
Abstract
Insight into the structure and inhibition mechanism of O-β-d-glucosidases by deoxa-pyranosylamine type inhibitors is provided by X-ray analysis of complexes between raucaffricine and strictosidine glucosidases and N-(cyclohexylmethyl)-, N-(cyclohexyl)- and N-(bromobenzyl)-β-d-gluco-1,5-deoxa-pyranosylamine. All inhibitors anchored exclusively in the catalytic active site by competition with appropriate enzyme substrates. Thus facilitated prospective elucidation of the binding networks with residues located at <3.9 Å distance will enable the development of potent inhibitors suitable for the production of valuable alkaloid glucosides, raucaffricine and strictosidine, by means of synthesis in Rauvolfia serpentina cell suspension cultures.
Introduction
Glycosidases constitute a large family of hydrolases displaying a broad range of biological functions in planta, including chemical defence, cell wall degradation, lignification, signalling and metabolism of natural productsCitation1–3. Their bio-significance spans all domains of life, thus bearing upon human health. In consequence, synthesis and development of potent glycosidase inhibitors is highly relevant to treatment of, among others, diabetes, lysosomal storage disorders and viral infectionsCitation4–6.
Deoxa-pyranosylamines, Footnote1dubbed also amino(hydroxymethyl)cyclopentanetriolsCitation7, boast structures related to those of well-established glycosidase inhibitors, mannostatins A and BCitation8–10. Selective inhibition of Golgi α-mannosidase II, effectively blocking the processing of influenza viral hemaglutinin as well as the oncogene induced changes in cell surface oligosaccharide structures, is but one example of the potent activity (and clinical applications thus implied) of the formerCitation11. Several chemical synthesis routes were developed, using α-d-mannosideCitation12, l-arabino-5-hexenoseCitation13, d-lyxoseCitation14 and d-glucoseCitation15 as starting materials to generate the hydrophilic core of the deoxa-pyranosylamine cyclopentane skeleton and to further exploit a variety of its derivativesCitation16. The inhibitory activities of the synthesized deoxa-pyranosylamines were tested against a range of commercially available glycoside hydrolases including representatives of microbial, plant and animal originCitation11–15, thereby facilitating the in-depth study of structure–activity relationships. These synthetic approaches allow generation of sets of inhibitors useful for the study of glycosidase active sites, thus providing preliminary insights into their molecular mechanisms.
Raucaffricine glucosidase (EC. 3.2.1.125, RG) and strictosidine glucosidase (EC. 3.2.1.105, SG) are glycoside hydrolases successfully cloned from Rauvolfia serpentinaCitation17–19, an ancient medicinal plant, traditionally used in the treatment of fever, insanity, inflammations and snake bitesCitation20. Both enzymes are involved in the biosynthetic pathway of ajmaline, one of the major active alkaloids of Rauvolfia, nowadays successfully applied in the therapy of arrhythmiaCitation21. Their substrates, strictosidine and raucaffricine, are highly valuable glucoalkaloids. The former acts as a biosynthetic precursor, common in the Apocynaceae family, of >2000 indole alkaloids, several of which boast substantial therapeutic potential (most notably, antineoplastic blockbusters, vinblastine and vincristine from Catharanthus roseusCitation22). In addition, in concert with raucaffricine, it is an excellent progenitor for the synthesis of novel alkaloids through the application of chemo-enzymatic approaches. Thus, a better understanding and further activity manipulation of the glucosidases of interest will facilitate the accumulation of the desired glucoalkaloid products.
In this study, we report on the analysis of 3D crystal structure complexes of the aforementioned inhibitors (specifically: N-(cyclohexylmethyl)-, N-(cyclohexyl)- and N-(bromobenzyl)-β-d-gluco-1,5-deoxa-pyranosylamine, 1, 2, and 3, respectively; ) with O-β-d-glucosidases, RG and SG. The obtained results now provide a unique visual and structural understanding of the binding and inhibitory properties of deoxa-pyranosylamine compounds.
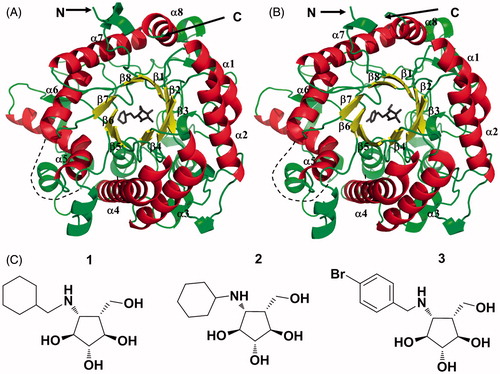
Figure 1. (A) Overall structure of N-His6-RG in complex with 1. (B) Overall structure of N-His6-SG in complex with 3. The α-helices, β-sheets of the barrel and loops resembling the (β/α)8 fold are depicted in red, yellow and green, respectively. Disordered loops are marked with dashed lines. (C) Chemical structures of inhibitors 1, 2 and 3.
