteres (sulfamates, sulfamides, etc.) are still the most investigated CAIsCitation18,Citation19,Citation23. Many low nanomolar sulfonamide CA IXCitation17,Citation18 or CA XIICitation24 inhibitors have been identified in the last few years. It has been recently shown that in vitro, in cell cultures and in animals with transplanted tumors, CA IX inhibition with sulfonamides led to a return to more normal extracellular pH values, with consequential delay of tumor growthCitation5,Citation6. Furthermore, in mice suffering from T lymphoma, it was observed that the inhibition of the overexpressed CA XII by sulfonamides decreased cell proliferation and induced apoptosisCitation16.
In this article we continue our interest in sulfonamide CAIs as potential anticancer agents, and report the synthesis of benzenesulfonamides incorporating amino acid scaffolds and guanidine moieties. These new compounds were investigated for the inhibition of the overexpressed in hypoxic tumors human CA (hCA) IX and XII isoforms over the physiologically dominant, cytosolic ones hCA I and II, in order to develop possibly isoform-selective CA IX/XII inhibitors as anticancer agentsCitation5.
Materials and methods
Chemistry
Anhydrous solvents and all reagents were purchased from Aldrich, Merck or Carlo Erba. All reactions involving air- or moisture-sensitive compounds were performed under nitrogen atmosphere using oven-dried glassware and syringes to transfer solutions. Nuclear magnetic resonance (1H-NMR and NOE-difference) spectra were determined in CDCl3 and DMSO-d6 on a Varian XL-400 (400 MHz) spectrometer (Germany). Chemical shifts (δ scale) are reported in parts per million downfield from tetramethylsilane used as an internal standard. The assignment of exchangeable protons (OH and NH) was confirmed by the addition of D2O. Electron ionization mass spectra (70 eV) were recorded on a Hewlett-Packard 5989 Mass Engine Spectrometer (Germany). Analytical thin-layer chromatography was performed on Merck silica gel F-254 plates (Darmstadt, Germany). For flash chromatography Merck Silica gel 60 was used with a particle size 0.040–0.063 mm (230–400 mesh ASTM). The synthesis and the full characterization of the compounds investigated here (4–25) as hCA inhibitors are reported elsewhereCitation25.
CA inhibition studies
An applied photophysics stopped-flow instrument has been used for assaying the CA catalyzed CO2 hydration activityCitation26. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM HEPES (pH 7.4) and 20 mM NaBF4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (10 mM) were prepared in distilled–deionized water and dilutions up to 0.01 nM were done thereafter with distilled–deionized water. Inhibitor and enzyme solutions were preincubated together for 15 min at RT prior to assay, in order to allow for the formation of the E-I complex. The inhibition constants were obtained by nonlinear least-squares method using PRISM 3, and the Cheng–Prusoff equation, as reported earlierCitation27,Citation28, and represent the mean from at least three different determinations. All CAs were recombinant proteins obtained as reported earlier by these groupsCitation27,Citation28.
Results and discussion
Chemistry
Sulfonamides derivates are known to be highly effective CAIsCitation1–3. However, their low water solubility represents one of the main weak points of this class of pharmacological agentsCitation29. This is probably due to the fact that the highly polar sulfamoyl moiety generally is attached to aromatic/heterocyclic rings and no water hydrophilic moieties are present in the molecule. Therefore, in this work we considered to add amino acyl moieties as tails to the scaffolds of aromatic sulfonamides, in order to increase the water solubility of such compoundsCitation30. We then used the tail approach for our drug design employing N-α-acetyl-l-lysine or γ-aminobutyric acid (GABA) as scaffolds, and the conversion of their terminal amino group to the guanidine one, for synthesizing compounds incorporating 4-aminoethyl/methyl-benzenesulfonamide as well as metanilamide/sulfanilamide as heads for binding to the zinc ion from the enzyme active site (Schemes 1 and 2, and ).
Scheme 1. Synthesis of N-α-acetyl-l-lysine sulfonamides 4–12.
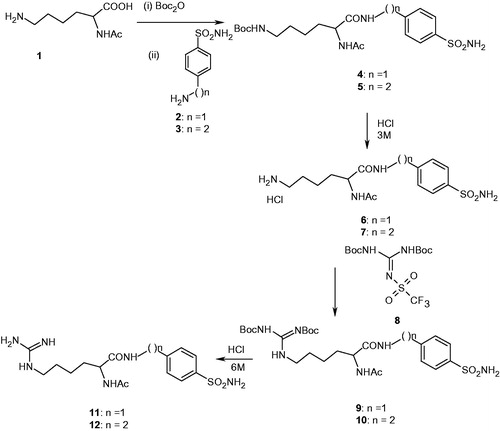
Scheme 2. Synthesis of GABA-containing sulfonamides 14–21.
