
Abstract
A series of umbelliferone analogues were synthesized and their inhibitory effects on the DPPH and mushroom tyrosinase were evaluated. The results showed that some of the synthesized compounds exhibited significant mushroom tyrosinase inhibitory activities. Especially, 2-oxo-2-[(2-oxo-2H-chromen-7-yl)oxy]ethyl-2,4-dihydroxybenzoate (4e) bearing 2,4-dihydroxy substituted phenyl ring exhibited the most potent tyrosinase inhibitory activity with IC50 value 8.96 µM and IC50 value of kojic acid is 16.69. The inhibition mechanism analyzed by Lineweaver–Burk plots revealed that the type of inhibition of compound 4e on tyrosinase was non-competitive. The docking study against tyrosinase enzyme was also performed to determine the binding affinity of the compounds. The compounds 4c and 4e showed the highest binding affinity with active binding site of tyrosinase. The initial structure activity relationships (SARs) analysis suggested that further development of such compounds might be of interest. The statistics of our results endorses that compounds 4c and 4e may serve as a structural template for the design and development of novel tyrosinase inhibitors.
Introduction
Tyrosinase or polyphenol oxidase (EC 1.14.18.1; PPO) is a binuclear copper containing enzyme present in biological systems. Two types of reactions are catalyzed by this enzyme in which molecular oxygen participates: (i) The orthohydroxylation of monophenols to o-diphenols (monophenolase activity) and (ii) the oxidation of o-diphenols to their corresponding o-quinones (diphenolase activity)Citation1. Roughly speaking, mushroom tyrosinases consist of three domains, of which the central domain contains two copper binding sites, called CuA and CuB. The enzyme seems to be almost universally distributed in animals, plants, fungi and bacteria but much is still unknown about its biological function.
Melanins high molecular weight brown-pigments are synthesized by polymerization of quinine because of their high reactivity and react with amino acids and proteins to enhance brown color of the pigmentCitation2. First two steps of biosynthesis of melanin are catalyzed by the tyrosinase, the over accumulation of melanin results in hyperpigmentation such as senile lentigo, melasma, freckles and pigmented acne scars is of particular concern to women. The treatment usually involves the use of medicines or medicinal cosmetics containing de-pigmenting agents or skin whitening agentsCitation3. Tyrosinase inhibitors are used for treatments of dermatological disorders related to melanin hyperaccumulation and are essential in cosmetics for depigmentationCitation4,Citation5. For example, age spots and freckle are caused by the accumulation of an excessive level of epidermal pigmentationCitation6.
Previous reports confirmed that tyrosinase was one of the main causes of most fruits and vegetables quality loss during post-harvest handling and processing, leading to faster degradation and shorter shelf lifeCitation7. Seo et al.Citation8 reviewed the importance of mushroom tyrosinase and defines its biochemical character and inhibition and activation by several inhibitors from natural and synthetic origin. Zhu et al.Citation9 also reported that tyrosinase enzyme has also been linked to Parkinson’s disease. Loizzo et al.Citation10 and ChangCitation11 reviewed the tyrosinase inhibitors from both natural and synthetic origin for their use in food and cosmetic industry. KhanCitation12 also reviewed the molecular design of tyrosinase inhibitors from synthetic origin.
The coumarins are especially abundant in the Umbelliferae and Rutaceae familiesCitation13 and also found in higher plants where they originate from phenylpropanoid pathwayCitation14. Some coumarins from natural sources have also been used as therapeutic agents for humansCitation15 and their antibacterial, antifungal and anticancer activities make these compounds attractive for further derivatization and for screening as therapeutic agentsCitation16. Umbelliferone also known as 7-hydroxy coumarin is a natural product widely distributed in many familiar plants Apiaceae, Asteraceae, Hydrangeaceae and Acanthaceae. Coumarin derivatives are of great interest due to their diverse structural features and versatile biological properties, such as anti-inflammatory, antioxidant, vasorelaxant, cytotoxic, anti-HIV, antitubercular and antimicrobialCitation17–22.
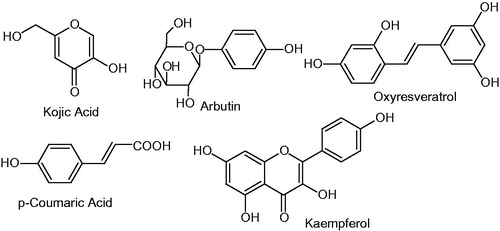
Coumarin derivatives have been synthesized and extensively studied for their biological response, but the tyrosinase inhibitor activities of this kind of compounds have hardly ever appeared in the literature. The literature survey revealed that clinically used tyrosinase inhibitors such as kojic acid, arbutine, oxyresveratrol, p-coumaric acid kaempferol, etc., all possess free phenolic hydroxyl moiety . The presence of hydroxy substituted phenyl ring is considered to be important in tyrosinase inhibitory activityCitation23,Citation24. Inspired by these findings, we synthesized the hydroxylated umbelliferone derivatives (4a–g) and (6a–b) in order to explore their inhibition mechanisms to offer a source for the development of new effective tyrosinase inhibitors. The title compounds 4a–g are synthesized by incorporating hydroxy substituted benzoic acids while compounds 6a–b by p-substituted cinnamic acids.
Figure 1. Structures of some mushroom tyrosinase inhibitors.
Experimental
All chemicals used for the synthesis of compounds were purchased from Sigma Chemical Co. (St. Louis, MO) Melting points were determined using a Digimelt MPA 160 (Holland, MI) melting point apparatus and are reported uncorrected. The FT-IR spectra were recorded with Shimadzu FTIR-8400S spectrometer (Kyoto, Japan, υ, cm−1). The 1H NMR and 13C NMR spectra (CDCl3) and (DMSO-d6) were recorded using a Bruker 400 MHz spectrometer. Chemical shifts (δ) are reported in ppm downfield from the internal standard tetramethylsilane (TMS). The purity of the compounds was checked by thin layer chromatography (TLC) on silica gel plate using n-hexane and ethyl acetate as mobile phase. The procedure for the synthesis of the desired compounds is depicted in Schemes 1 and 2.
