
Abstract
Several (thiazol-2-yl)hydrazone derivatives from 2-, 3- and 4-acetylpyridine were synthesized and tested against human monoamine oxidase (hMAO) A and B enzymes. Most of them had an inhibitory effect in the low micromolar/high nanomolar range, being derivatives of 4-acetylpyridine selective hMAO-B inhibitors also at low nanomolar concentrations. The structure–activity relationship, as confirmed by molecular modeling studies, proved that the pyridine ring linked to the hydrazonic nitrogen and the substituted aryl moiety at C4 of the thiazole conferred the inhibitory effects on hMAO enzymes. Successively, the strongest hMAO-B inhibitors were tested toward acetylcholinesterase (AChE) and the most interesting compound showed activity in the low micromolar range. Our results suggest that this scaffold could be further investigated for its potential multi-targeted role in the discovery of new drugs against the neurodegenerative diseases.
Introduction
Monoamine oxidases A and B (MAO-A and -B) are flavin adenine dinucleotide (FAD)-depending enzymes present in the outer side of mitochondrial membrane. They are encoded by different genes which are located on the X chromosome and could derive from a common gene, because of their identical exon–intron organization. MAO enzymes can be found in brain, liver, blood platelets, placenta and gastrointestinal tract, but their action and concentration in these tissues are different: MAO-A activity predominates in the placenta and the gut, whereas MAO-B in the blood platelets, the liver and in the human basal gangliaCitation1. In the brain, MAO-A is expressed in catecholaminergic neurons of the cortex while MAO-B is present in serotonergic neurons. Both enzymes catalyze the oxidative deamination of endogenous neurotransmitters: even if they both degrade dopamine and p-tyramine, MAO-A mostly oxidizes serotonin, noradrenaline and adrenaline, whereas MAO-B generally metabolizes the small amines like benzylamine and phenethylamine. This metabolic pathway of endogenous amines is responsible for the production of highly reactive peroxides causing oxidative damage and neurodegenerationCitation2,Citation3. Furthermore, it has been demonstrated that an over-expression of MAO-B isoform is involved in the onset of neurodegenerative diseases such as Alzheimer’s (AD) and Parkinson’s disease (PD), whereas the deregulation of MAO-A activity in the cortex could be the cause of mental disorders like depression, due to the reduction of catecholaminergic neurotransmittersCitation4. Specifically, in patients affected by AD there are structural alterations of numerous cerebral neuronal circuits especially in the hippocampus and cortical areas, characterized by the formation of senile plaques and neurofibrillary tangles. The degeneration of subcortical cholinergic and monoaminergic systems also occurs, playing an important role in the onset of AD due to the fact that these degenerated neurons, with long and poorly myelinated axons, innervate the cerebral areas involved in attention and memory acquisition and retrievalCitation5. On the other side, PD is the second most common neurodegenerative disease caused by the depletion of dopamine in nigrostriatal neurons which are involved in the control of motor function in the basal ganglia: the loss of dopaminergic neurons in the substantia nigra provokes bradykinesia, rigidity, hypokinesia and akinesiaCitation6,Citation7. Considering all these features and the involvement of a deregulated metabolism of monoamine transmitters in the onset of neurodegenerative and mental disorders, the development of new specific MAO inhibitors endowed with a selective activity and low toxicity is an open challengeCitation8–14.
Currently, there are three FDA-approved drugs based on the “one drug, one target” paradigm (donepezil, galantamine and rivastigmine) that improve symptoms by inhibiting acetylcholinesterase evidencing the limitations of this strategy for the complex nature of AD. Conversely, the most current innovative therapeutic approach is based on the “one drug, multiple targets” paradigm, which suggests the use of compounds with multiple activities at different target sitesCitation15,Citation16. MTDL (multi-target directed ligand) strategy targeting MAO-B represents a promising approach to treat complex and multifaceted neurodegenerative diseasesCitation17. This approach mainly includes, in AD, dual inhibitors of monoamine oxidase and acetylcholinesterase (AChE).
The therapeutic potential of MAO-B inhibitors in AD has been also proposed due to their validated neuroprotective/neurorestorative and cognitive enhancing abilities besides their positive effect on monoaminergic transmission. In detail, scaffolds containing a propargylamine moiety are of particular interest due to their reported beneficial actionsCitation18,Citation19.
In a previous work we have demonstrated that the concurrent inhibition of this enzyme and the binding to the 5-HT3 receptor can lead to interesting compoundsCitation20. AChE is a serine hydrolase mainly found at neuromuscular junctions and cholinergic brain synapsesCitation21. AD is associated with a loss of cholinergic neurons in the brain and a decreased level of AChE, thus the major therapeutic goal in the AD treatment strategies is the inhibition of brain AChECitation22.
