
Abstract
The inhibition of two human cytosolic carbonic anhydrase (hCA, EC 4.2.1.1) isozymes I and II, with some 3,4-dihydroxypyrrolidine-2,5-dione and 3,5-dihydroxybenzoic acid derivatives, were investigated by using the esterase assay, with 4-nitrophenyl acetate (4-NPA) as substrate. Compounds 10–13 showed KI values in the range of 112.7–441.5 μM for hCA I and of 3.5–10.76 μM against hCA II, respectively. These hydroxyl group containing compounds generally were competitive inhibitors. Some hydroxyl group containing compounds investigated here showed effective hCA II inhibitory effects, in the same range as the clinically used sulfonamide acetazolamide, and might be used as leads for generating enzyme inhibitors possibly targeting other CA isoforms which have not been yet assayed for their interactions with such agents.
Introduction
Carbonic anhydrase (CA, EC 4.2.1.1) enzymes are involved in important physiological and pathological functions, such as pH and CO2 homeostasis, respiration and transport of between metabolizing tissues and the lungs, ion secretion in different tissues/organs and biosynthetic reactions (e.g. gluconeogenesis, lipogenesis and ureagenesis)Citation1. CA isoforms are found in a variety of tissues where they participate in several important biological processes such as acid–base balance, respiration, carbon dioxide and ion transport, bone resorption, ureagenesis, gluconeogenesis, lipogenesis and electrolyte secretion. Many CA isozymes involved in these processes are important therapeutic targets with the potential to be inhibited/activated for the treatment of a range of disorders such as edema, glaucoma, obesity, cancer, epilepsy and osteoporosisCitation1–5.
Pyrrolidine derivatives are widely used as prodrugs. Notably, 5-lactams such as pyrrol-2-one and its derivatives have illustrated anti-tumor activity. Recently, N-substituted maleimides and 5-ylidene pyrrol-2(5H)-ones have received growing attentionCitation6–8, due to their biological characteristics and potentials for organic synthesis as key compounds for synthesis of bio-active molecules, which are Oteromycin, Talaroconvolutin A, Azaspirene, Fusarin, Clausenamide, and so onCitation9,Citation10.
Our groups recently investigated the interaction of 12 mammalian CA isozymes with several types of phenolic compounds, such as catechol, resorcinol, a series of phenols, a series of phenolic acids and some other derivativesCitation11–14. Here, we extend these earlier investigations to series of phenolic compounds, some of which are widely used as antioxidant food additives or as drugs. Among the various natural or unnatural phenolic compounds with antioxidant properties, these compounds are very active in quenching reactive oxygen speciesCitation13. They are reported to possess anti-cancer, anti-carcinogenic, anti-mutagenic, anti-bacterial, anti-viral or anti-inflammatory activitiesCitation12,Citation13. Phenol, phenolic compounds and hydroxybenzoic acid derivatives are widely used as prodrugs or drugs. Salicylic acid is known for its ability to ease aches, pains, and reduce fevers. These medicinal properties, particularly fever relief, have been known since ancient times, and it was used as an anti-inflammatory drugCitation12,Citation13.
In the present study, we have purified CA I and II (hCA I and hCA II) from human erythrocytes and examined the in vitro inhibition effects of above mentioned hydroxyl group containing compounds on these enzymes, using the esterase activity of hCA I and II, with 4-NPA as substrate.