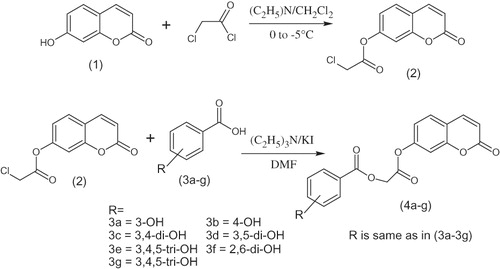
Scheme 1. Synthesis of coumarin analogues (4a–g).
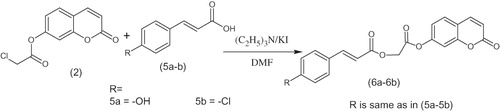
Scheme 2. Synthesis of coumarin analogues (6a–b).
Reagents
Mushroom tyrosinase was purchased from Sigma (St. Louis, MO); l-DOPA and umbelliferone were purchased from Sigma (St. Louis, MO). Stock solutions of the reducing substrates were prepared in phosphate buffer (20 mM, pH 6.8).
Synthesis of umbelliferone chloroacetyl derivative (2)
A mixture of 7-hydroxy coumarin (1) (0.01 mol), triethylamine (0.01 mol) in anhydrous dichloromethane (25 mL) was cooled in an ice salt mixture to 0–5 °C. To this reaction mixture, chloroacetyl chloride (0.01 mol) in dry dichloromethane was added drop wise with constant stirring over a period of 1 h maintaining the temperature constant. The reaction mixture was then stirred at room temperature for further 5 h, washed with 5% HCl, 5% sodium hydroxide and finally with brine solution. The organic layer was dried over anhydrous magnesium sulfate, filtered and the solvent was removed under reduced pressure to afford the corresponding umbelliferone chloroacetyl derivative (2).
2-oxo-2H-chromen-7-yl chloroacetate (2)
White solid; reaction time, 6 h; yield, 85%; melting point, 162–164 °C; Rf 0.64 (n-hexane:ethyl acetate 2:1), FTIR νmax cm−1: 3079 (sp2 C–H), 2973 (sp3 C–H), 1766 (C=O ester), 1728 (C=O, lactone), 1615 (C=C aromatic); 1H NMR (CDCl3, δ ppm): 7.73 (d, J = 9.2 Hz, 1H, H-4), 7.54 (d, J = 4.4 Hz, 1H, H-5), 7.19 (d, J = 2.4 Hz, 1H, H-8), 7.13 (dd, J = 2.4, 6.0 Hz, 1H, H-6), 6.26 (d, J = 9.6 Hz, 1H, H-3), 4.36 (s, 2H, –CH2); 13C NMR (CDCl3, δ ppm): 165.2 (C=O ester), 160.0 (C-2), 154.7 (C-7), 152.5 (C-10), 142.6 (C-4), 128.7 (C-5), 117.8 (C-9), 117.1 (C-6), 116.5 (C-8), 110.1 (C-3), 40.6 (CH2).
Synthesis of title compounds 4a–g and 6a–b
A mixture of umbelliferone chloroacetyl derivative (2) (0.01 mol), hydroxy substituted benzoic acids (3a–g) (0.01 mol), triethyl amine (0.01 mol), potassium iodide (0.01 mol) in dimethyl formamide (25 mL) was stirred overnight at room temperature (Scheme 1). The reaction mixture was poured into finely crushed ice with stirring and extracted with ethyl acetate (4 × 25 mL). The combined organic layer was washed with 5% HCl, 5% sodium hydroxide and finally with brine solution. The organic layer was dried over anhydrous magnesium sulfate, filtered and the solvent was removed under reduced pressure to afford the final products (4a–g). The title compounds 4a–g were purified by recrystallization in a mixture of hexane and ethyl acetate. The same procedure was used for the preparation of compounds 6a and 6b (Scheme 2).
2-oxo-2-[(2-oxo-2H-chromen-7-yl)oxy]ethyl-3-hydroxybenzoate (4a)
Solid; reaction time, 24 h; yield, 73%; melting point, 184–186 °C; Rf 0.42 (n-hexane:ethyl acetate 2:1), FTIR νmax cm−1: 3126 (O–H), 3014 (sp2 C–H), 2873 (sp3 C–H), 1705 (C=O ester), 1680 (C=O lactone), 1589 (C=C aromatic), 1148 (C–O, ester); 1H NMR (DMSO-d6, δ ppm): 7.81 (dd, J = 2.0, 2.1 Hz, 1H, H-2′), 7.80 (m, 2H, H-4′, H-6′), 7.66 (t, J = 4.0 Hz, 1H, H-5′), 7.58 (d, J = 1.2 Hz, 1H, H-8), 7.55 (d, J = 2.8 Hz, 1H, H-4), 7.53 (d, J = 4.8 Hz, 1H, H-5), 6.81 (dd, J = 2.4, 2.0 Hz, 1H, H-6), 6.21 (d, J = 9.2 Hz, 1H, H-3), 5.28 (s, 2H, –CH2), 2.50 (s, 1H, 3′-OH); 13C NMR (DMSO-d6, δ ppm): 177.7 (C-1″), 161.7 (C=O ester), 160.8 (C-2), 155.9 (C-3′), 153.1 (C-7), 144.9 (C-10), 143.8 (C-4), 139.3 (C-1′), 132.2 (C-5′), 130.1 (C-5), 119.3 (C-6′), 116.5 (C-4′), 113.6 (C-6), 111.8 (C-2′), 111.7 (C-8), 107.5 (C-6′), 102.6 (C-3), 40.2 (CH2).