Taking into account our recent studies for the development of new MAO inhibitors in which we explained that a pyridine ring linked at N1 of the hydrazine moiety, together with the simultaneous presence of an aromatic substituent at C4 of the thiazole nucleus, could confer an inhibitory activity on hMAO enzymesCitation23,Citation24, we designed 36 (thiazol-2-yl)hydrazone derivatives of acetylpyridine regioisomers. Supported by molecular recognition studies we evaluated: the influence of 2-, 3- and 4-acetylpyridines bound to the hydrazonic nitrogen; the effect of several electron-withdrawing groups (chloride, bromide and iodide), electron-donating groups (methoxy and methyl) and the non-substituted phenyl ring as substituents of the aryl moiety linked at C4 of the pharmacophoric nucleus. Furthermore, keeping in mind our study on coumarin derivatives as hMAO inhibitorsCitation25,Citation26, we also evaluated the effect on hMAO inhibition due to the presence of this ring. In addition, considering the structural analogy among our compounds and some cholinesterase inhibitors described in the literatureCitation27, we have selected the most potent and selective hMAO-B inhibitors to define their activity also towards acetylcholinesterase.
Materials and methods
Chemistry
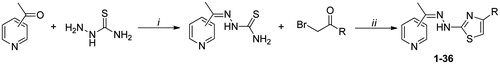
(4-Substituted-thiazol-2-yl)hydrazone compounds (1–36) were synthesized in high yields as reported in our previous communicationsCitation28–32. Acetylpyridine regioisomers reacted directly with thiosemicarbazide in ethanol in presence of catalytic amounts of acetic acid at room temperature. Then, the resulting thiosemicarbazones were reacted in ethanol with the appropriate α-bromoacetophenone through Hantzsch cyclization to give (4-substituted-thiazol-2-yl)hydrazone derivatives. All the synthesized products were washed with n-hexane, petroleum ether and diethyl ether and purified by chromatography (ethyl acetate/petroleum ether) before characterization by spectroscopic methods and elemental analysis.
Biochemistry
In vitro inhibition studies of hMAO-A and hMAO-B
The biological evaluation of the test drugs on hMAO activity was investigated by measuring their effects on the production of hydrogen peroxide (H2O2) from p-tyramine (a common substrate for both hMAO-A and hMAO-B), using the Amplex® Red MAO assay kit and microsomal MAO isoforms prepared from insect cells (BTI-TN-5B1-4) infected with recombinant baculovirus containing cDNA inserts for hMAO-A or hMAO-B. The production of H2O2 catalyzed by MAO isoforms can be detected using 10-acetyl-3,7-dihydroxyphenoxazine (Amplex® Red reagent), a non-fluorescent and highly sensitive probe that reacts with H2O2 in the presence of horseradish peroxidase to produce a fluorescent product, resorufin. In this study, hMAO activity was evaluated using the above method following the general procedure previously described by usCitation33. The test drugs (new compounds and reference inhibitors) themselves were unable to react directly with the Amplex® Red reagent, which indicates that these drugs do not interfere with the measurements.
In our experiments and under our experimental conditions, hMAO-A displayed a Michaelis constant (Km) equal to 457.17 ± 38.62 µM and a maximum reaction velocity (Vmax) in the control group of 185.67 ± 12.06 nmol p-tyramine/min/mg protein whereas hMAO-B showed a Km of 220.33 ± 32.80 µM and a Vmax equal to 24.32 ± 1.97 nmol p-tyramine/min/mg protein (n = 5). Most tested drugs concentration-dependently inhibited this enzymatic control activity ().
Table 1. Inhibitory activity of 2-acetylpyridine derivatives 1–12 towards hMAO enzymes.
Table 2. Inhibitory activity of 3-acetylpyridine derivatives 13–24 towards hMAO enzymes.
Table 3. Inhibitory activity of 4-acetylpyridine derivatives 25–36 and reference drugs towards hMAO enzymes.