Materials and methods
Chemicals
The CNBr-activated Sepharose 4B, protein assay reagents, p-aminobenzene sulphonamide, l-tyrosine, 4-nitrophenylacetate (4-NPA) and chemicals for electrophoresis were purchased from Sigma-Aldrich Co. (St Louis, MO). All other chemicals were of analytical grade and obtained from either Sigma or Merck (Darmstadt, Germany). Reagents for syntheses of compounds 10–13; L-(+)-tartaric acid, benzylamine, aniline, cyclohexylamine, 3,5-dihydroxybenzoic acid and triethylamine were purchased from Aldrich and (benzotriazol-1-yloxy) tris (dimethylamino) phosphonium hexafluorophosphate (BOP reagent) from Alfa Aesar (Ward Hill, MA). Other common chemicals and solvents are commercially available and were used after distillation or treatment with drying agents. Progress of reactions was monitored by thin layer chromatography (Merck, TLC Silica gel 60 F254) in a suitable solvent system. Column chromatography was performed on Merck Silica Gel 60 (70–230 mesh). Melting points were determined with a Reichert thermovar micro melting point apparatus and are uncorrected. Nuclear magnetic resonance (NMR) spectra were taken on a Bruker Ultra Shield Plus 400 operating at 400 MHz for 1H- and 100 MHz for Citation13C-nuclei. NMR spectra were recorded in deutorated dimethyl sulfoxide (DMSO-d6) solvent using TMS as internal standard and chemical shifts referred to δ were expressed in parts per million, ppm. FT-IR spectra were measured with a Perkin–Elmer BXII ATR spectrometer. Elemental analyses were carried out by a Carlo Erba 1106 instrument.
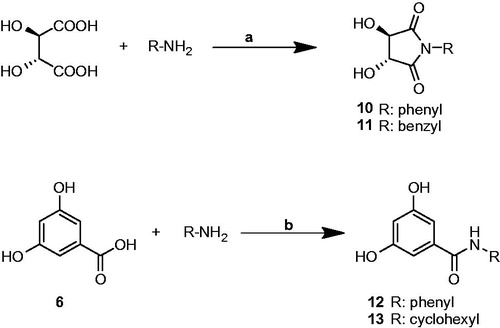
Synthesis of (3R, 4R)-1-benzyl-3,4-dihydroxy-2,5-dioxopyrrolidine (10)
Benzylamine (5.35 g, 0.05 mol) and L-(+)-tartaric acid (7.5 g, 0.05 mol) were refluxed in xylene overnight with a Dean-Stark apparatus. The reaction mixture was cooled to ambient temperature and resulting crystalline product was filtered off. After successive washing with portions of hexane, crude product was recrystallized from ethanol to give product as pale yellow solid, 83%, m.p. 198–200 °C (200–201 °C)Citation15. FT-IR (cm−1): 3189 br, 2885, 1710, 1452, 1390, 1155, 1099, 1003. 1H-NMR (400 MHz, DMSO-d6): δ 7.35–7.23 (m, 5H, ArH), 6.30 (d, J = 5.24 Hz, 2H, –OH), 4.55 (d, J = 7.22 Hz, 2H, –CH, H3 and H4), 4.38 (d, J = 4.62 Hz, 2H, –PhCH2). Citation13C NMR (100 MHz, DMSO-d6): δ 174.6, 135.9, 128.5, 127.5, 127.5, 74.5, 41.1. Anal. found: C, 59.51; H, 5.07; N, 6.27. Calcd for C11H11NO4: C, 59.73; H, 5.01; N, 6.33.
Synthesis of (3R, 4R)-1-phenyl-3,4-dihydroxy-2,5-dioxopyrrolidine (11)
Freshly distilled aniline (4.65 g, 0.05 mol) and L-(+)-tartaric acid (7.5 g, 0.05 mol) were refluxed in xylene with a Dean-Stark apparatus. After overnight reaction, the resulting crystalline product was filtered off and washed several times with hexane. Recrystallization from ethanol provided white crystalline product with 87% yield, m.p. 248–250 °C (249–250 °C).Citation16 FT-IR (cm−1): 3338 br, 3064, 1710, 1497, 1393, 1182, 1103, 999. 1H-NMR (400 MHz, DMSO-d6): δ 7.51–7.39 (m, 3H, ArH), 7.32–7.30 (m, 2H, ArH), 6.39 (dd, J = 4.64 Hz and J = 1.78 Hz, 2H, –OH), 4.57 (dd, J = 4.69 Hz and J = 1.65 Hz, 2H, –CH, H3 and H4). Citation13C-NMR (100 MHz, DMSO-d6): δ 174.0, 132.0, 128.9, 128.3, 126.9, 74.4. Anal. found: C, 58.11; H, 5.51; N, 6.54. Calcd for C10H9NO4: C, 57.97; H, 4.38; N, 6.76.