Methods
Enzyme preparation
RG- and SG-encoding genes were heterologously expressed in Escherichia coli. The resulting proteins, tagged with a His6-motif at their N-termini, were isolated, purified and crystallized as reported previouslyCitation18,Citation23,Citation24. Protein concentrations were determined by the Bradford methodCitation25.
Measurements of Ki values
To determine the Ki values, simultaneous nonlinear regression (SNLR) was appliedCitation26. The substrate gradient concentrations were: 0.2, 0.4, 0.8, 1.2, 1.6 and 2.0 mM for raucaffricine and 30, 60, 90, 120 and 180 μM for strictosidine. Quantitative kinetic assays were performed at the optimal pH of both enzymes (pH 5, sodium citrate). Enzymes and inhibitors were mixed. After 15 min, substrates were added to enzyme-inhibitor mixtures. The assayed samples (100 μL) were incubated at 30 °C for 30 min. The hydrolysis of raucaffricine was monitored at 258 nm by HPLC (Agilent 1200, Beijing, China) with a LiChrospher 250 × 4 mm, 5 µm 60 RP-select B column (Merck, Darmstadt, Germany), injection volume of 50 μL and flow rate of 1.0 mL× min−1; the hydrolysis of strictosidine was determined by LC-MS (Agilent 1290 HPLC-6224 TOF Spectrometer, Beijing, China) with a C18 column (100 × 3.0 mm, 3.5 μm) (Agilent, Beijing, China), injection volume of 20 μL and flow rate of 0.2 mL × min−1, using linear gradients (5–95%, solvent B in A; B: CH3OH (Tedia, Shanghai, China), 0.1% formic acid (Tedia, Shanghai, China), A: H2O, 0.1% formic acid).
X-ray structure determination of RG-1, SG-2 and SG-3 ligand complexes: data collection, complex formation, model building and refinement
Data collection for structure determination of the RG-1 complex was performed at the Beamline X11 of EMBL-Hamburg (Germany) under cryo-conditions. Prior to data collection, crystals of RG were cryo-protected with 30 mM inhibitor 1 in 25% glycerol. RG crystals were soaked for maximum of 20 s and then flash-cooled in liquid nitrogen at 100 K. For RG-1, 360 images were collected (1° rotation per frame) with a MAR CCD detector at the wavelength of 0.8150 Å. The data were processed using the DENZO software, scaled by means of the SCALEPACKCitation27 and indexed in I centred orthorhombic space group I222. Diffraction data for structure determination of SG-2 and SG-3 complexes were collected at Beamline BL17U1 of SSRF-Shanghai (China) under cryo-conditions. Prior to data collection, the SG-2 complex was prepared by soaking the SG crystals in 5 mM inhibitor 2 solution for 60 min, then cryo-protected with 5 mM 2 in 25% glycerol. For the SG-3 complex, SG crystals in the hanging drop were treated with inhibitor 3 powder overnight, then soaked in 40 mM solution of 3 in 20% glycerol for 10 s and flash-cooled to 100 K. Three-hundred and sixty images were collected with 0.5° rotation per frame using a Q315CCD detector at the wavelength of 0.97915 Å and 1.004350 Å for SG-2 and SG-3, respectively. The data were processed using the XDS software and scaled by means of the XSCALECitation28 in primitive tetragonal space group P4212. All statistical data are summarized in .
The structure of RG-1 was determined by the molecular replacement method employing the software pipeline AUTO-RICKSHAWCitation29 and using the refined native RG structure (PDB code, 4ATD) as a search model. REFMAC5Citation30 was used for refinement of atomic coordinates and atomic displacement parameters. For manual and semi-automated model building of the protein, the solvent and the inhibitor, COOTCitation31 was utilized. The structures of SG-2 and SG-3 were resolved by means of the molecular replacement technique and using the native SG structure (PDB code, 2JF7) as a search model. Refinement and model building were carried out as explained above. The overall B-factor of water molecules for SG-3 was much lower than that characteristic of protein/inhibitor atoms (). This may have had two reasons: (a) intensity of low resolution reflections was overloaded due to X-ray over-exposure of the protein crystals and/or (b) intensity of the reflections was, most likely, underestimated during data processing. MolProbity and PROCHECKCitation32 were applied for structure validation. All figures were obtained with PyMOLCitation33.