In vitro inhibition studies of AChE
Electric eel AChE (EC 3.1.1.7), acetylthiocholine iodide, 5,5′-dithio-bis-(2-nitrobenzoic acid) (DTNB) and donepezil, used as reference standard, were purchased from Sigma-Aldrich (Milan, Italy). All other chemical and biological reagents and solvents used were of the highest analytical, commercially available grade. We determined the inhibitory activity by the spectrophotometric method of EllmanCitation34 based on the reaction of released thiocholine to give a colored product with a chromogenic reagent DTNB, to evaluate the inhibition of AChE. The study was carried out as previously describedCitation35 using a double beam UV–Vis lambda 40 Perkin Elmer spectrophotometer and optical polystyrene cuvettes (10 × 10 × 45 mm, 340–800 nm optical transparency). AChE was daily tested to evaluate the effective enzymatic activity. Tested compounds were dissolved in DMSO or H2O/DMSO. The final DMSO content of the assay mixture (<0.05%) had no effect on enzyme activity. Each measure was repeated at least in triplicate. In order to calculate the percent inhibition, each compound (final concentration 3 µM) was mixed in a polystyrene cuvette with 3.0 mL of 0.1 M (pH 7.4) phosphate buffer containing DTNB (0.25 mM) and AChE (0.083 U mL−1).
The reaction was initiated by the addition of substrate, acetylthiocholine iodide (final concentration 100 µM), then changes in the absorbance at 412 nm at 25 °C were recorded between 0.5 and 1.5 min after substrate addition. The percent inhibition was calculated using the following equation:
where Ai and Ac represent the change in the absorbance in the presence of inhibitor and without inhibitor, respectively.
To calculate the Ki value for compound 26, the hydrolysis rates (v) were measured at various acetylthiocholine (75–150 µM) and inhibitor (0.5–2.5 µM) concentrations. The data were analyzed with the enzyme kinetic module of SigmaPlot, version 8.02 a (Sigma-Aldrich), in order to find the best fitting model of inhibition, as indicated by calculated linear regression coefficient r2. Its Ki value was obtained according to Dixon’s method, reporting the reciprocal of the hydrolysis rate versus the inhibitor concentrations.
Results and discussion
We designed 36 (thiazol-2-yl)hydrazone derivatives of 2-, 3- and 4-acetylpyridine as potential human MAO inhibitors. According to the biological results reported in , with the only exception of products 1, 17 and 22 that were not active, all the compounds have shown to be hMAO inhibitors in low micromolar/high nanomolar range and most of the 4-acetylpyridine derivatives displayed a selective inhibitory activity at low nanomolar concentrations. As regards 2-acetylpyridine derivatives (), the presence of a methyl group (compound 4), a methoxy group (8) and a further phenyl moiety (9) at the para position of the aromatic ring conferred a discrete selectivity towards B isoform. In detail, compound 4 had an IC50 of 2.39 µM against hMAO-A and 880.30 nM against hMAO-B, compound 8 possessed an IC50 of 6.18 µM against hMAO-A and 1393.10 nM against hMAO-B, while the IC50 values of compound 9 towards hMAO-A and -B were 4.38 µM and 995.88 nM, respectively. Regarding 3-acetylpyridine derivatives (), the selectivity towards hMAO-B of compounds containing a methyl group (16), a methoxy group (20) and a further phenyl moiety (21) at C4 position of the aryl moiety increased. Moreover, for compound 20 the concentration required for the inhibitory effect on hMAO-B diminished (IC50 of 427.54 nM). On the contrary, the presence of the coumarin ring in 2- and 3-acetylpyridines 12 and 24 conferred a high hMAO-A selectivity with an IC50 of 4.76 and 6.03 µM respectively, which were better than that of the reference drug moclobemide (361.38 µM). Finally, 4-acetylpyridine derivatives reported in have proven to be the most active and selective inhibitors. In this series, 4-chlorophenyl (29) and 2,4-dichlorophenyl (34) derivatives showed a great selectivity of action towards B isoform at very low concentrations (22.59 and 62.59 nM, respectively). Also derivatives 26, 27 and 30, which contained 2-, 3- and 4-bromophenyl substituents respectively, were selectively active against hMAO-B (4.24 nM for compound 26, 5.02 nM for 27 and 73.52 nM for 30). Compound 27 was more selective (ratio = 910.36) than the other two bromophenyl regioisomers as well as compounds 32 and 33 with respect to their corresponding 2- and 3-acetylpyridine derivatives. The 4-methylphenyl substituted compound 28 was not only one of the most efficacious products (12.79 nM), but also the most selective human MAO-B inhibitor of this scaffold (ratio = 2105.55, which is similar to that of the reference drug R-(−)-deprenyl, that had an IC50 of 19.60 nM). Finally, unlike its 2- and 3-acetylpyridine analogues (selective MAO-A inhibitors with ratio <0.06), the coumarin derivative of 4-acetylpiridine (36) possessed a reversed selectivity (ratio = 3.80). These biological results demonstrated the importance of the pyridine ring linked at the hydrazonic N1 and of a substituted aryl moiety at C4 of the thiazole nucleus for the development of new hMAO inhibitors. Moreover, 4-acetylpyridine derivatives could be further investigated for their potential role in the management of neurodegenerative diseases caused by hMAO-B over-expression.
