
Abstract
A small series of C-glycosides containing the phenol moiety was tested for the inhibition of the β-class carbonic anhydrases (βCAs, EC 4.2.1.1) from Brucella suis. Many compounds showed activities in the micromolar or submicromolar range and excellent selectivity for pathogen CAs over human isozymes. Glycosides incorporating the 3-hydroxyphenyl moiety showed the best inhibition profile, and therefore this functionality represents lead for the development of novel anti-infectives with a new mechanism of action.
Introduction
Brucella spp. are facultative intracellular pathogens responsible of widespread zoonosis, known as brucellosis or Malta fever. Brucellae are Gram-negative α-proteobacteria, infecting various vertebrates, from fish to primates. Brucellosis is difficult to fight, as these bacteria have developed strategies to evade immune recognition by the host. The bacterium is able to cause enormous losses in agriculture and is endemic in several areas such as the Mediterranean Europe, Middle East and Latin America.
Among the many antibacterial drug targets available so far from bacterial genomics studies, metalloenzymes are highly attractive as they provide an excellent opportunity for mechanism-based drug discovery of novel classes of antibioticsCitation1,Citation2. In this framework, carbonic anhydrases have recently emerged as promising anti-infective targets. Indeed, several bacterial β-class carbonic anhydrases (β-CA) representatives have been cloned and characterized in some pathogens such as, among others, Helicobacter pylori and Mycobacterium tuberculosisCitation3,Citation4.
The genome of the bacterial pathogen Brucella suis contains two CAs belonging to the β-class: bsCA I and bsCA IICitation5,Citation6. These two CAs were shown to be catalytically efficient, with activity for the CO2 hydration reaction similar to that of the human (h) isoform hCA II and are inhibited by many sulfonamides/sulfamatesCitation2. Furthermore, certain sulfonamide carbonic anhydrase inhibitors (CAIs) were shown to inhibit the bacterial growth in cell culturesCitation7.
The use of glycomimetics in the design of CAIs has proven to be a successful approach and now constitutes one of the most attractive ways to develop new generations of effective and selective inhibitorsCitation8–10. Winum’s group has previously reported that N-(4-sulfamoylphenyl)-β-D-glycopyranosylamines are very efficient inhibitors of the B. suis growthCitation7. Recently. our group has applied the “sugar approach” to the preparation of inhibitors of the M. tuberculosis β-CAs, which leads to the identification of the first mtCAs inhibitor with antimycobacterial activityCitation11,Citation12. Exploring alternative chemotypes to the usual CAIs, we developed a novel series of C-glycosides containing the methoxyaryl scaffold and investigated them as inhibitors against human and bacterial isozymes of carbonic anhydrase, allowing us to identify six potent and highly selective inhibitors of bsCA I and IICitation13,Citation14.
In the search of non-sulfonamide CAIs belonging to different classes of compounds, we report in this study the synthesis of a series of C-glycosides incorporating the phenol moiety, and their inhibitory activity against the off-target hCA I and II, and B. suis β-CAs.
Materials and methods
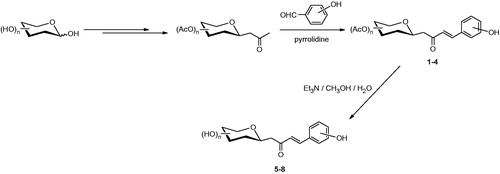
C-glycosides 1–8 were previously described and have been prepared by aldol reaction of aryl aldehydes with per-O-acetylated C-glucosyl or C-galactosyl ketones and subsequent deprotection using triethylamine in methanol/waterCitation11.
An Applied Photophysics (Surrey, UK) stopped-flow instrument has been used for assaying the CA-catalyzed CO2 hydration activityCitation15. Phenol red (at a concentration of 0.02 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5) as buffer, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled-deionized water, and dilutions up to 0.01 nM were done thereafter with distilled–deionized water. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E-I complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3, and the Cheng–Prusoff equationCitation16 as reported earlier and represent the mean from at least three different determinations.