Synthesis of 3,5-dihydroxy-N-(benzyl)benzamide (12)
3,5-dihydroxybenzoic acid (1.54 g, 10 mmol, 1 eq.) was dissolved in 20 ml of acetonitrile. To solution, triethylamine (4.18 ml, 30 mmol, 3 eq.), (Benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP) (4.42 g, 10 mmol, 1 eq.) and benzylamine (1.07 g, 10 mmol, 1 eq.) were added. The mixture was stirred at room temperature for 6 h. The precipitate was filtered off and then washed with acetonitrile. The filtrate was evaporated under vacuum, taken up with ethyl acetate then washed with 20 ml of 2N HCl solution, 20 ml of saturated NaHCO3 solution and with 20 ml of a saturated solution of NaCl. The product was purified by column chromatography on silica gel eluting with mixture of ethyl acetate-hexane (2:1) to yield a white solid (65%), m.p. 213–215 °C. FT-IR (cm−1): 3380, 3301, 3035, 1655, 1536, 1154, 997. 1H-NMR (400 MHz, DMSO-d6): δ 9.48 (s, 2H, –OH), 8.83 (t, J = 6.03 Hz, 1H, –NH), 7.33–7.19 (m, 5H, ArH), 6.70(d, J = 2.15 Hz, 2H, H2 and H6), 6.35 t, J = 2.15 Hz, 1H, H4), 4.40(d, J = 6.03 Hz, 2H, PhCH2). Citation13C-NMR (100 MHz, DMSO-d6): δ 166.5, 158.3, 139.9, 136.6, 128.2, 127.1, 126.6, 105.4, 105.1, 42.5. anal. found: C, 68.87; H, 5.64; N, 5.60. Calcd for C14H13NO3: C, 69.12; H, 5.39; N, 5.76.
Synthesis of 3,5-dihydroxy-N-(cyclohexyl)benzamide (13)
In a flask, 3,5-dihydroxybenzoic acid (1.54 g, 10 mmol, 1 eq.) was dissolved in 20 ml of acetonitrile, and triethylamine (4.18 ml, 30 mmol, 3 eq.), (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP) (4.42 g, 10 mmol, 1 eq.) and cyclohexylamine (0.99 g, 10 mmol, 1 eq.) were added. The mixture was stirred at room temperature for 6 h and then the precipitate was filtered off by washing with acetonitrile. The filtrate was evaporated under vacuum and taken up with ethyl acetate. Successive washings with 20 ml of a solution of 2N HCl, 20 ml of saturated NaHCO3 solution and 20 ml of a saturated solution NaCl provided the crude. The product was purified by chromatography on silica (eluent: EtOAc/hexane: 2/1) to yield a white solid (69%). Melting point 203–205 °C. FT-IR (cm−1): 3382, 3281, 1636, 1553, 1149, 858. 1H-NMR (400 MHz, DMSO-d6): δ 9.39 (s, 2H, –OH), 7.97 (t, J = 8.02 Hz, 1H, –NH), 6.64 (d, J = 2.14 Hz, 2H, H2 and H6), 6.32(t, J = 2.10 Hz, 1H, H4), 1.76–1.04 (m, 11H, aliphatic hydrogens). Citation13C-NMR (100 MHz, DMSO-d6): δ 165.6, 158.1, 137.1, 105.4, 104.8, 48.1, 32.3, 25.2, 24.9. Anal. found: C, 66.81; H, 7.45; N, 5.87. Calcd for C13H17NO3: C, 66.36; H, 7.28; N, 5.95.
CA purification assay
Purification of two human CA isozymes (hCA I and hCA II) were previously described with a simple one-step method by a Sepharose-4Baniline-sulfanilamide affinity column chromatography Citation17,Citation18.