To better understand the molecular mechanism of hMAO-A and hMAO-B inhibition, docking simulations were performed for the most active compound 26 and its 2- and 3-acetylpyridine analogues, compounds 14 and 2, respectively. Both E and Z isomers of the 3D molecules were docked into the binding pocket of the enzymes in order to estimate the ligand binding affinity by means of Glide SP scoring function (Gscore) in kcal/molCitation36. For each compound the corresponding final score has been considered as the average of E and Z isomers Gscore and, in according to the biological data, confirmed the better accommodation into the hMAO-B active site with respect the hMAO-A one (). On the basis of not discriminant Gscore values, for the best pose of docked compounds the binding energy (dG Bind) by Prime MM-GBSA utilityCitation37 were also calculated and, in agreement with the experimental IC50 data, compound 26 showed the best dG Bind value for the hMAO-B isoform, corresponding to −66.94 kcal/mol.
Table 4. Scoring values (Gscore) and binding energy values (dG Bind) after GlideSP docking and Prime-MMGBSA simulations, for the best pose of ligands 2, 14 and 26 within the hMAO-A and hMAO-B active sites.
Despite the different inhibitory activity of compounds 26, 14 and 2, in terms of IC50 values, the configuration of the double bond and the substitution to the pyridine ring did not remarkably influence the E and Z isomers recognition into the hMAO-B catalytic site: in all cases the bromophenyl substituent was oriented toward the FAD cofactor and the pyridine was located at the entrance site (Figure S1). This binding mode is common to the E, but not to the Z isomers, docked into the hMAO-A lipophilic cage: the configuration of the double bond caused the orientation of pyridine ring toward the FAD with the bromophenyl substituent located at the gate of the catalytic site (Figure S2).
As reported in our previous work, amino acid residues in the area opposite to the flavin: hMAO-A ILE180, ASN181, PHE208, SER209 and ILE335 with the corresponding hMAO-B LEU171, CYS172, ILE199, SER200 and TYR326, respectively, strongly modify shape and size of the active site and, as a consequence, the different substrate recognitionCitation30. The hMAO-A binding pocket is shorter and wider than the longer and narrower pocket of hMAO-BCitation38 and its smaller cavity volume could explain the better biological data of most of the derivatives 1–36 () for the hMAO-B isoform, as corroborated by molecular modeling studies.
However, the docking simulation and the best pose of ligands in complex with the hMAO-B crystallographic model, downloaded from the Protein Data Bank (PDB) siteCitation39 with the code 2V5ZCitation40, was unable to explain the molecular basis of the low nanomolar IC50 value for the 4-acetylpyridine 26 better than the micromolar values of its analogues 14 and 2: the superimposition of the compounds displayed a strong alignment of pyridine and bromophenyl rings into the catalytic site and no different hydrophobic ligand–target interactions (Figure S1).
In order to further investigate the stereochemical requirement for optimal hMAO-B inhibition, both E and Z isomers of compounds 26 and 2 were submitted to a molecular dynamic (MD) of 5 ns carried out by Desmond packageCitation41. The “Simulation Event Analysis” panel was used to calculate the fluctuation of energy during the dynamic simulation, considering as final score (MDE) the average of E and Z isomers energy values: the derivative 26 with the MDE value of −59.88 kcal/mol showed the best theoretical result (). Therefore, to display interactions of ligands during the MD with the amino acid residues lining the catalytic site of the target, “Simulation Interaction Diagram” panel was used. The MD results were submitted to graphical inspection and the last frames are reported in and .
Figure 1. The dynamic simulation last frame of both E (A) and Z (B) isomers of compound 26 and their H-bond contacts into the hMAO-B binding site. The inhibitor E and Z isomers are rendered as carbon-pink sticks, the pocket amino acids are labeled as carbon-blue sticks and FAD co-factor as carbon-cyan sticks. Target is described as blue cartoon and all non-carbon atoms are colored according to atom types. For the colored version of this figure, please refer to the online version of the article.