2-oxo-2-[(2-oxo-2H-chromen-7-yl)oxy]ethyl-4-hydroxybenzoate (4b)
Solid; reaction time, 24 h; yield, 78%; melting point, 177–179 °C; Rf 0.44 (n-hexane:ethyl acetate 2:1), FTIR νmax cm−1: 3093 (O–H), 2962 (sp2 C–H), 2852 (sp3 C–H), 1772 (C=O ester), 1710 (C=O lactone), 1602 (C=C aromatic), 1159 (C–O, ester); 1H NMR (DMSO-d6, δ ppm): 7.92 (dd, J = 9.2, 1.6 Hz, 2H, H-2′, H-6′), 7.52 (d, J = 8.4, 1H, H-4), 6.92 (dd, J = 5.6, 1.2 Hz, 2H, H-3′, H-5′), 6.81 (dd, J = 6.4, 2.0 Hz, 1H, H-6), 6.73 (d, J = 2.0 Hz, 1H, H-8), 6.51 (d, J = 9.6 Hz, 1H, H-5), 6.21 (d, J = 9.6 Hz, 1H, H-3), 5.18 (s, 2H, –CH2), 2.51 (s, 1H, 4′-OH); 13C NMR (DMSO-d6, δ ppm): 173.4 (C-1″), 162.4 (C=O ester), 160.8 (C-2), 155.9 (C-4′), 149.1 (C-7), 144.9 (C-10), 143.4 (C-4), 135.3 (C-1′), 132.3 (C-3′, C-5′), 130.1 (C-5), 114.4 (C-6), 113.4 (C-8), 111.8 (C-2′, C-6′), 110.5 (C-9), 102.5 (C-3), 40.5 (CH2).
2-oxo-2-[(2-oxo-2H-chromen-7-yl)oxy]ethyl-3,4-dihydroxybenzoate (4c)
Solid; reaction time, 24 h; yield, 71%; melting point, 226–228–186 °C; Rf 0.39 (n-hexane:ethyl acetate 2:1), FTIR νmax cm−1: 3126 (O–H), 2868 (sp2 C–H), 2827 (sp3 C–H), 1712 (C=O ester), 1681 (C=O lactone), 1567 (C=C aromatic), 1128 (C–O, ester); 1H NMR (DMSO-d6, δ ppm): 7.86 (d, J = 9.6 Hz, 1H, H-6′), 7.80 (d, J = 8.6 Hz, 1H, H-2′), 7.48 (d, J = 8.6 Hz, 1H, H-4), 7.39 (d, J = 7.2 Hz, 1H, H-5), 6.88 (d, J = 6.2 Hz, 1H, H-5′), 6.82 (dd, J = 6.4, 2.4 Hz, 1H, H-6), 6.73 (d, J = 2.4 Hz, 1H, H-8), 6.21 (d, J = 9.6 Hz, 1H, H-3), 4.90 (s, 2H, –CH2), 3.01 (s, 2H, 2′, 4′-OH); 13C NMR (DMSO-d6, δ ppm): 173.6 (C-1″), 162.4 (C=O ester), 161.5 (C-2), 156.2 (C-4′), 150.2 (C-3′), 148.1 (C-7), 144.6 (C-10), 136.8 (C-4), 136.3 (C-1′), 133.2 (C-5′), 129.2 (C-5), 124.3 (C-6′), 113.1 (C-4′), 111.6 (C-6), 110.9 (C-2′), 108.7 (C-8), 105.5 (C-6′), 102.0 (C-3), 49.2 (CH2).
2-oxo-2-[(2-oxo-2H-chromen-7-yl)oxy]ethyl-3,5-dihydroxybenzoate (4d)
Solid; reaction time, 24 h; yield, 78%; melting point, 221–223 °C; Rf 0.40 (n-hexane:ethyl acetate 2:1), FTIR νmax cm−1: 3170 (O–H), 3010 (sp2 C–H), 2962 (sp3 C–H), 1775 (C=O ester), 1679 (C=O lactone), 1567 (C=C aromatic), 1130 (C–O, ester); 1H NMR (DMSO-d6, δ ppm): 7.84 (d, J = 9.6 Hz, 1H, H-4), 7.68 (d, J = 8.4 Hz, 1H, H-5), 7.38 (dd, J = 1.6, 2.0 Hz, 2H, H-2’,6’), 7.03 (dd, J = 1.2, 1.6 Hz, 1H, H-4′), 6.84 (dd, J = 8.0, 2.4 Hz, 1H, H-6), 6.73 (d, J = 2.0 Hz, 1H, H-8), 6.21 (d, J = 9.6 Hz, 1H, H-3), 4.88 (s, 2H, –CH2), 3.32 (s, 2H, 3′, 5′-OH); 13C NMR (DMSO-d6, δ ppm): 166.3 (C-1″), 162.2 (C=O ester), 161.7 (C-2), 158.5 (C-3′, C-5′), 155.8 (C-7), 144.6 (C-10), 143.5 (C-4), 129.2 (C-1′), 115.3 (C-2′, C-6′), 113.1 (C-5), 111.7 (C-4′), 110.9 (C-6), 109.7 (C-8), 102.0 (C-3), 59.2 (CH2).
2-oxo-2-[(2-oxo-2H-chromen-7-yl)oxy]ethyl-2,4-dihydroxybenzoate (4e)
Solid; reaction time, 24 h; yield, 64%; melting point, 204–206 °C; Rf 0.45 (n-hexane:ethyl acetate 2:1), FTIR νmax cm−1: 3125 (O–H), 2924 (sp2 C–H), 2852 (sp3 C–H), 1709 (C=O ester), 1677 (C=O lactone), 1603 (C=C aromatic), 1128 (C–O, ester); 1H NMR (DMSO-d6, δ ppm): 8.09 (d, J = 9.6 Hz, 1H, H-6′), 7.94 (d, J = 9.6 Hz, 1H, H-4), 7.53 (d, J = 8.4 Hz, 1H, H-5), 7.22 (d, J = 2.4 Hz, 1H, H-3′), 6.80 (dd, J = 6.4, 2.4 Hz, 1H, H-5′), 6.73 (d, J = 2.0 Hz, 1H, H-8), 6.50 (dd, J = 9.6, 2.4 Hz, 1H, H-6), 6.33 (d, J = 2.0 Hz, 1H, H-8), 6.21 (d, J = 9.6 Hz, 1H, H-3), 4.60 (s, 2H, –CH2), 3.34 (s, 2H, 2′, 4′-OH); 13C NMR (DMSO-d6, δ ppm): 176.5 (C-1″), 161.7 (C=O ester), 160.8 (C-2), 155.9 (C-2′, C-4′), 144.9 (C-7), 143.8 (C-10), 142.3 (C-4), 132.6 (C-1′), 130.9 (C-5), 113.6 (C-6′), 111.8 (C-3′, C-5′), 111.7 (C-6), 107.7 (C-8), 102.6 (C-3), 58.2 (CH2).