Growth experiments with B. suis were performed as follows: For inoculation, bacteria, from a stationary phase, overnight culture in tryptic soy broth, were washed once with minimal medium prior to a dilution 1:100 in 3 ml of minimal mediumCitation17. Growth was performed under shaking at 170 rpm/37 °C in the absence or in the presence of various drugs at a final concentration of 100 µM or 200 µM. The growth of the bacteria was followed by measuring the optical density at 600 nm for a period of eight days.
Results and discussion
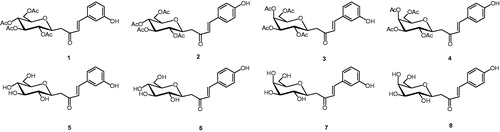
A set of C-cinnamoyl glycosides () was synthesized as outlined in Scheme 1 and described previously by usCitation11. C-cinnamoyl glycosides 1–4 have been prepared by aldol condensation of β-C-glucosyl and β-C-galactosyl ketones with 3-hydroxy or 4-hydroxybenzaldehyde at room temperature in the presence of pyrrolidine as catalyst. The O-acetate protecting groups of the carbohydrate moiety were next removed using triethylamine in methanol/water to afford the deprotected C-glycosides 5–8.
Figure 1. Peracetylated C-glycosides (1–4) and fully deprotected derivatives (5–8).
Scheme 1. Preparation of C-cinnamoyl glycosides 1–8.
The inhibitory activity of the C-glycosides 1–8 against human CA I and CA II and the purified pathogen β-CAs, bsCA I and bsCA II, are listed in . These inhibition data were acquired using a stopped flow assay that monitors the physiological reaction, that is, the CA-catalyzed hydration of CO2.
Table 1. Inhibition of hCA isoforms I, II and bsCA 1 and 2 with C-glycosides 1–8 and acetazolamide (AAZ; 5-acetamido-1,3,4-thiadiazole-2-sulfonamide) as standard, by a stopped-flow CO2 hydrase assay*.
A number of structure–activity relationships were identified in this study and are summarized as follows.
All investigated C-glycosides 1–8 were efficient, micromolar hCA I and hCA II inhibitors with inhibition constants in the range of 3.6–9.3 µM (CA I) and of 3.1–8.8 µM (CA II).
B. suis enzyme, bsCA I, was inhibited by the C-glycosides investigated in this study in micromolar or submicromolar range, with Ki values in the range of 0.68–7.92 µM. It is significant to note that the protected C-glycosides containing the 3-hydroxyphenyl moiety (1 and 3) showed to be the most efficient inhibitors of bsCA I.
The inhibition profile for bsCA II lays in two distinct groups. The first group includes compounds 1, 3, 5 and 7 with Kis of 0.63–0.83 µM, while glycosides 2, 4, 6 and 8 (second group) were less effective bsCA II inhibitors with Kis in the range 2.68–4.85 µM. It is important to note that the position of hydroxyl in the aromatic ring influences the inhibition profile, with the 3-hydroxyphenyl derivatives been more effective inhibitors. Neither the stereochemistry presented by the differing carbohydrate moiety nor the nature of the carbohydrate hydroxyl groups, either as the C-glycosides (5–8) or less polar and bulkier acetylated sugar (1–4), impacted to alter enzyme inhibition characteristics.
Selectivity for inhibiting the B. suis isozymes (bs CA I and II) over the human cytosolic forms (hCA I and II) is a key issue when designing bacterial CAIsCitation2. As can be seen in , several compounds showed better activity profile against β-CAs over α-CAs, which is highly desirable when only the bacterial isoforms would be targeted. The selectivity ratios listed in show that protected C-glycosides 1 and 3 were up to 12-fold selective for Brucella CAs over human CA I and CA II and thus may represent leads for better discriminating the inhibition of β-CAs from this pathogen. This observation provides a compelling opportunity to explore the 3-hydroxyphenyl moiety scaffold in the development of potent and selective glycosyl inhibitors for the bs CAs. Clearly, the deprotected glycosides containing this scaffold (5 and 7) were less effective in this respect. C-glycosides incorporating the 4-hydroxyphenyl moiety (2, 4, 6 and 8) showed almost no selectivity and are not useful in the design of selective inhibitors.