Results and discussion
Inhibition of raucaffricine and SGs
Inhibition constants of the investigated compounds 1, 2, and 3 towards RG and SG were determined using their corresponding natural substrates. According to previous reports, 1 and other structurally related compounds exhibited competitive inhibition for >10 tested glycosidasesCitation12,Citation13, binding as the protonated speciesCitation11,Citation34. In this study, the SNLR method was applied to determine the relevant glucosidase Ki valuesCitation26 (Figure S1, Supplemental Information). Although the three inhibitors share the basic deoxa-pyranosylamine structure, compounds 1 and 3 proved significantly more efficient, as compared to 2, in arresting the activity of both RG and SG. Moreover, all investigated compounds exhibited much stronger inhibition against RG relative to SG (). Garnering comprehensive information delineating the binding features of 1, 2 and 3 with RG and SG will enhance the understanding of the differences in kinetic data thus obtained, and may be used to develop more specific inhibitors for each of the hereby investigated enzymes of interest.
Table 1. Inhibition constants of N-(cyclohexylmethyl)-, N-(cyclohexyl)- and N-(bromobenzyl)-β-d-gluco-1,5-deoxa-pyranosylamine (1, 2 and 3) against RG and SG.
Table 2. Data collection and structure refinement statistics of RG-1, SG-2 and SG-3 complexes.
X-ray analysis of enzyme-ligand complex structures
For the structural determination of RG in complex with inhibitor 1 (PDB code, 3ZJ6), X-ray data were collected at 100 K to 2.4 Å. The asymmetric unit (AU) contained two enzyme molecules, each of 468 residues. Concurrently, the complex structures of SG with ligands 2 (PDB code, 3ZJ7) and 3 (PDB code, 3ZJ8), also harbouring two enzyme molecules in the AU, revealed the highest resolution of 2.5 and 3.0 Å, respectively (). Physiologically, the enzymes act as monomer.
The complex structures described herein, RG-1, SG-2 and SG-3, and those reported previously for the enzymes of interestCitation23,Citation24,Citation35 overlapped, with a main-chain root mean square deviation of <0.3 Å, indicating that inhibitor binding had only marginal structural impact on the glucosidases.
The overall structures of RG-1, SG-2 and SG-3 complexes all adopted the (β/α)8 TIM barrel conformation – the typical fold of glycoside hydrolase family 1 (GH-1) enzymesCitation36. Therein, inhibitors 1, 2 and 3 settled in the centre of the barrel scaffold, blocking the active sites of RG and SG (, Figure S2, Supplemental Information).
As depicted in , the nitrogen atom of 1 was positioned between the two catalytic residues characteristic of RG, Glu-186 and Glu-420. Superimposition of RG-1, SG-2 and SG-3 structures with their previously obtained RG-glucose counterpart (PDB code, 4ATL) clearly showed that the positioning of the hydroxyl groups of the inhibitors closely corresponded to that of the enzyme product, glucose (, oxygen atoms of –OH groups in the glycone moieties are indicated with arrows). Furthermore, superimposed complexes of RG-1 and the inactive mutant, RG-Glu186Gln-substrate (PDB code, 3U57; ), as well as SG-2, SG-3 and SG-Glu207Gln-strictosidine (PDB code, 2JF6; ) showed the inhibitors occupying binding pockets of the investigated glucosidases. Moreover, all hydroxyl groups of the inhibitors formed strong networks of hydrogen bonds in the enzyme active centres with residues highly conserved in all structurally elucidated members of the GH-1 family (, Table S1, Supplemental information). These results indicate that inhibitors 1, 2 and 3 successfully mimic the glucose moiety of natural RG and SG substrates, thus competing for binding within the catalytic centres of the hydrolases of interest.
Figure 2. Divergent stereo-ribbon representation of RG in complex with 1. The acid/base Glu-186, the nucleophile Glu-420 and 1 are depicted as “ball-and-stick” structures. Electron density of the 2Fo-Fc map was contoured at 1σ.