CA activity assay and kinetic studies
CA activity was assayed by following the change in absorbance at 348 nm of 4-NPA to 4-nitrophenolate ion over a period of 3 min at 25 °C using a spectrophotometer (Shimadzu UV–VIS, Kyoto, Japan) according to the method described by Verpoorte et al.Citation19 The inhibitory effects of compounds 1–13 and AZA were examined. All compounds were tested in triplicate at each concentration used. Control cuvette activity in the absence of inhibitor was taken as 100%. For each inhibitor an Activity %−[Inhibitor] graph was drawn. To determine KI values, three different inhibitor concentrations were tested; In these experiments, 4-NPA was used as substrate at five different concentrations (0.15–0.75 mM). The Lineweaver–Burk curves were drawnCitation20. Regression analysis graphs were drawn for IC50 using inhibition % values by a statistical package (SPSS for windows, version 10.0; SPSS Inc., Chicago, IL) on a computer (Student’s t-test; n: 3).
Results and discussion
The rationale of investigating phenols as CA inhibitors (CAIs) is due to the fact that the simple phenol (PhOH) has been shown to be the only competitive inhibitor with CO2 as substrate for the main isoform of CA, i.e. human CA II (hCA II)Citation14. The X-ray crystal structure for the adduct of hCA II with phenolCitation14. The phenyl moiety of phenol was found to lay in the hydrophobic part of the hCA II active site, where CO2, the physiologic substrate of the CAs, binds in the precatalytic complex, explaining thus the behavior of phenol as a unique CO2 competitive inhibitor. Only recently, our groups investigated the interactions of some simple phenols, salicylic acid derivatives, some antioxidant phenolic compounds, some natural product polyphenols and phenolic acids with all mammalian isozymes, CA I–XVCitation11–14, evidencing some low micromolar/submicromolar inhibitors as well as the possibility to design isozyme selective CAIs. Indeed, the inhibition profile of various isozymes with this class of agents is very variable, with inhibition constants ranging from the millimolar to the submicromolar range for many simple phenolsCitation11–13.
(3R, 4R)-3,4-dihydroxy-2,5-dioxopyrrolidine derivatives (10 and 11) were synthesizedCitation15,Citation16 by the reaction of L-(+)-tartaric acid with primary amines. Benzamide structures (12 and 13) were established (Scheme 1) by coupling 3,5-dihydroxybenzoic acid with primary amines using BOP (benzotriazol-1-yloxy-tris (dimethylamino)-phosphoniumhexafluorophosphate) as coupling reagent. Hydroxyl groups of benzoic acid were not protected since BOP coupling is safely mediated in the presence of unprotected alkyl and phenolic hydroxyl groups in many examplesCitation21–23.
Scheme 1. Reagents and conditions. (a) xylene reflux; (b) acetonitrile, rt, TEA, BOP reagent.
We report here the first study on the inhibitory effects of compounds 10–13 on the esterase activity of hCA I and II. The previous reports by Innocenti et al.Citation11 investigated other phenol derivatives (including catechol and resorcinol) by using a stopped flow, CO2 hydration assay for monitoring CA inhibition. Data of show the following regarding inhibition of hCA I and II with compounds 10–13, by an esterase assayCitation19, with 4-NPA as substrate:
Table 1. hCA I and II inhibition data some compounds, by an esterase assay with 4-NPA as substrateCitation19.
(i) Against the slow cytosolic isozyme hCA I, compounds 10–13 behave as good inhibitors (), with KI values in the range of 112.7–441.5 μM similarly to the structurally related compounds 1–8 and acetazolamide (AZA) (KIs of 0.55–4003 µM). It is interesting to note that the compounds 10 and 11 were much better hCA I inhibitors as compared to the corresponding compounds 12 and 13 from which they were prepared. Kinetic investigations (Lineweaver–Burk plots, data not shown) indicate that similarly to sulfonamides and inorganic anionsCitation3–5,Citation17–20, all the investigated natural compounds act as competitive inhibitors with 4-NPA as substrate, i.e. they bind in different regions of the active site cavity as compared to the substrate. However, the binding site of 4-NPA itself is unknown, but it is presumed to be in the same region as that of CO2, the physiological substrate of this enzymeCitation11–14.