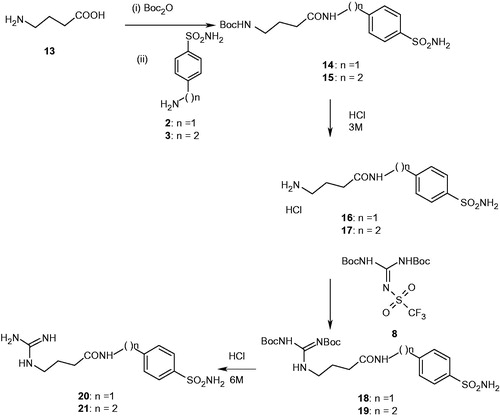
Chart 1. Structure of compounds 22–25 and the clinically used sulfonamide acetazolamide AAZ.
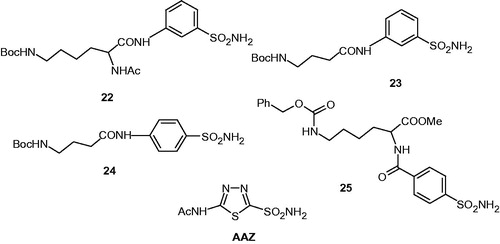
The first group of compounds were obtained starting from N-α-acetyl-l-lysine 1, followed by the protection of the ɛ-amino group with the Boc moiety, and the coupling with benzenesulfonamides 2 or 3 to give the ɛ-Boc-α-acetyl-protected-lysyl sulfonamides 4 and 5 (Scheme 1). After removing of the Boc (tert-butyloxycarbonyl) protecting group in 3 M HCL, the hydrochlorides 6, 7 were treated with N,N′-di-Boc-N″-trifluoromethane-sulfonylguanidine 8 for converting the terminal amino moiety to the di-Boc-protected guanidines 9 and 10. Deprotection of the guanidine moiety in the presence of acid led to the guanidine-substituted sulfonamides 11 and 12 (Scheme 1).
The remaining derivatives were synthesized using a similar approach. In this case GABA 13 was used as starting material following by similar derivatization reactions, compounds 14–21 incorporating guanidine–GABA moieties were thus obtained (Scheme 2). Furthermore, several metanilamide (22, 23), sulfanilamide (24) and 4-carboxy-benzenesulfonamide (25) derivatives incorporating lysyl- or GABA moieties were also obtained by following the same synthetic procedure ().
CA inhibition
Inhibition data with the new group of sulfonamides 4–25 reported here, against hCA isoforms hCA I, II (cytosolic) and hCA IX, XII (transmembrane and tumor associated) are shown in . Therefore, the following structure–activity relationship can be observed from data reported in mentioned table:
The new sulfonamides reported here were generally ineffective or medium potency inhibitors against the cytosolic slow isoform hCA I, with KIs values ranging between 88.9 and 6030 nM. However, only two sulfonamides, 14 and 23, both containing Boc–GABA moieties, showed to be effective inhibitors against this isoform, with KIs of 19.8–22.1 nM. For all substitution patterns, generally the 4-aminoethyl-benzenesulfonamides were more effective hCA I inhibitors compared with the corresponding 4-aminomethylbenzenesulfonamide derivates ().
The physiologically dominant human isoform hCA II was effectively inhibited by derivatives 14 and 23–25. Indeed, these compounds showed KIs in the range of 4.4–29.4 nM, of the same order of magnitude as acetazolamide (AAZ; KIs of 12 nM). These compounds again incorporate the Boc-GABA moieties, except 25 which is a derivative of 4-carboxybenzenesulfonamide to which the derivatization was achieved at the α-amino moiety, whereas the ɛ-amino one was protected by the benzyloxycarbonyl group. Some of the derivatives reported here, such as 4, 5, 12, 15, 16, 18 and 19, were medium potency hCA II inhibitors (KIs in the range of 44.0–91.9 nM). The remaining compounds showed to be ineffective inhibitors against hCA II, with KIs in the range of 384–5100 nM ().