2-oxo-2-[(2-oxo-2H-chromen-7-yl)oxy]ethyl-2,6-dihydroxybenzoate (4f)
Solid; reaction time, 24 h; yield, 74%; melting point, 190–192 °C; Rf 0.46 (n-hexane:ethyl acetate 2:1), FTIR νmax cm−1: 3103 (O–H), 2912 (sp2 C–H), 2853 (sp3 C–H), 1678 (C=O ester), 1601 (C=O lactone), 1566 (C=C aromatic), 1122 (C–O, ester); 1H NMR (DMSO-d6, δ ppm): 7.92 (d, J = 9.2 Hz, 1H, H-4), 7.54 (d, J = 8.4 Hz, 1H, H-5), 7.18 (m, 3H, H-3′,4′,5′), 6.85 (dd, J = 9.6, 2.4 Hz, 1H, H-6), 6.79 (d, J = 2.4 Hz, 1H, H-8), 6.20 (d, J = 9.2 Hz, 1H, H-3), 5.20 (s, 2H, –CH2), 4.09 (s, 2H, 2′,6′-OH); 13C NMR (DMSO-d6, δ ppm): 180.8 (C-1″, 166.4 (C=O ester), 161.3 (C-2), 155.6 (C-2′, C-6′), 150.1 (C-7), 144.9 (C-10), 142.8 (C-4), 136.1 (C-4′),132.4 (C-1′) 130.1 (C-3′, C-5′), 119.5 (C-5), 111.9 (C-6), 111.8 (C-8), 102.5 (C-3), 48.5 (CH2).
2-oxo-2-[(2-oxo-2H-chromen-7-yl)oxy]ethyl-3,4,5-trihydroxybenzoate (4g)
Solid; reaction time, 24 h; yield, 52%; melting point, 230–232 °C; Rf 0.42 (n-hexane:ethyl acetate 2:1), FTIR νmax cm−1: 3124 (O–H), 2914 (sp2 C–H), 2829 (sp3 C–H), 1705 (C=O ester), 1678 (C=O lactone), 1566 (C=C aromatic), 1129 (C–O, ester); 1H NMR (DMSO-d6, δ ppm): 7.87 (d, J = 0.4 Hz, 1H, H-2′, H-6′), 7.85 (d, J = 0.4 Hz, 1H, H-8), 7.48 (d, J = 8.8 Hz, 1H, H-4), 6.82 (dd, J = 6.0, 2.4 Hz, 1H, H-6), 6.73 (d, J = 2.4 Hz, 1H, H-5), 6.21 (d, J = 9.6 Hz, 1H, H-3), 4.90 (s, 2H, –CH2), 3.90 (s, 1H, –OH); 13C NMR (DMSO-d6, δ ppm): 178.7 (C-1″), 162.3 (C=O ester), 161.7 (C-2), 155.6 (C-3′, 4′,5′), 144.6 (C-7), 141.7 (C-10), 138.3 (C-4), 131.6 (C-1′), 129.2 (C-2′, C-6′), 113.1 (C-5), 111.7 (C-6), 110.9 (C-8), 102.0 (C-3), 48.2 (CH2).
2-oxo-2-[(2-oxo-2H-chromen-7-yl)oxy]ethyl(2E)-3-(4-hydroxyphenyl)prop-2-enoate (6a)
Solid; reaction time, 24 h; yield, 76%; melting point, 201–203 °C; Rf 0.45 (n-hexane:ethyl acetate 2:1), FTIR νmax cm−1: 3166 (O–H), 2931 (sp2 C–H), 2843 (sp3 C–H), 1678 (C=O ester), 1624 (C=O lactone), 1601 (C=C aliphatic) 1567 (C=C aromatic), 1128 (C–O, ester); 1H NMR (DMSO-d6, δ ppm): 8.08 (d, J = 9.6 Hz, 1H, H-4), 7.93 (d, J = 9.2 Hz, 2H, H-2′, H-6′), 7.68 (d, J = 3.2 Hz, 1H, H-5), 7.53 (d, J = 8.4 Hz, 2H, H-3′, H-5′), 6.83 (d, J = 2.4 Hz, 1H, H-8), 6.78 (dd, J = 6.0, 2.4 Hz, 1H, H-6), 6.49 (d, J = 16.0 Hz, 1H, H-1″), 6.31 (d, J = 16.0 Hz, 1H, H-2″), 6.20 (d, J = 9.2 Hz, 1H, H-3), 5.07 (s, 2H, –CH2), 2.52 (s, 1H, –OH); 13C NMR (DMSO-d6, δ ppm): 162.3 (C-ester), 161.7 (C-3″, C=O), 160.8 (C-2), 155.9 (C-4′), 146.3 (C-7), 144.9 (C-10), 132.1 (C-1′), 131.3 (C-4), 130.1 (C-2′, C-6′), 116.3 (C-5), 115.8 (C-2″), 113.6 (C-3′, C-5′), 111.8 (C-6), 111.7 (C-8), 102.6 (C-3), 36.2 (CH2).
2-oxo-2-[(2-oxo-2H-chromen-7-yl)oxy]ethyl(2E)-3-(4-chlorophenyl)prop-2-enoate (6b)
Solid; reaction time, 24 h; yield, 84%; melting point, 154–156 °C; Rf 0.47 (n-hexane:ethyl acetate 2:1), FTIR νmax cm−1: 3083 (sp2 C–H), 2867 (sp3 C–H), 1716 (C=O ester), 1692 (C=O lactone), 1615 (C=C aliphatic), 1568 (C=C aromatic), 1123 (C–O, ester); 1H NMR (DMSO-d6, δ ppm): 7.77 (d, J = 16.0 Hz, 1H, H-1″), 7.70 (d, J = 9.2 Hz, 1H, H-4), 7.54 (d, J = 6.0 Hz, 1H, H-5), 7.51 (dd, J = 4.0, 2.4 Hz, 2H, H-2′, H-6′), 7.39 (dd, J = 4.8, 1.6 Hz, 2H, H-3′, H-5′), 7.21 (d, J = 2.0 Hz, 1H, H-8), 7.14 (dd, J = 6.0, 2.4 Hz, 1H, H-6), 6.53 (d, J = 16.0 Hz, 1H, H-2″), 6.43 (d, J = 9.6 Hz, 1H, H-3), 5.03 (s, 2H, –CH2); 13C NMR (DMSO-d6, δ ppm): 165.8 (C=O ester), 160.1 (C-3″, C=O), 154.7 (C-2), 152.4 (C-4′), 145.3 (C-7), 142.6 (C-10), 136.8 (C-1′), 132.5 (C-3′, C-5′), 129.4 (C-2′, C-6′), 129.3 (C-4), 128.6 (C-5), 118.0 (C-2″), 117.0 (C-6), 116.4 (C-8), 110.2 (C-3), 60.7 (CH2).