Figure 1. Structure of tested compounds.
(ii) A better inhibitory activity has been observed with compounds 10–13 investigated here for the inhibition of the rapid cytosolic isozyme hCA II (). Structure–activity relationship (SAR) is thus quite sharp for this small series of hydroxylic compounds: the 2,5-dioxopyrrolidine containing compounds 10 and 11 are ineffective leads, with two 3, 5 dihidroxy moieties is already a submicromolar hCA II inhibitor. This trend is maintained when different groups are present in the meta position to the phenol OH moiety, such as in resorcinol. The best hCA II inhibitor in this series of derivatives were compounds 10 and 11, which with a KI of 8.0–3.5 µM. It must be stressed that KIs measured with the esterase method are always in the micromolar range because hCA I and II are weak esterases.Citation24–27
In a recent study, it was reported that catechol and resorcinol Citation26 a simple compound lack of the sulfonamide, sulfamate, or related functional groups which are typically found in all known CAIs – act as a CAI inhibitor, and could represent the starting point for a new class of inhibitors that may have advantages for patients with sulfonamide allergiesCitation24–30. The sulfonamide zinc-binding group is thus superior to the thiol one (from the thioxolone hydrolysis product) for generating CAIs with a varied and sometimes isozyme-selective inhibition profile against the mammalian enzymes. However, it is critically important to explore further classes of potent CAIs in order to detect compounds with a different inhibition profile as compared to the sulfonamides and their bioisosteres and to find novel applications for the inhibitors of these widespread enzymes.
Conclusion
Compounds 10–13 used in this study affect the activity of CA isozymes due to the presence of the different functional groups (OH, phenyl, benzyl, and cyclohexyl) present in their aromatic scaffold. Our findings here indicate thus another class of possible CAIs of interest, in addition to the well-known sulfonamides/sulfamates/sulfamides, the phenols/biphenyl diphenols bearing bulky ortho moieties in their molecules. Indeed, some hyroxylic compounds investigated here showed effective hCA I and II inhibitory activity, in the low micromolar range, by the esterase method which usually gives KI-s an order of magnitude higher as compared to the CO2 hydrase assayCitation30–45. These findings point out that substituted hydroxylic compounds may be used as leads for generating potent CAIs eventually targeting other isoforms which have not been assayed yet for their interactions with such agents.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the article.
References
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81
- Hilvo M, Baranauskiene L, Salzano AM, et al. Biochemical characterization of CA IX, one of the most active carbonic anhydrase isozymes. J Biol Chem 2008;283:27799–809
- Supuran CT, Scozzafava A. Carbonic anhydrase inhibitors and their therapeutic potential. Expert Opin Ther Pat 2000;10:575–600
- Alterio V, Di Fiore A, D’Ambrosio K, Supuran CT, De Simone G. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68
- Cavdar H, Ekinci D, Talaz O, et al. α-Carbonic anhydrases are sulfatases with cyclic diol monosulfate esters. J Enzyme Inhib Med Chem 2012;27:148–54
- Kiren S, Hong X, Levertt CA, Padwa A. A facile synthesis of 5-alkoxypyrrol-2(5H)-ones using a modified aza-Achmatowicz oxidation. Tetrahedron 2009;65:6720–9