The transmembrane isoform, hCA IX was poorly inhibited by most of the derivatives reported here. Generally the GABA derivatives were better hCA IX inhibitors compared with the N-α-acetyl-l-lysines possessing similar sulfonamide and protecting group scaffolds (). Indeed, the compounds 4, 5, 7, 9–12 and 22 have shown high KIs values in the range of 1373–2474 nM (except for the compound 16 with KI of 1015 nM, which contains a GABA moiety). Although this tumor-associated isoform, hCA IX, was slightly better inhibited by the derivatives 14, 17, 18, 20, 21 and 23 containing a GABA moiety (KIs of 517.8–862.2 nM) and derivatives 6 which contain N-α-acetyl-l-Lysine (KI of 876.4 nM), the most efficient hCA IX inhibitors were 15, 19 and 24, which showed inhibition constants between 65.7 and 95.7 nM. The best activity is correlated with the presence of derivatized GABA moieties and 4-amino-ethylbenzenesulfonamide (15 and 19) and 4-aminobenzenesulfonamide (24) head group. However, the best hCA IX inhibitor was compound 25, (containing a different scaffold, as mentioned above) with KI value of 47.7 nM, which was anyhow a weaker inhibitor compared with the clinically used inhibitor AAZ (KI of 25 nM).
The second transmembrane isoform associated with tumors, hCA XII, was much better inhibited by the new sulfonamides investigated here, compared with hCA IX. Several low nanomolar hCA XII inhibitors are reported here, such as 14, 18, 20, 24 and 25 derivatives with KIs in the range of 4.8–12.3 nM. Although many of the highly effective hCA XII inhibitors reported here have not possessed excellent selectivity ratios for inhibiting this transmenbrane isoform over the cytosolic ones, the most interesting CA XII inhibitor listed here was derivate 20 which has shown selectivity ratios as high as 139 against hCA I and 67 against hCA II (). The remaining derivatives 10, 15, 16, 19 and 23 have showed moderate inhibitory potency against this isoform (KIs in the range of 26.5–69 nM).
Table 1. Inhibition data against human isoforms hCA I, II (cytosolic) and IX, XII (transmembrane and tumor-associated) of sulfonamides 4–25 reported here and AAZ (as standard inhibitor) by a stopped-flow CO2 hydrase assayCitation26.
The least effective hCA XII inhibitors were derivatives 4, 5, 7, 9, 12, 17, 21 and 23, which showed KIs in the range of 126.2–413.7 nM, being thus low potency inhibitors of this isoform compared with the clinically used inhibitor AAZ.
Conclusions
Two series of benzenesulfonamides incorporating amino acid scaffolds (GABA or the N-α-acetyl-l-lysine) were explored here as a new generation of selective tumor-associated hCA XII inhibitors. The compounds investigated contain guanidine groups at the terminal amino moiety which confer them higher water solubility. Most of the new compounds were medium potency or ineffective as inhibitors of the human CAs, hCA I, II and IX, but they showed significant inhibition potency against the transmembrane isoform hCA XII, with efficacy in nanomolar range.
Therefore, considering the selectivity ratios of some of these derivatives for inhibiting the tumor-associated hCA XII over the physiologically dominant hCA I and II isoforms, these compounds may constitute interesting tools for the development of new anticancer agents.
Declaration of interest
C.T.S. reports conflict of interest as author of many patents on CA inhibitors. The authors alone are responsible for the content and writing of this article. This project was funded by a 7th FP EU grant (METOXIA).