Free radical scavenging assay
Radical scavenging activity was determined by modifying method by 2,2-diphenyl-1-picrylhydrazyl (DPPH) assayCitation25,Citation26. The assay solution consisted of 100 µL of (150 µM) 2,2-diphenyl-1-picrylhydrazyl (DPPH), 20 µL of increasing concentration of test compounds and the volume was adjusted to 200 µL in each well with DMSO. This reaction mixture was then incubated for 30 min at room temperature. Ascorbic acid (Vitamin C) was used as a reference inhibitor. The measurements were carried out by using a micro plate reader (OPTIMax, tunable) at 517 nm. The reaction rates were compared and the percent inhibition due to the presence of tested inhibitors was calculated. Each concentration was analyzed in three independent experiments. The IC50 values were determined after serial dilution by the Data analysis and graphing software Origin 8.6, 64-bit (OriginLab Corporation, Northampton, MA).
Mushroom tyrosinase inhibition assay
The mushroom tyrosinase was used for in vitro bioassays as described previously with some modificationsCitation27,Citation28. Briefly, 140 μL of phosphate buffer (20 mM, pH 6.8), 20 μL of mushroom tyrosinase (30 U/mL) and 20 μL of the inhibitor solution were placed in the wells of a 96-well micro plate. After pre-incubation for 10 min at room temperature, 20 μL of L-DOPA (3,4-dihydroxyphenylalanine; 0.85 mM) was added and the plate was further incubated at 25 °C for 20 min. Subsequently, the absorbance of dopachrome was measured at 492 nm using a micro plate reader. Kojic acid was used as a reference inhibitor and phosphate buffer was used as negative control. Each concentration was analyzed in three independent experiments. The IC50 values were determined by the data analysis and graphing software Origin 8.6, 64-bit. The percent of inhibition of tyrosinase reaction was calculated as following:
Here, the B and S are the absorbances for the blank and samples.
Kinetic analysis of the inhibition of tyrosinase
A series of experiments were performed to determine the inhibition kinetics by following this method: for example, for each of five different inhibitor concentrations of compound 4e (0.00, 0.007, 0.014, 0.028 and 0.056 mM, respectively), l-DOPA concentration was varied (0.5, 1.0, 1.5 and 2.0 mM). Pre-incubation and measurement time was the same as discussed above in the “Mushroom tyrosinase inhibition assay” section. Maximal initial velocity was determined from initial linear portion of absorbance up to 5 min after addition of mushroom tyrosinase at a 15 s interval. The inhibition type on the enzyme was assayed by Lineweaver–Burk plots of inverse of velocities (1/V) versus inverse of substrate concentration 1/[S] mM−1, and the inhibition constant Ki was determined by the second plots of the apparent Km/Vmax or 1/Vmax versus the concentration of compound 4e.
Molecular modeling
The experimental X-ray structure of human tyrosinase has not been determined till now. To overcome this problem, a homology model was made with the crystal structure of mushroom tyrosinase (PDB accession code 2ZWE) taken from the research collaborator for structural bioinformatics (RCSB) Protein Data Bank. The attached ligand and water molecules were removed from the model using the AutoDock Tools 1.5.6 (La Jolla, CA). The charges were stabilized and polar hydrogen molecules were added to the protein structure. The ligand molecules were also prepared for docking using the same program. Among the synthesized coumarin analogues compounds 4a, 4c, 4e and 6a were selected for docking studies. The binding pocket of tyrosinase has already been defined with the residues surrounding the dicopper ions since they are implied to the recognition of the l-DOPA substrateCitation29. The values of additional starting conformations per molecule and maximum number of poses per ligand were both expanded to 40 to increase the accuracy of binding mode. Other parameters were based on the software default setting. Molecular docking was, then, carried out by providing appropriate search space details to AutoDock Vina 1.1.2 (La Jolla, CA). The results were obtained in the form of binding affinities in kcal/mol. The docking process was replicated 10 times on 10 separate computing systems to decrease the biased experimentation. The binding modes were visualized using Discovery Studio 4.0 (San Diego, CA). Both the two- and three-dimensional images were studied to ascertain the interacting molecules in the study.
Results and discussion
Chemistry
The umbelliferone derivatives were synthesized by following the previously described methodCitation30 with slight modification shown in Schemes 1 and 2. Chloroacetyl derivative (2) was synthesized by esterification of 7-hydroxyl group of umbelliferrone with chloroacetyl chloride in the presence of anhydrous methylene chloride. The title compounds 4a–g and 6a–b were prepared by simple nucleophilic substitution at 2 with hydroxy substituted benzoic acids (3a–g) and para-substituted cinnamic acids (5a–b), respectively. In the presence of triethylamine as catalyst, the reaction was carried out at room temperature for 24 h. The yields of these compounds were from moderate to good. All of the target compounds possess hydroxyl moieties except compound 6b in which halogen substituent is present to evaluate the role of halogens in tyrosinase inhibition. The structures of all of the synthesized compounds were confirmed by FTIR, 1H and 13C NMR spectroscopic data.
Bioassay for inhibition of tyrosinase activity
The kojic acid is a competitive tyrosinase inhibitor and exhibited a mixed inhibitory effect on diphenolase activity of mushroom tyrosinaseCitation31. Therefore, we used kojic acid as the standard inhibitor of tyrosinase for comparison purposes. In this study, the effect of coumarin derivatives (4a–g) and (6a–b) on the inhibition of tyrosinase activity was measured at six different inhibitor concentrations. When l-DOPA solution was incubated with tyrosinase at 25 °C, its color turned to red-brown because of the formation of dopachrome. In the presence of kojic acid and coumarin derivatives, the color intensity of the solution became faint as the result of enzyme activity inhibition. The IC50 values for each of kojic acid and compounds 4a–g and 6a–b are presented in which shows that the standard kojic acid is more active than all the synthesized coumarin derivatives except compound 4e.