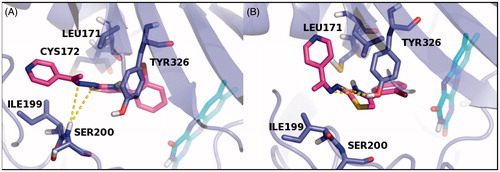
Figure 2. The dynamic simulation last frame of both E (A) and Z (B) isomers of compound 2 and their H-bond contacts into the hMAO-B binding site. The inhibitor E and Z isomers are rendered as carbon-yellow sticks, the pocket amino acids are labeled as carbon-blue sticks and FAD co-factor as carbon-cyan sticks. Target is described as blue cartoon and all non-carbon atoms are colored according to atom types. For the colored version of this figure, please refer to the online version of the article.
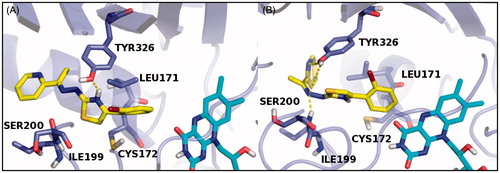
The MD analysis of compound 26 revealed that both the isomers, during the simulation, are able to establish not only hydrophobic contacts with critical amino acid residues of the binding site, but polar contacts too. Moreover, the E isomer hydrazonic moiety was involved in two H-bonds with SER200 residue, while the same molecule portion in the Z isomer established two H-bonds with TYR326 (). On the other hand, the MD analysis of compound 2 showed for the E isomer thiazole ring (tautomeric form) a polar contact with TYR326,while the hydrazonic portion and the pyridine of the Z isomer were involved in several H-bond contacts with SER200 and TYR326 amino acid residues (). On the basis of these results, we concluded that the pyridine ring linked to the hydrazonic nitrogen and the substituted aryl moiety at C4 of the pharmacophoric thiazole conferred a great inhibitory effect on hMAO-B enzyme.
Finally, the most potent and selective hMAO-B inhibitors (26–29 and 34) were chosen to perform the enzyme inhibition tests of AChE (). Among them, compound 26 was found as the most active compound with inhibition values of 50% at 3 µM. The inhibition percentages of 27–29 and 34 showed these compounds as inactive at the tested concentration, with values ranging from 2% (compound 34) to 7% (compound 28). The type of inhibition and Ki value were determined only for compound 26. Our data show that 26 is a competitive inhibitor with Ki = 1.82 ± 0.55 µM (r2 = 0.96) as also corroborated by molecular docking study.
Table 5. Experimental and theoretical inhibition data of derivatives 26–29, 34 and reference drug Donepezil towards AChE target.
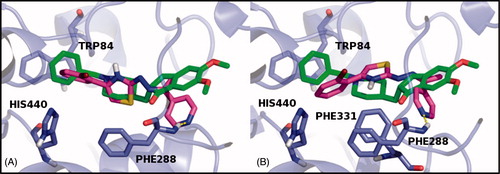
The X-ray model of AChE with the PDB code 1EVECitation42, co-crystallized with Donepezil, was used to generate docking ligand poses of all compounds tested in vitro. Scoring values Gscore and Embrace Free Energy (EFE)Citation43 calculated as the average of E and Z isomers values are reported in . According to the biological results, in silico data showed compound 26 with the acceptable Gscore value of −7.81 kcal/mol and the best EFE value of −63.98 kcal/mol but in all cases less than the positive control Donepezil values (Gscore = −9.89 kcal/mol; EFE = −253.67 kcal/mol). Both the isomers of 26 were stabilized, into the binding pocket, through a hydrophobic interaction between the thiazole and PHE288 amino acid residue, involved in the H-bond contact with the pyridine ring of the molecule, too. The E isomer established an additional non-polar contact with the THP84, common binding residue within the AChE–Donepezil complex, while the Z isomer best pose was stabilized by PHE331 (). Therefore, the 4-pyridine ring linked to the hydrazonic nitrogen also conferred the inhibitory activity on the AChE enzyme in addition to selective hMAO-B inhibition and could be considered for future investigations.
Figure 3. The docking best pose of both E (A) and Z (B) isomers of compound 26 and their H-bond contact into the AChE binding site, superimposed to Donepezil co-crystallized inhibitor. The E and Z isomers are rendered as carbon-pink sticks, Donepezil as carbon-green sticks and the pocket amino acids are labeled as carbon-blue sticks. Target is described as blue cartoon and all non-carbon atoms are colored according to atom types. For the colored version of this figure, please refer to the online version of the article.