Table 2. Selectivity ratios of Ki for β-CAs compared to human α-CA isozymes I and II for the C-glycosides 1–8*.
We have investigated the effect of our compounds on the growth of B. suis in cell cultures. C-glycosides 1–8 showed no significant inhibition of the bacterial growth after eight days of culture both at the concentration of 100 µM as well as at 200 µM. Although the C-glycosides 1–8 are lipophilic, they do not penetrate the bacterial cell walls. This could be explained in terms of their topological polar surface area (TPSA)Citation18,Citation19. Molecules with a TPSA greater than 140 A2 are likely to have low capacity for penetrating cell membranes, while those with TPSA ≤ 60 A2 have good passive permeability properties. The calculated TPSA for the protected C-glycoside 1–4 is 152 A2, while for the deprotected compounds 5–8, the calculated are somewhat lower (127 A2). This molecular property shows that all compounds fall within the range indicative of molecules with poor membrane permeability.
In conclusion, we have investigated the enzyme inhibition profile of a series of C-glycosides incorporating the phenol moiety (compounds 1–8) against a panel of CAs encompassing the human α-CAs I and II and the pathogenic B. suis enzymes. Inhibition of bacterial CAs is indeed a topic of great interest, with the possibility to uncover novel mechanisms of pathogenesis or classes of antibiotics with new characteristics, distinct of the classical agents for which drug resistance problems emergedCitation20–29. The 3-hydroxyphenyl glycosides preferentially inhibited pathogen CAs over human CAs showing that this moiety therefore represents a lead for the development of novel anti-infectives with a new mechanism of action. The tested compounds showed no inhibition of the bacterial growth probably due to their large sizes.
Declaration of interest
The authors report no declarations of interest.
This work was financed, in part, by an EU grant (Dynano) to CTS, CNRS/CNR (CoopIntEER program, grant no. 131999) to J-Y. W. and by UNLP and CONICET (Argentina). P. A. C. is member of the Scientific Research Career of CONICET.
References
- Lopez M, Köhler S, Winum JY. Zinc metalloenzymes as new targets against the bacterial pathogen Brucella. J Inorg Biochem 2012;111:138–45
- Capasso C, Supuran CT. Anti-infective carbonic anhydrase inhibitors: a patent and literature review. Expert Opin Ther Pat 2013;23:693–704
- Nishimori I, Onishi S, Takeuchi H, Supuran CT. The α and β classes carbonic anhydrases from Helicobacter pylori as novel drug targets. Curr Pharm Des 2008;14:622–30
- Nishimori I, Minakuchi T, Maresca A, et al. The β-carbonic anhydrases from Mycobacterium tuberculosis as drug targets. Curr Pharm Des 2010;16:3300–9
- Joseph P, Turtaut F, Ouahrani-Bettache S, et al. Cloning, characterization, and inhibition studies of a β-carbonic anhydrase from Brucella suis. J Med Chem 2010;53:2277–85
- Joseph P, Ouahrani-Bettache S, Montero JL, et al. A new β-carbonic anhydrase from Brucella suis, its cloning, characterization, and inhibition with sulfonamides and sulfamates, leading to impaired pathogen growth. Bioorg Med Chem 2011;19:1172–8
- Vullo D, Nishimori I, Scozzafava A, et al. Inhibition studies of a β-carbonic anhydrase from Brucella suis with a series of water soluble glycosyl sulfanilamides. Bioorg Med Chem Lett 2010;20:2178–82
- Colinas PA. Novel glycomimetics: anomeric and N-glycosyl sulfonamides. Curr Org Chem 2012;16:1670–6