Table 1. The inhibitory effects of coumarin analogues (4a–g) and (6a–b) on mushroom tyrosinase activity: (substrate: L-DOPA).
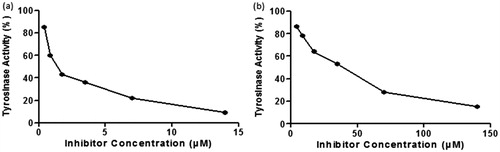
From results, it was observed that compounds 4e, 4c and 6b exhibited potent inhibition of mushroom tyrosinase with an IC50 values ranged from 8.96 to 118.48 µM. Especially, compound 4e and 4c possessing 2,4-dihydroxy and 3,4-dihydroxy phenyl scaffold showed more potent inhibitory activities than the other compounds. Rest of the coumarin analogues showed lower or negligible activities against tyrosinase compared to standard kojic acid. The IC50 value of umbelliferone was also mentioned in table taken from literature and it was found that all of umbelliferone analogues have high potential to inhibit the activity of tyrosinase than umbelliferone. Excellent enzyme inhibition was exhibited by compound 4e as it is more potent than the standard kojic acid. From , it was observed that compound 4e produced same enzyme inhibition at a lower dose than the reference kojic acid which indicates its higher potency than kojic acid.
Figure 2. Effect of compound 4e (a) and kojic acid (b) on the diphenolase activity against mushroom tyrosinase for the catalysis of L-DOPA at 25 °C.
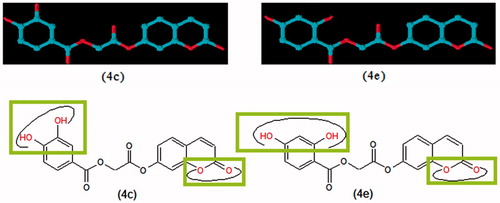
The following general structural features of SAR can be deduced from compounds 4a–g and 6a–b. Compounds 4a–g possess the hydroxyl substituted benzoic acid moiety while (6a–b) have hydroxyl and chloro-substituted cinnamic acid scaffolds. When activities of these scaffolds possessing different substituents viz. monohydroxy, dihydroxy and chloro compared, then it was observed that coumarin analogues with dihydroxy phenyl rings exhibited more potent inhibitory activities than the monohydroxy phenyl rings. In addition, the activities of coumarin derivatives with 2,4- and 3,4-dihydroxy substituted phenyl have different activities than 3,5-dihydroxy phenyl ring. The presence of 4-hydroxy along with the presence of 2 or 3 hydroxy groups on phenyl ring play very important role to form a more tight interaction with mushroom tyrosinase. We also explore the role of halogen atom for the inhibition of activity of tyrosinase enzyme in this study because compound 6b possess 4-chloro while 6a have 4-hydroxy substituted cinnamic acid scaffold. The compound 6b exhibited less enzyme inhibition than compounds with dihydroxy substituted phenyl rings but it showed higher activity than compound 6a which possess 4-hydoxy substituted cinnamic acid residue. Further investigation to explore the type of interactions of halogen with tyrosinase enzyme is needed. The initial structure–activity relationships (SARs) analysis recommended that additional progress in these structural motifs might be of interest. The depicted the three-dimensional view and highlighted the functionalities involved in the tyrosinase inhibition. Our results propose that compounds 4c and 4e may serve as a structural template for the design and development of novel tyrosinase inhibitors.
Figure 3. Three-dimensional view of compounds 4c and 4e with highlighted functionalities involve in enzyme binding.
Kinetic studies
To understand the inhibitory mechanism of compound 4e on mushroom tyrosinase kinetic studies were conducted. The results were presented in and . From these kinetic studies, inhibition mechanism of compound 4e was determined to be non-competitive inhibitor of mushroom tyrosinase from . The inhibition constant Ki (0.0215 mM) was determined from . The value of 1/Vmax is increased to a new value while that of Km remains same which indicated that compound 4e simply lowers the concentration of functional enzyme by a non-competitively binding mode at enzyme. Mushroom tyrosinase having central copper containing active binding sites where most of the competitive inhibitors bind. The non-competitive behavior of compound 4e can be verified by the presence of 2,4-dihydroxy substituted phenyl ring a different structural feature as compared to substrate l-DOPA which possess ortho-diphenol scaffold.
Figure 4. (a) Lineweaver–burk plots for the inhibition of compound 4e on mushroom tyrosinase for catalysis of L-DOPA. Inhibitor concentrations were 0, 0.007, 0.014, 0.028 and 0.056 mM. The (b) represents the secondary plot of 1/Vmax versus concentration of compound 4e to determine the inhibition constant (Ki).
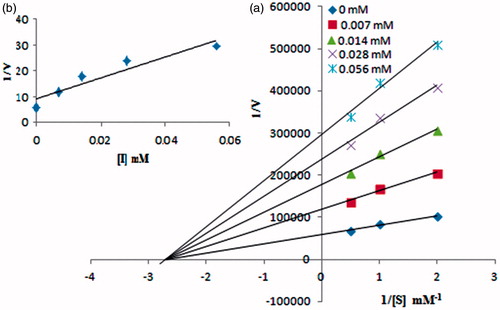
Table 2. Kinetic analysis of compound 4e.
Molecular bocking study
The three-dimensional structure of tyrosinase enables us to gain a better understanding of the tyrosinase inhibition mechanism. Considering compounds 4a, 4c, 4e and 6a having mono- and di-hydroxy substituted phenyl rings, we selected them as representative compounds to study the interaction mode by docking. The docking results were obtained in the form of binding affinities in kcal/mol and average docking values are presented in . The docking results show that compounds 4c and 4e possess highest binding affinity with the enzyme. The potential ligand–protein interaction of compounds 4a, 4c, 4e and 6a with the active site of tyrosinase (generated using Discovery Studio 4.0 are presented in ), respectively. Both two- and three-dimensional views of the ligand–protein interaction are portrayed in figures.