Experimental
Chemistry
Starting materials and reagents used in the synthetic procedures were obtained from commercial suppliers and were used without further purification. Melting points (mp) were determined by the capillary method on an FP62 apparatus (Mettler-Toledo) and are uncorrected. Temperatures are reported in °C. 1H spectra were recorded on 400 MHz Bruker spectrometer using DMSO-d6 as the solvent. Chemical shifts are reported in parts per million (δ) downfield from the signal of tetramethylsilane (TMS) which was added to the deuterated solvent. The assignment of exchangeable protons (NH) was confirmed by the addition of D2O. Coupling constants J are expressed in hertz (Hz). Elemental analyses for C, H and N were determined with a Perkin-Elmer 240 B microanalyzer and the analytical results were ≥95% purity for all compounds. All reactions were monitored by TLC performed on 0.2 mm thick silica gel plates (60 F254 Merck). Visualization was carried out under ultra-violet irradiation (254 nm). Preparative flash column chromatography was carried out on silica gel (230–400 mesh, G60 Merck). Organic solutions were dried over anhydrous sodium sulfate. Concentration and evaporation of the solvent after reaction or extraction were carried out on a rotary evaporator (Büchi Rotavapor); where given, systematic compound names are those generated by ChemDraw ultra following IUPAC conventions.
General procedure for the synthesis of derivatives 1–36
The appropriate 2-, 3- or 4-acetylpyridine (50 mmol) was dissolved in 50 mL of ethanol and magnetically stirred with equimolar amounts of thiosemicarbazide for 24 h at room temperature with acetic acid as the catalyst. The resulting thiosemicarbazone precipitating from reaction mixture was filtered and washed with solvents (n-hexane, petroleum ether and diethyl ether). The obtained thiosemicarbazone (50 mmol) reacted with an equimolar quantity of the corresponding α-bromoacetophenone dissolved in ethanol (50 mL) under magnetic stirring at room temperature. The reaction was monitored until completion by TLC. The resulting (thiazol-2-yl)hydrazone was filtered, washed with petroleum ether and diethyl ether, and, if necessary, purified by chromatography on silica gel (ethyl acetate: n-hexane, 3:1), to give all compounds in high yields (78–99%). In preceding works, products 4, 8, 16, 20, 28, 32Citation44, 7, 19, 31Citation4Citation5, 12, 24, 36Citation4Citation6, 18 and 21Citation4Citation7, 5, 17, 29Citation48 have already been synthesized and characterized for other purposes and were tested in this paper to ensure a complete structure–activity relationship (Scheme 1).
Scheme 1. Synthesis of (thiazol-2-yl)hydrazone derivatives 1–36. Reagents and conditions: (i) EtOH, acetic acid (cat.), rt; (ii) EtOH, rt. For substituents (R) see .
Characterization data for new compounds
1-(4-(2-Chlorophenyl)thiazol-2-yl)-2-(1-(pyridin-2-yl)ethylidene)hydrazine (1): m.p. 235–237 °C; 78% yield; 1H NMR (400 MHz, DMSO-d6): δ 2.49 (s, 3H, CH3), 7.40–7.53 (m, 3H, Ar), 7.55 (s, 1H, thiazole), 7.60–7.63 (m, 1H, Ar), 8.00–8.10 (m, 2H, Ar), 8.67–8.69 (m, 2H, Ar), 12.73 (bs, 1H, NH, D2O exch.); 13C NMR (101 MHz, DMSO-d6): δ 13.98, 102.34, 123.98, 125.12, 125.78, 126.15, 127.19, 129.99, 130.67, 132.68, 135.67, 142.24, 146.43, 148.34, 152.89, 170.80. Anal. Calcd for C16H13ClN4S: C, 58.44; H, 3.98; N, 17.04. Found: C, 58.22; H, 4.12; N, 16.89.
1-(4-(2-Bromophenyl)thiazol-2-yl)-2-(1-(pyridin-2-yl)ethylidene)hydrazine (2): m.p. 210–211 °C; 80% yield; 1H NMR (400 MHz, DMSO-d6): δ 2.41 (s, 3H, CH3), 7.44–7.53 (m, 2H, Ar), 7.55 (s, 1H, thiazole), 7.59–7.63 (m, 2H, Ar), 8.00–8.10 (m, 2H, Ar), 8.67–8.69 (m, 2H, Ar), 12.17 (bs, 1H, NH, D2O exch.); 13C NMR (101 MHz, DMSO-d6): δ 13.78, 101.67, 123.56, 124.68, 125.01, 125.95, 127.44, 130.67, 130.99, 133.13, 135.02, 144.56, 146.97, 148.32, 154.01, 171.03. Anal. Calcd for C16H13BrN4S: C, 51.48; H, 3.51; N, 15.01. Found: C, 51.60; H, 3.73; N, 15.20.