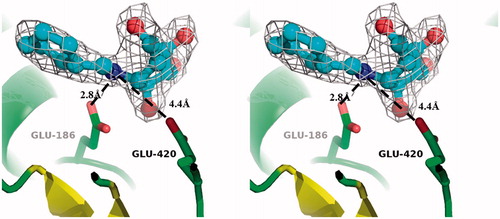
Figure 3. (A) Superimposed 1, 2 and 3 (yellow, cyan and magenta, respectively) with glucose (black), as anchored within the elucidated RG-1, SG-2, SG-3 and RG-glucose complexes; the hydroxyl groups of inhibitors overlap with those of glucose. (B) Rotation of A by 90°. (C) Superimposed complexes of RG-1 and RG-Glu186Gln-dihydroraucaffricine (red) in RG binding pocket (gray). (D) Superimposed SG-2, SG-3 and SG-Glu207Gln-strictosidine (blue) complexes in SG binding pocket (gray). Oxygen atoms of –OH groups in the glycone moieties are indicated with arrows. (E) Chemical structures of RG and SG substrates.
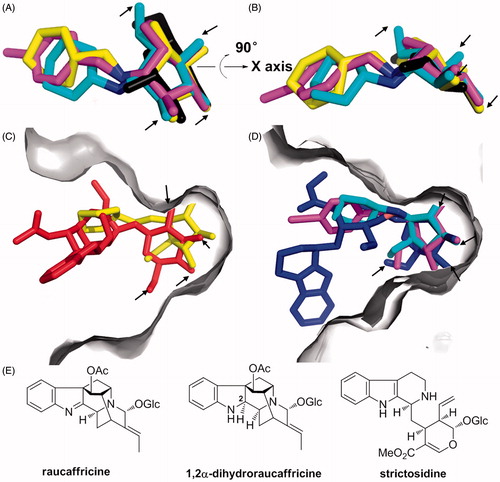
Figure 4. Binding networks between inhibitors and enzyme residues at ≤3.9 Å in the investigated ligand structure complexes. (A) His6-RG-1. (B) His6-SG-2. (C) His6-SG-3.
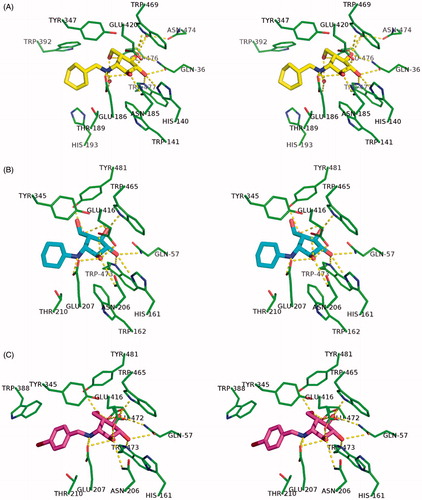
Since the enzyme-cinching modes of the hydrophilic inhibitor elements proved mostly similar, the interactions between their hydrophobic counterparts and the RG- and SG-binding pockets turned out to be especially important for efficient differentiation in deoxa-pyranosylamine inhibition efficacyCitation37. Four hydrophobic residues, Trp-392, Tyr-347, His-193 and Thr-189 were found within only 4 Å of the inhibitor-specific cyclohexyl group in the RG-1 complex (). In contrast, only two to three amino acids exhibited hydrophobic interaction with compounds 2 and 3 in SG-2 and SG-3 (, Figure S3, Supplemental information). As for inhibitor 2, its cyclohexyl moiety was only slightly stabilized due to very weak interactions with Thr-210 and, possibly, Asn-214 residues. Thus, the resulting higher flexibility of 2 contributed to the observed loss of inhibitory activity (). The remarkably higher affinity of all the tested inhibitors (1–3) towards RG, as compared to SG, was probably due to the different spatial distribution of the aglycone-binding residues, especially Trp-392 and Trp-388, in respective glucosidases (Figure S3, Supplemental information). Hence, the inhibitors could be anchored more tightly within the active centre of the former.
The possible role of the lipophilic N-substituents inducing entropy changes – by dispelling water from the active site with increase of translational entropy – cannot be specified yet; this would necessitate higher-resolution crystal structure analyses in order to provide precise representations of crystal water molecules.
Conclusions
In summary, the first 3D crystal complexes of RG-1, SG-2 and SG-3 demonstrated that deoxa-pyranosylamine type inhibitors anchored precisely to the active centres of both investigated glucosidases, providing structural evidence for their competitive inhibition mechanism, with the hydrophobic moieties of the compounds of interest exhibiting most pronounced influence on their efficacy. Detailed interaction maps between the inhibitors and the adjacent amino acids of RG and SG now afford the first structural basis for further optimization of deoxa-pyranosylamines. It should be noted that the enzymatic activity of RG was suppressed by its product, glucose, only at molar concentrations in R. serpentina cell suspension cultures, indicating that glucose itself is a weak glucosidase inhibitorCitation38,Citation39.