Figure 5. The potential ligand–protein interaction of compound 4a with the active site of tyrosinase (PDB ID 2ZWE) generated using Discovery Studio 4.0. The (A) and (B) show the three-dimensional docking of the compound in the binding pocket. Dashed lines indicate the interactions between the ligand and the amino acids of the receptor. The (C) shows the two dimensional interaction patterns. The legend inset represents the type of interaction between the ligand atoms and the amino acid residues of the protein.
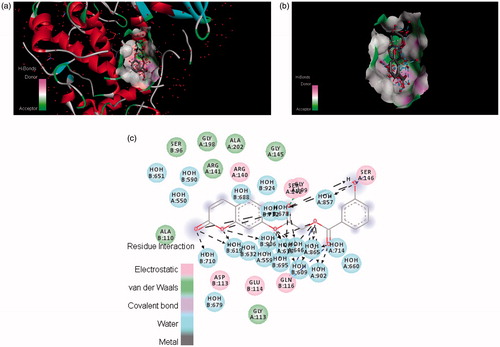
Figure 6. The potential ligand–protein interaction of compound 4c with the active site of tyrosinase (PDB ID 2ZWE) generated by using Discovery Studio 4.0. The (A) and (B) show the three-dimensional docking of the compound in the binding pocket. Dashed lines indicate the interaction between the ligand and the amino acids of the receptor. The (C) shows the two dimensional interaction patterns. The legend inset represents the type of interaction between the ligand atoms and the amino acid residues of the protein.
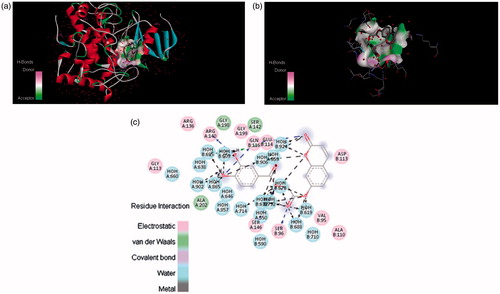
Figure 7. The potential ligand–protein interaction of compound 4e with the active site of tyrosinase (PDB ID 2ZWE) generated using Discovery Studio 4.0. The (A) and (B) show the three-dimensional docking of the compound in the binding pocket. Dashed lines indicate the interaction between the ligand and the amino acids of the receptor. The (C) shows the two dimensional interaction patterns. The legend inset represents the type of interaction between the ligand atoms and the amino acid residues of the protein.
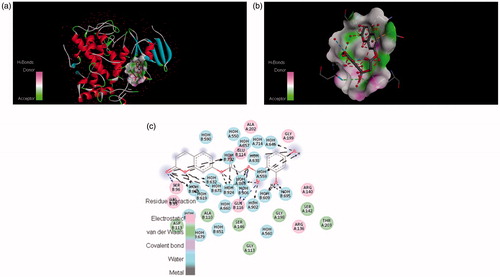
Figure 8. The potential ligand–protein interaction of compound 6a with the active site of tyrosinase (PDB ID 2ZWE) generated using Discovery Studio 4.0. The (A) and (B) show the three-dimensional docking of the compound in the binding pocket. Dashed lines indicate the interaction between the ligand and the amino acids of the receptor. The (C) show the two dimensional interaction patterns. The legend inset represents the type of interaction between the ligand atoms and the amino acid residues of the protein.
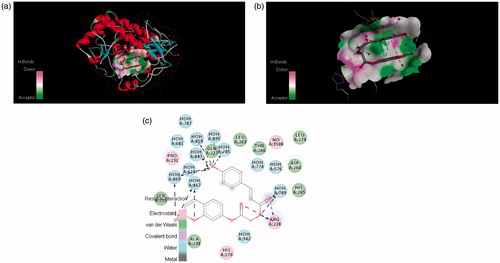
Table 3. The average binding energy calculated after docking of compounds 4a, 4c, 4e and 6a.
In , it was found that compound 4c formed π–π stacks between lactonic phenyl ring of the inhibitors and ASP113 of tyrosinase. There were hydrogen bonds between the 3- and 4-position of phenyl ring of (4c) and the ARG140 and GLN116 residue of the active site of tyrosinase. This result further confirmed that hydroxyl groups in the phenyl ring play a crucial role in the tyrosinase inhibitory activity. The lactonic carbonyl in case of (4e) also involved in hydrogen bonding with SER96 and carboxylic carbonyl form hydrogen bonding with GLN116 amino acid residue of the enzyme . Both of the phenolic hydroxyls present at 2 and 4 position of phenyl in compound 4e tightly bound by HOH609, HOH646 and HOH695 of the target protein.
DPPH radical scavenging assay
The free radical scavenging activity was determined because most of the tyrosinase inhibitors possess antioxidant activity. The interaction of the synthesized compounds 4a–g and 6a–b with the stable free radical DPPH indicates their radical scavenging ability. The synthesized compounds exhibited no radical scavenging potential at low concentration 20 µM but as the concentration is increased up to 100 µM then compound 4f showed antioxidant activity. presented the results and it was found that the interactions of the synthesized coumarin analogues are concentration dependent. All of the synthesized compounds show smaller interactions when compared to the standard ascorbic acid.
Table 4. DPPH free radical scavenging activity of the coumarin analogues (4a–g) and (6a–b).
Conclusion
In conclusion, we describe the synthesis and tyrosinase inhibitory activity of coumarin derivatives (4a–g) and (6a–b). The title compound (4e) showed the excellent tyrosinase inhibitory activity among all the synthesized compounds. The in silico docking results also support our findings. The kinetic analysis of compound 4e demonstrated that it is non-competitive tyrosinase inhibitor with same value of inhibition constant Ki regardless of the concentration. The position of free hydroxyl group –OH on the side chain phenyl ring of compound 4e plays very significant role in masking tyrosinase activity.
Declaration of interest
This work (Grants No. C0036335) was supported by Business for Cooperative R & D between Industry, Academy, and Research Institute and funded by Korea Small and Medium Business Administration in 2012.