- Winum JY, Colinas PA, Supuran CT. Glycosidic carbonic anhydrase IX inhibitors: a sweet approach against cancer. Bioorg Med Chem 2013;21:1419–26
- Colinas PA, Núñez NA, Bravo RD. Sulfonamidoglycosylation of methyl glycosides employing perchloric acid supported on silica gel. J Carbohydr Chem 2008;27:141–7
- Riafrecha LE, Rodríguez OM, Vullo D, et al. Synthesis of C-cinnamoyl glycosides and their inhibitory activity against mammalian carbonic anhydrases. Bioorg Med Chem 2013;21:1489–94
- Buchieri MV, Riafrecha LE, Rodríguez OM, et al. Inhibition of the β-carbonic anhydrases from Mycobacterium tuberculosis with C-cinnamoyl glycosides: identification of the first inhibitor with anti-mycobacterial activity. Bioorg Med Chem Lett 2013;23:740–3
- Riafrecha LE, Rodríguez OM, Vullo D, et al. Attachment of carbohydrates to methoxyaryl moieties leads to highly selective inhibitors of the cancer associated carbonic anhydrase isoforms IX and XII. Bioorg Med Chem 2014;22:5308–14
- Riafrecha LE, Vullo D, Supuran CT, Colinas PA. C-glycosides incorporating the 6-methoxy-2-naphthyl moiety are selective inhibitors of fungal and bacterial carbonic anhydrases. J Enzyme Inhib Med Chem. [Epub ahead of print]. DOI:10.3109/14756366.2014.967233
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73
- Yung-Chi C, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 1973;22:3099–108
- Hanna N, Ouahrani-Bettache S, Drake KL, et al. Global Rsh-dependent transcription profile of Brucella suis during stringent response unravels adaptation to nutrient starvation and cross-talk with other stress responses. BMC Genomics 2013;14:459
- Rodríguez OM, Maresca A, Témpera CA, et al. N-β-Glycosyl sulfamides are selective inhibitors of the cancer associated carbonic anhydrase isoforms IX and XII. Bioorg Med Chem Lett 2011;21:4447–50
- Ertl P, Rohde B, Selzer P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J Med Chem 2000;43:3714–17
- Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Disc 2008;7:168–81
- Supuran CT. Carbonic anhydrases: from biomedical applications of the inhibitors and activators to biotechnologic use for CO2 capture. J Enzyme Inhib Med Chem 2013;28:229–30
- Capasso C, Supuran CT. Sulfa and trimethoprim-like drugs-antimetabolites acting as carbonic anhydrase, dihydropteroate synthase and dihydrofolate reductase inhibitors. J Enzyme Inhib Med Chem 2014;29:379–87
- Bilginer S, Unluer E, Gul HI, et al. Carbonic anhydrase inhibitors. Phenols incorporating 2- or 3-pyridyl-ethenylcarbonyl and tertiary amine moieties strongly inhibit Saccharomyces cerevisiae β-carbonic anhydrase. J Enzyme Inhib Med Chem 2014;29:495–9
- Supuran CT. Bacterial carbonic anhydrases as drug targets: towards novel antibiotics? Front Pharmacol 2011;2:34
- Del Prete S, De Luca V, Scozzafava A, et al. Biochemical properties of a new α-carbonic anhydrase from the human pathogenic bacterium Vibrio cholerae. J Enzyme Inhib Med Chem 2014;29:23–7
- Maresca A, Vullo D, Scozzafava A, Supuran CT. Inhibition of the alpha- and beta- carbonic anhydrases from the gastric pathogen Helycobacter pylori with anions. J Enzyme Inhib Med Chem 2013;28:388–91
- Maresca A, Scozzafava A, Vullo D, Supuran CT. Dihalogenated sulfanilamides and benzolamides are effective inhibitors of the three β-class carbonic anhydrases from Mycobacterium tuberculosis. J Enzyme Inhib Med Chem 2013;28:384–7
- Maresca A, Carta F, Vullo D, Supuran CT. Dithiocarbamates strongly inhibit the beta-class carbonic anhydrases from Mycobacterium tuberculosis. J Enzyme Inhib Med Chem 2013;28:407–11