1-(4-(3-Bromophenyl)thiazol-2-yl)-2-(1-(pyridin-2-yl)ethylidene)hydrazine (3): m.p. 229–231 °C; 82% yield; 1H NMR (400 MHz, DMSO-d6): δ 2.42 (s, 3H, CH3), 7.36–7.39 (m, 2H, Ar), 7.50–7.51 (m, 2H, Ar), 7.60 (s, 1H, thiazole), 7.87–7.89 (d, J = 8.0 Hz, 1H, Ar), 8.09–8.16 (m, 2H, Ar), 8.67–8.68 (m, 1H, Ar), 11.73 (bs, 1H, NH, D2O exch.); 13C NMR (101 MHz, DMSO-d6): δ 13.99, 101.24, 124.01, 124.12, 124.99, 125.14, 127.96, 132.02, 133.13, 133.68, 134.80, 144.36, 146.90, 147.98, 155.10, 170.50. Anal. Calcd for C16H13BrN4S: C, 51.48; H, 3.51; N, 15.01. Found: C, 51.21; H, 3.32; N, 14.83.
1-(4-(4-Bromophenyl)thiazol-2-yl)-2-(1-(pyridin-2-yl)ethylidene)hydrazine (6): mp 239–241 °C; 80% yield; 1H NMR (400 MHz, DMSO-d6): δ 2.48 (s, 3H, CH3), 7.51 (m, 1H, Ar), 7.60–7.62 (m, 2H, Ar), 7.63 (s, 1H, thiazole), 7.82–7.84 (m, 1H, Ar), 7.85 (m, 1H, Ar), 8.14 (m, 2H, Ar), 8.53 (m, 1H, Ar), 11.77 (bs, 1H, NH, D2O exch.); 13C NMR (101 MHz, DMSO-d6): δ 13.89, 100.78, 122.12, 122.98, 124.34, 125.46, 126.79, 130.98, 131.54, 144.67, 147.32, 148.29, 153.66, 170.19. Anal. Calcd for C16H13BrN4S: C, 51.48; H, 3.51; N, 15.01. Found: C, 51.70; H, 3.73; N, 15.19.
1-(4-(4-Biphenyl)thiazol-2-yl)-2-(1-(pyridin-2-yl)ethylidene)hydrazine (9): m.p. 208–210 °C; 79% yield; 1H NMR (400 MHz, DMSO-d6): δ 2.44 (s, 3H, CH3), 7.38–7.50 (m, 5H, Ar), 7.53 (s, 1H, thiazole), 7.72–7.79 (m, 4H, Ar), 7.98–8.14 (m, 3H, Ar), 8.65–8.67 (m, 1H, Ar), 11.84 (bs, 1H, NH, D2O exch.); 13C NMR (101 MHz, DMSO-d6): δ 13.77, 101.23, 124.16, 124.97, 125.01, 125.55, 126.51, 127.66, 128.23, 130.98, 131.54, 133.80, 135.33, 147.23, 148.44, 148.97, 154.33, 171.44. Anal. Calcd for C22H18N4S: C, 71.32; H, 4.90; N, 15.12. Found: C, 71.10; H, 5.07; N, 15.29.
1-(4-(2,4-Dichlorophenyl)thiazol-2-yl)-2-(1-(pyridin-2-yl)ethylidene)hydrazine (10): m.p. 198–200 °C; 85% yield; 1H NMR (400 MHz, DMSO-d6): δ 2.45 (s, 3H, CH3), 7.22–7.25 (m, 1H, Ar), 7.32 (s, 1H, thiazole), 7.39–7.43 (m, 1H, Ar), 7.53–7.55 (m, 1H, Ar), 8.10–8.17 (m, 3H, Ar), 8.73–8.75 (m, 1H, Ar), 11.87 (bs, 1H, NH, D2O exch.); 13C NMR (101 MHz, DMSO-d6): δ 13.95, 100.21, 123.33, 124.45, 125.02, 126.22, 127.98, 130.43, 130.78, 135.89, 136.22, 142.34, 146.67, 148.22, 152.09, 170.11. Anal. Calcd for C16H12Cl2N4S: C, 52.90; H, 3.33; N, 15.42. Found: C, 53.11; H, 3.50; N, 15.28.