For a more efficient and simple production platform of valuable alkaloid glucosides, such as strictosidine (biosynthetic precursor of ∼2000 indole alkaloids, ) or raucaffricine (chemoenzymatic significance)Citation40 in plant cell culture systems, chemical synthesis of highly active and more specific deoxa-pyranosylamines may become of crucial importance in the near future.
Figure 5. Future production strategy of strictosidine by inhibition of SG in R. serpentina cell suspension cultures.
Supplementary material available online
Supplementary Figures S1–S3 and Table S1.
Supplemental Material.pdf
Download PDF (1.4 MB)Acknowledgements
We appreciate the support of Fonds der Chemischen Industrie (Frankfurt/Main, Germany), K. P. Chao HighTech Foundation of Zhejiang University (Hangzhou, China) and Deutsche Forschungsgemeinschaft (Bad Godesberg, Germany). We, moreover, gratefully acknowledge the assistance of the EMBL Beamline (DESY, Hamburg, Germany) and Beamline BL17U1 (Shanghai Synchrotron Radiation Facility, China) staff teams.
Declaration of interest
The authors report no declarations of interest
Notes
1Amino(hydroxymethyl)cyclopentanetriols structurally relate to pyranosylamines but for the ring-oxygen excision; hence, we term them “deoxa-pyranosylamines”. Thus, all stereodescriptors for hexopyranoses, as introduced by Fischer21, can be conveniently applied.
References
- Bellincampi D, Camardella L, Delcour JA, et al. Potential physiological role of plant glycosidase inhibitors. Biochim Biophys Acta 2004;1696:265–74
- Minic Z. Physiological roles of plant glycoside hydrolases. Planta 2008;227:723–40
- Morant AV, Jørgensen K, Jørgensen C, et al. Beta-Glucosidases as detonators of plant chemical defence. Phytochemistry 2008;69:1795–813
- Schmidt DD, Frommer W, Müller L, et al. Alpha-Glucosidase inhibitors. New complex oligosaccharides of microbial origin. Naturwissenschaften 1977;64:535–6
- Asano N. Glycosidase inhibitors: update and perspectives on practical use. Glycobiology 2003;13:93R–104R
- Butters TD, Dwek RA, Platt FM. Imino sugar inhibitors for treating the lysosomal glycosphingolipidoses. Glycobiology 2005;15:43R–52R
- Kleban M. Diastereoselektive Synthese von Aminocyclopentanpolyolen “Desoxapyranosylamine” als neue Klasse von Glykosidase-Inhibitoren. Doctoral Dissertation, Universität Stuttgart (Germany), 1996
- Aoyagi T, Yamamoto T, Kojiri K, et al. Mannostatins A and B: new inhibitors of alpha-d-mannosidase, produced by Streptoverticillium verticillus var. quintum ME3-AG3: taxonomy, production, isolation, physico-chemical properties and biological activities. J Antibiot (Tokyo) 1989;42:883–9
- Tropea JE, Kaushal GP, Patushak J, et al. Mannostatin A, a new glycoprotein-processing inhibitor. Biochemistry 1990;29:10062–9
- Kleban M, Kautz U, Greul JN, et al. Vitamin B12 catalysis of zinc-mediated 6-deoxy-6-iodopyranoside fragmentation: a mild and convenient preparation of ω-unsaturated hexose derivatives (5-hexenoses). Synthesis 2000;7:1027–33
- Kawatkar SP, Kuntz DA, Woods RJ, et al. Structural basis of the inhibition of Golgi alpha-mannosidase II by mannostatin A and the role of the thiomethyl moiety in ligand-protein interactions. J Am Chem Soc 2006;128:8310–19
- Kleban M, Hilgers P, Greul JN, et al. Amino(hydroxymethyl)cyclopentanetriols, an emerging class of potent glycosidase inhibitors – Part I: synthesis and evaluation of beta-D-pyranoside analogues in the manno, gluco, galacto, and GlcNAc series. ChemBioChem 2001;2:365–8
- Greul JN, Kleban M, Schneider B, et al. Amino(hydroxymethyl)cyclopentanetriols, an emerging class of potent glycosidase inhibitors–Part II: synthesis, evaluation, and optimization of beta-D-galactopyranoside analogues. ChemBioChem 2001;2:368–70
- Leroy E, Reymond JL. Anomer-selective inhibition of glycosidases using aminocyclopentanols. Org Lett 1999;1:775–7