References
- Sánchez-Ferrer A, Rodríguez-López JN, García-Cánovas F, GarcíaCarmona F. Tyrosinase: a comprehensive review of its mechanism. Biochim Biophys Acta 1995;1247:1–11
- Matsuura R, Ukeda H, Sawamura M. Tyrosinase inhibitory activity of citrus essential oils. J Agric Food Chem 2006;54:2309–13
- Tripathi RK, Hearing VJ, Urabe K, et al. Mutational mapping of the catalytic activities of human tyrosinase. J Biol Chem 1992;267:23707–12
- Schallreuter KU, Hasse S, Rokos H, et al. Cholesterol regulates melanogenesis in human epidermal melanocytes and melanoma cells. Exp Dermatol 2009;18:680–8
- Wood JM, Decker H, Hartmann H, et al. Senile hair graying: H2O2-mediated oxidative stress affects human hair colour by blunting methionine sulfoxide repair. FASEB J 2009;23:2065–75
- Thanigaimalai P, Hoang TAL, Lee KC, et al. Structural requirement(s) of N-phenylthioureas and benzaldehyde thiosemicarbazones as inhibitors of melanogenesis in melanoma B16 cells. Bioorg Med Chem Lett 2010;20:2991–3
- Yi W, Cao R, Peng W, et al. Synthesis and biological evaluation of novel 4-hydroxybenzaldehyde derivatives as tyrosinase inhibitors. Eur J Med Chem 2010;45:639–46
- Seo SY, Sharma VK, Sharma N. Mushroom tyrosinase: recent prospects. J Agric Food Chem 2003;51:2837–53
- Zhu YJ, Zhou HT, Hu YH, et al. Antityrosinase and antimicrobial activities of 2-phenylethanol, 2-phenylacetaldehyde and 2-phenylacetic acid. Food Chem 2011;124:298–302
- Loizzo MR, Tundis R, Menichini F. Natural and synthetic tyrosinase inhibitors as antibrowning agents: an update. Compr Rev Food Sci Food Safety 2012;11:378–98
- Chang TS. An updated review of tyrosinase inhibitors. Int J Mol Sci 2009;10:2440–75
- Khan MTH. Molecular design of tyrosinase inhibitors: a critical review of promising novel inhibitors from synthetic origins. Pure Appl Chem 2007;79:2277–95
- Masamoto Y, Murata Y, Baba K, et al. Inhibitory effects of esculetin on melanin biosynthesis. Biol Pharm Bull 2004;27:422–5
- Bourgaud F, Hehn A, Larbat R, et al. Biosynthesis of coumarins in plants a major pathway still to be unravelled for cytochrome P450 enzymes. Phytochem Rev 2006;5:293–308
- Mueller RL. First-generation agents: aspirin, heparin and coumarins. Best Pract Res Clin Haematol 2004;17:23–53
- Khode S, Maddi V, Aragade P, et al. Synthesis and pharmacological evaluation of a novel series of 5-(substituted)aryl-3-(3-coumarinyl)-1-phenyl-2-pyrazolines as novel anti-inflammatory and analgesic agents. Eur J Med Chem 2009;44:1682–8
- Belluti F, Fontana G, Bo LD, et al. Design, synthesis and anticancer activities of stilbene-coumarin hybrid compounds: identification of novel proapoptotic agents. Bioorg Med Chem 2010;18:3543–50
- Chimenti F, Bizzarri B, Bolasco A, et al. Synthesis and antihelicobacter pylori activity of 4-(coumarin-3-yl)thiazol-2-ylhydrazone derivatives. J Heterocycl Chem 2010;47:1269–74
- Kostova I. Coumarins as inhibitors of HIV reverse transcriptase. Curr HIV Res 2006;4:347–63
- Neyts J, Clercq DE, Singha R, et al. Structure-activity relationship of new anti-hepatitis C virus agents: heterobicycle-coumarin conjugates. J Med Chem 2009;52:1486–90
- Ostrov DA, Prada JAH, Corsino PE, et al. Discovery of novel DNA gyrase inhibitors by high-throughput virtual screening. Antimicrob Agents Chemother 2007;51:3688–98
- Roussaki M, Kontogiorgis CA, Litina DH, et al. A novel synthesis of 3-aryl coumarins and evaluation of their antioxidant and lipoxygenase inhibitory activity. Bioorg Med Chem Lett 2010;20:3889–92
- Neelakantan S, Raman PV, Tinabaye A. A new and convenient synthesis of 4-methyl-3-phenylcoumarins and 3-phenylcoumarins. Indian J Chem Sect B 1982;21:256–57
- Rescigno A, Sollai F, Pisu B, et al. Tyrosinase inhibition: general and applied aspects. J Enzyme Inhib Med Chem 2002;17:207–18
- Reddy SAM, Mudgal J, Bansal P, et al. Antioxidant, anti-inflammatory and anti-hyperglycaemic activities of heterocyclic homoprostanoid derivatives. Bioorg Med Chem 2011;19:384–92
- Abourashed EA, Mikell JR, Khan IA. Bioconversion of silybin to phase I and II microbial metabolites with retained antioxidant activity. Bioorg Med Chem 2012;20:2784–8
- Seo WD, Ryu YB, Curtis-Long MJ, et al. Evaluation of anti-pigmentary effect of synthetic sulfonylamino chalcone. Eur J Med Chem 2010;45:2010–17
- Saghaie L, Pourfarzam M, Fassihi A, Sartippour B. Synthesis and tyrosinase inhibitory properties of some novel derivatives of kojic acid. Res Pharm Sci 2013;8:233–42
- Matoba Y, Kumagai T, Yamamoto A, et al. Crystallographic evidence that the dinuclear copper center of tyrosinase is flexible during catalysis. J Biol Chem 2006;281:8981–90
- Chu W, Tu Z, McElveen E, et al. Synthesis and in vitro binding of N-phenyl piperazine analogs as potential dopamine D3 receptor ligands. Bioorg Med Chem 2005;13:77–87
- Noh JM, Kwak SY, Seo HS, et al. Kojic acid-amino acid conjugates as tyrosinase inhibitors. Bioorg Med Chem Lett 2009;19:5586–9
- Vilar S, Quezada E, Alcaide C, et al. Quantitative structure vasodilatory activity relationship. Synthesis and “in silico” and “in vitro” evaluation of resveratrol-coumarin hybrids. QSAR Comb Sci 2007;26:317–32