- Clayden J, Watson DW, Helliwel M, Chambers M. Beta-lactams or gamma-lactams by 4-exo-trig or 5-endo-trig anionic cyclisation of lithiated acrylamide derivatives. Chem Commun 2003;25:82–3
- Alizadeh A, Rezvanian A, Zhu LG. One-pot synthesis of 3-benzoyl-5-hydroxy-4-(alkylamino)-5-phenyl-1-(arylsulfonyl)-1,5-dihydro-2H-pyrrol-2-ones via multicomponent reaction. Helv Chim Acta 2007;90:2414–20
- Snider BB, Neubert SB. A novel biomimetic route to the 3-Acyl-5-hydroxy-3-pyrrolin-2-one and 3-Acyl-3,4-epoxy-5-hydroxy-pyrrolidin-2-one ring systems. J Org Chem 2004;69:8952–5
- Feng Z, Li X, Zheng G, Huang L. Synthesis and activity in enhancing long-term potentiation (LTP) of clausenamide stereoisomers. Bioorg Med Chem Lett 2009;19:2112–15
- Innocenti A, Vullo D, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: Interactions of phenols with the 12 catalytically active mammalian isoforms (CA I-XIV). Bioorg Med Chem Lett 2008;18:1583–7
- Cakmak R, Durdagi S, Ekinci D, et al. Design, synthesis and biological evaluation of novel nitroaromatic compounds as potent glutathione reductase inhibitors. Bioorg Med Chem Lett 2011;21:5398–402
- Sentürk M, Gülçin I, Dastan A, et al. Carbonic anhydrase inhibitors. Inhibition of human erythrocyte isozymes I and II with a series of antioxidant phenols. Bioorg Med Chem 2009;17:3207–11
- Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2010;20:3467–74
- Gonsalves AMAR, Serra MES, Murtinho D, et al. Pyrrolidine-based amino alcohols: novel ligands for the enentioselective alkylation of benzaldehyde. J Molec Cat A: Chem 2003;195:1–9
- De Simone G, Alterio V, Supuran CT. Exploiting the hydrophobic and hydrophilic binding sites for designing carbonic anhydrase inhibitors. Expert Opin Drug Discov 2013;8:793–810
- Akin Kazancioglu E, Guney M, Senturk M, Supuran CT. Simple methanesulfonates are hydrolyzed by the sulfatase carbonic anhydrase activity. J Enzyme Inhib Med Chem 2012;27:880–5
- Ekinci D, Cavdar H, Talaz O, et al. NO-releasing esters show carbonic anhydrase inhibitory action against human isoforms I and II. Bioorg Med Chem 2010;18:3559–63
- Verpoorte JA, Mehta S, Edsall JT. Esterase activities of human carbonic anhydrases B and C. J Biol Chem 1967;242:4221–9
- Lineweaver H, Burk D. The determination of enzyme dissociation constants. J Am Chem Soc 1934;56:658–66
- Fournier A, Wang CT, Felix AM. Applications of BOP reagent in solid phase synthesis: Advantages of BOP reagent for difficult couplings exemplified by a synthesis of [Ala15]-GRF-(1-29)-NH2. Int J Pep Pro Res 1988;31:86–97
- Sawayama AM, Tanaka H, Wandless TJ. The total synthesis of ustiloxin D and considerations on the origin of selectivity of the asymmetric allylic alkylation. J Org Chem 2004;69:8810–20
- Howe J, Quibell M, Johnson T. A new generation of reversible backbone-amide protection for the solid phase synthesis of difficult sequences. Tetrahedron Lett 2000;41:3997–4001
- Oztürk Sarikaya SB, Topal F, Sentürk M, et al. In vitro inhibition of a-carbonic anhydrase isozymes by some phenolic compounds. Bioorg Med Chem Lett 2011;21:4259–62
- Alp C, Ekinci D, Gültekin MS, et al. A novel and one-pot synthesis of new 1-tosyl pyrrol-2-one derivatives and analysis of carbonic anhydrase inhibitory potencies. Bioorg Med Chem 2010;18:4468–74
- Sentürk M, Gülçin I, Beydemir S, et al. In vitro inhibition of human carbonic anhydrase I and II isozymes with natural phenolic compounds. Chem Biol Drug Des 2011;77:494–9
- Ekinci D, al-Rashida M, Abbas G, et al. Chromone containing sulfonamides as potent carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:744–7
- Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72
- Masini E, Carta F, Scozzafava A, Supuran CT. Antiglaucoma carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Pat 2013;23:705–16
- Ekinci D, Cavdar H, Durdagi S, et al. Structure-activity relationships for the interaction of 5,10-dihydroindeno[1,2-b]indole derivatives with human and bovine carbonic anhydrase isoforms I, II, III, IV and VI. Eur J Med Chem 2012;49:68–73
- Ekinci D, Ceyhun SB, Senturk M, et al. Characterization and anions inhibition studies of an α-carbonic anhydrase from the teleost fish Dicentrarchus labrax. Bioorg Med Chem 2011;19:744–8
- Ekinci D, Kurbanoglu NI, Salamci E, et al. Carbonic anhydrase inhibitors: inhibition of human and bovine isoenzymes by benzenesulphonamides, cyclitols and phenolic compounds. J Enzyme Inhib Med Chem 2012;27:845–8
- Balaydin HT, Durdagi S, Ekinci D, et al. Inhibition of human carbonic anhydrase isozymes I, II and VI with a series of bisphenol, methoxy and bromophenol compounds. J Enzyme Inhib Med Chem 2012;27:467–75
- Supuran CT. Carbonic anhydrases: from biomedical applications of the inhibitors and activators to biotechnologic use for CO2 capture. J Enzyme Inhib Med Chem 2013;28:229–30
- Durdagi S, Scozzafava G, Vullo D, et al. Inhibition of mammalian carbonic anhydrases I-XIV with grayanotoxin III: solution and in silico studies. J Enzyme Inhib Med Chem 2014;29:469–75
- Bilginer S, Unluer E, Gul HI, et al. Carbonic anhydrase inhibitors. Phenols incorporating 2- or 3-pyridyl-ethenylcarbonyl and tertiary amine moieties strongly inhibit Saccharomyces cerevisiae β-carbonic anhydrase. J Enzyme Inhib Med Chem 2014;29:495–9
- Ekinci D, Karagoz L, Ekinci D, et al. Carbonic anhydrase inhibitors: in vitro inhibition of α isoforms (hCA I, hCA II, bCA III, hCA IV) by flavonoids. J Enzyme Inhib Med Chem 2013;28:283–8
- Supuran CT, Maresca A, Gregáň F, Remko M. Three new aromatic sulfonamide inhibitors of carbonic anhydrases I, II, IV and XII. J Enzyme Inhib Med Chem 2013;28:289–93
- Alp C, Maresca A, Alp NA, et al. Secondary/tertiary benzenesulfonamides with inhibitory action against the cytosolic human carbonic anhydrase isoforms I and II. J Enzyme Inhib Med Chem 2013;28:294–8
- Del Prete S, De Luca V, Scozzafava A, et al. Biochemical properties of a new α-carbonic anhydrase from the human pathogenic bacterium Vibrio cholerae. J Enzyme Inhib Med Chem 2014;29:23–7
- Singh S, Supuran CT. 3D-QSAR CoMFA studies on sulfonamide inhibitors of the Rv3588c β-carbonic anhydrase from Mycobacterium tuberculosis and design of not yet synthesized new molecules. J Enzyme Inhib Med Chem 2014;29:449–55
- Maresca A, Vullo D, Scozzafava A, Supuran CT. Inhibition of the alpha- and beta- carbonic anhydrases from the gastric pathogen Helycobacter pylori with anions. J Enzyme Inhib Med Chem 2013;28:388–91
- Maresca A, Scozzafava A, Vullo D, Supuran CT. Dihalogenated sulfanilamides and benzolamides are effective inhibitors of the three β-class carbonic anhydrases from Mycobacterium tuberculosis. J Enzyme Inhib Med Chem 2013;28:384–7
- Maresca A, Carta F, Vullo D, Supuran CT. Dithiocarbamates strongly inhibit the beta-class carbonic anhydrases from Mycobacterium tuberculosis. J Enzyme Inhib Med Chem 2013;28:407–11
- Allouche F, Chabchoub F, Carta F, Supuran CT. Synthesis of aminocyanopyrazoles via a multi-component reaction and anti-carbonic anhydrase inhibitory activity of their sulfamide derivatives against cytosolic and transmembrane isoforms. J Enzyme Inhib Med Chem 2013;28:343–9