References
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81
- Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72
- Alterio V, Di Fiore A, D'Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68
- Carta F, Garaj V, Maresca A, et al. Sulfonamides incorporating 1,3,5-triazine moieties selectively and potently inhibit carbonic anhydrase transmembrane isoforms IX, XII and XIV over cytosolic isoforms I and II: solution and X-ray crystallographic studies. Bioorg Med Chem 2011;19:3105–19
- Krall N, Pretto F, Decurtins W, et al. A small-molecule drug conjugate for the treatment of carbonic anhydrase IX expressing tumors. Angew Chem Int Ed Engl 2014;53:4231–5
- Neri D, Supuran CT. Interfering with pH regulation in tumors as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77
- Supuran CT. Carbonic anhydrase inhibitors and activators for novel therapeutic applications. Future Med Chem 2011;3:1165–80
- De Simone G, Alterio V, Supuran CT. Exploiting the hydrophobic and hydrophilic binding sites for designing carbonic anhydrase inhibitors. Expert Opin Drug Discov 2013;8:793–810
- Proescholdt MA, Merrill MJ, Stoerr EM, et al. Function of carbonic anhydrase IX in glioblastoma multiforme. Neuro Oncol 2012;14:1357–66
- McIntyre A, Patiar S, Wigfield S, et al. Carbonic anhydrase IX promotes tumour growth and necrosis in vivo and inhibition enhances anti-VEGF therapy. Clin Cancer Res 2012;18:3100–11
- Hussain SA, Ganesan R, Reynolds G, et al. Hypoxia-regulated carbonic anhydrase IX expression is associated with poor survival in patients with invasive breast cancer. Br J Cancer 2007;96:104–9
- Barnett DH, Sheng S, Charn TH, et al. Estrogen receptor regulation of carbonic anhydrase XII through a distal enhancer in breast cancer. Cancer Res 2008;68:3505–15
- Haapasalo J, Hilvo M, Nordfors K, et al. Identification of an alternatively spliced isoform of carbonic anhydrase XII in diffusely infiltrating astrocytic gliomas. Neuro Oncol 2008;10:131–8
- Said HM, Hagemann C, Carta F, et al. Hypoxia induced CA9 inhibitory targeting by two different sulfonamide derivatives including acetazolamide in human glioblastoma. Bioorg Med Chem 2013;21:3949–57
- Battke C, Kremmer E, Mysliwietz J, et al. Generation and characterization of the first inhibitory antibody targeting tumour-associated carbonic anhydrase XII. Cancer Immunol Immunother 2011;60:649–58
- Lounnas N, Rosilio C, Nebout M, et al. Pharmacological inhibition of carbonic anhydrase XII interferes with cell proliferation and induces cell apoptosis in T-cell lymphomas. Cancer Lett 2013;333:76–88
- Dubois L, Lieuwes NG, Maresca A, et al. Imaging of CA IX with fluorescent labelled sulphonamides distinguishes hypoxic and (re)-oxygenated cells in a xenograft tumour model. Radiother Oncol 2009;92:423–8
- Cecchi A, Hulikova A, Pastorek J, et al. Carbonic anhydrase inhibitors. Sulfonamides inhibit isozyme IX mediated acidification of hypoxic tumors. Fluorescent sulphonamides design as probes of membrane-bound carbonic anhydrase isozymes involvement in tumorigenesis. J Med Chem 2005;48:4834–41
- Švastová E, Hulíková A, Rafajová M, et al. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett 2004;577:439–45
- Vullo D, Franchi M, Gallori E, et al. Carbonic anhydrase inhibitors. Inhibition of cytosolic isozymes I and II and transmembrane, cancer-associated isozyme IX with anions. J Enzym Inhib Med Chem 2003;18:403–6
- Innocenti A, Vullo D, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Interactions of phenols with the 12 catalytically active mammalian isoforms (CA I–XIV). Bioorg Med Chem Lett 2008;18:1583–7
- Maresca A, Temperini C, Vu H, et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–62
- Vullo D, Franchi M, Gallori E, et al. Carbonic anhydrase inhibitors. Inhibition of the tumor-associated isozyme IX with aromatic and heterocyclic sulphonamides. Bioorg Med Chem Lett 2003;13:1005–9
- Carradori S, De Monte C, D'Ascenzio M, et al. Salen and tetrahydrosalen derivatives act as effective inhibitors of the tumor-associated carbonic anhydrase XII – a new scaffold for designing isoform-selective inhibitors. Bioorg Med Chem Lett 2013;23:6759–63
- Ceruso M, Del Prete S, Alothman Z, et al. Sulfonamides with potent inhibitory action and selectivity against the α-carbonic anhydrase from Vibrio cholera. ACS Med Chem Lett 2014;5:826–30
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73
- Supuran CT, Mincione F, Scozzafava A, et al. Carbonic anhydrase inhibitors. Part 52. Metal complexes of heterocyclic sulfonamides: a new class of strong topical intraocular pressure-lowering agents with potential use as antiglaucoma drugs. Eur J Med Chem 1998;33:247–54
- Wilkinson BL, Bornaghi LF, Houston TA, et al. Carbonic anhydrase inhibitors: inhibition of isozymes I, II and IX with triazole-linked O-glycosides of benzene sulphonamides. J Med Chem 2007;50:1651–7
- Carta F, Scozzafava A, Supuran CT. Sulfonamides (RSO2NH2): a patent review 2008–2012. Expert Opin Ther Pat 2012;22:747–58
- Scozzafava A, Briganti F, Mincione G, et al. Carbonic anhydrase inhibitors. Synthesis of water-soluble, amino acyl/dipeptidyl sulfonamides possessing long lasting-intraocular pressure lowering properties via the topical route. J Med Chem 1999;42:3690–700