- Boss O, Leroy E, Blaser A, Reymond JL. Synthesis and evaluation of aminocyclopentitol inhibitors of beta-glucosidases. Org Lett 2000;2:151–4
- Blaser A, Reymond JL. Stereoselective inhibition of alpha-L-fucosidases by N-benzyl aminocyclopentitols. Org Lett 2000;2:1733–6
- Warzecha H, Obitz P, Stöckigt J. Purification, partial amino acid sequence and structure of the product of raucaffricine-O-beta-D-glucosidase from plant cell cultures of Rauwolfia serpentina. Phytochemistry 1999;50:1099–109
- Warzecha H, Gerasimenko I, Kutchan TM, Stöckigt J. Molecular cloning and functional bacterial expression of a plant glucosidase specifically involved in alkaloid biosynthesis. Phytochemistry 2000;54:657–66
- Gerasimenko I, Sheludko Y, Ma X, Stöckigt J. Heterologous expression of a Rauvolfia cDNA encoding strictosidine glucosidase, a biosynthetic key to over 2000 monoterpenoid indole alkaloids. Eur J Biochem 2002;269:2204–13
- Sahu BN. Rauvolfia serpentina (sarpagandha), Vol. 2-Chemistry, and Pharmacology-R. 8vo. New Delhi: Today and Tomorrow’s Printers and Publishers; 1979:45–7
- Fischer F, Vonderlin N, Zitron E, et al. Inhibition of cardiac Kv1.5 and Kv4.3 potassium channels by the class Ia anti-arrhythmic ajmaline: mode of action. Naunyn Schmiedebergs Arch Pharmacol 2013;386:991–9
- Noble RL. The discovery of the vinca alkaloids – chemotherapeutic agents against cancer. Biochem Cell Biol 1990;68:1344–51
- Barleben L, Panjikar S, Ruppert M, et al. Molecular architecture of strictosidine glucosidase: the gateway to the biosynthesis of the monoterpenoid indole alkaloid family. Plant Cell 2007;19:2886–97
- Xia L, Ruppert M, Wang M, et al. Structures of alkaloid biosynthetic glucosidases decode substrate specificity. ACS Chem Biol 2012;7:226–34
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–54
- Kakkar T, Boxenbaum H, Mayersohn M. Estimation of Ki in a competitive enzyme-inhibition model: comparisons among three methods of data analysis. Drug Metab Dispos 1999;27:756–62
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 1997;276:307–26
- Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr 2010;66:125–32
- Panjikar S, Parthasarathy V, Lamzin VS, et al. Auto-rickshaw: an automated crystal structure determination platform as an efficient tool for the validation of an X-ray diffraction experiment. Acta Crystallogr D Biol Crystallogr 2005;61:449–57
- Murshudov GN, Skubák P, Lebedev AA, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 2011;67:355–67
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 2004;60:2126–32
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst 1993;26:283–91
- DeLano WL. The PyMOL molecular graphics system. San Carlos, CA: DeLano Scientific; 2002
- Varrot A, Tarling CA, Macdonald JM, et al. Direct observation of the protonation state of an imino sugar glycosidase inhibitor upon binding. J Am Chem Soc 2003;125:7496–7
- Xia L, Rajendran C, Ruppert M, et al. High speed X-ray analysis of plant enzymes at room temperature. Phytochemistry 2013;91:88–92
- Henrissat B, Davies G. Structural and sequence-based classification of glycoside hydrolases. Curr Opin Struct Biol 1997;7:637–44
- Dickson LG, Leroy E, Reymond JL. Structure-activity relationships in aminocyclopentitol glycosidase inhibitors. Org Biomol Chem 2004;2:1217–26
- Schübel H, Ruyter CM, Stöckigt J. Improved production of raucaffricine by cultivated Rauwolfia cells. Phytochemistry 1989;28:491–4
- Ketudat Cairns JR, Esen A. β-Glucosidases. Cell Mol Life Sci 2010;67:3389–405: “Free glucose is a poor inhibitor (typically, Ki = 100–200 mM) because glucose must be distorted toward the half-chair conformation for binding to the glycone-binding site, which is thought to require a portion of the energy of aglycone binding”
- see Figure S1 in Supporting Information of24