1-(4-(2,4-Dimethoxyphenyl)thiazol-2-yl)-2-(1-(pyridin-2-yl)ethylidene)hydrazine (11): m.p. 218–220 °C; 81% yield; 1H NMR (400 MHz, DMSO-d6): δ 2.40 (s, 3H, CH3), 3.79 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 7.15–7.17 (m, 1H, Ar), 7.23 (s, 1H, thiazole), 7.37–7.40 (m, 1H, Ar), 7.48–7.50 (m, 1H, Ar), 8.00–8.10 (m, 3H, Ar), 8.64–8.66 (m, 1H, Ar), 11.58 (bs, 1H, NH, D2O exch.); 13C NMR (101 MHz, DMSO-d6): δ 13.87, 57.23, 57.44, 101.01, 121.89, 123.34, 124.02, 125.46, 127.21, 130.97, 131.33, 135.61, 135.89, 142.63, 146.32, 148.44, 151.11, 170.74. Anal. Calcd for C18H18N4O2S: C, 61.00; H, 5.12; N, 15.81. Found: C, 60.82; H, 4.97; N, 15.64.
1-(4-(2-Chlorophenyl)thiazol-2-yl)-2-(1-(pyridin-3-yl)ethylidene)hydrazine (13): m.p. 230–232 °C; 97% yield; 1H NMR (400 MHz, DMSO-d6): δ 2.43 (s, 3H, CH3), 7.38–7.40 (d, J = 8.0 Hz, 2H, Ar), 7.44 (s, 1H, thiazole), 7.66–7.74 (m, 2H, Ar), 8.14–8.16 (d, J = 8.0 Hz, 2H, Ar), 8.94–9.13 (m, 1H, Ar), 9.31 (s, 1H, Ar), 11.95 (bs, 1H, NH, D2O exch.); 13C NMR (101 MHz, DMSO-d6): δ 13.91, 100.22, 123.18, 123.97, 124.44, 125.56, 127.12, 129.78, 130.21, 132.44, 135.34, 143.01, 146.36, 147.53, 152.35, 170.20. Anal. Calcd for C16H13ClN4S: C, 58.44; H, 3.98; N, 17.04. Found: C, 58.71; H, 4.09; N, 16.90.
1-(4-(2-Bromophenyl)thiazol-2-yl)-2-(1-(pyridin-3-yl)ethylidene)hydrazine (14): m.p. 220–221 °C; 91% yield; 1H NMR (400 MHz, DMSO-d6): δ 2.39 (s, 3H, CH3), 7.08 (s, 1H, thiazole), 7.26–7.28 (d, J = 8.0 Hz, 1H, Ar), 7.29–7.30 (m, 1H, Ar), 7.36–7.37 (m, 1H, Ar), 7.64–7.75 (m, 1H, Ar), 8.38–8.40 (d, J = 8.0 Hz, 2H, Ar), 8.74–8.75 (m, 1H, Ar), 9.16 (s, 1H, Ar), 12.00 (bs, 1H, NH, D2O exch.); 13C NMR (101 MHz, DMSO-d6): δ 13.66, 100.77, 122.18, 124.02, 125.11, 126.88, 128.77, 130.43, 130.99, 131.17, 135.21, 144.76, 146.41, 147.22, 152.44, 170.11. Anal. Calcd for C16H13BrN4S: C, 51.48; H, 3.51; N, 15.01. Found: C, 51.25; H, 3.20; N, 15.17.
1-(4-(3-Bromophenyl)thiazol-2-yl)-2-(1-(pyridin-3-yl)ethylidene)hydrazine (15): m.p. 250–252 °C; 86% yield; 1H NMR (400 MHz, DMSO-d6): δ 2.40 (s, 3H, CH3), 7.38–7.42 (m, 1H, Ar), 7.50–7.53 (m, 1H, Ar), 7.58 (s, 1H, thiazole), 7.89–7.91 (m, 2H, Ar), 8.10–8.11 (m, 1H, Ar), 8.74 (m, 2H, Ar), 9.06 (s, 1H, Ar), 11.68 (bs, 1H, NH, D2O exch.); 13C NMR (101 MHz, DMSO-d6): δ 13.87, 100.20, 123.00, 124.78, 126.34, 126.97, 127.13, 131.78, 132.45, 133.66, 136.79, 145.45, 145.32, 148.22, 154.87, 171.56. Anal. Calcd for C16H13BrN4S: C, 51.48; H, 3.51; N, 15.01. Found: C, 51.59; H, 3.29; N, 14.77.
1-(4-(2,4-Dichlorophenyl)thiazol-2-yl)-2-(1-(pyridin-3-yl)ethylidene)hydrazine (22): m.p. 225–227 °C; 90% yield; 1H NMR (400 MHz, DMSO-d6): δ 2.35 (s, 3H, CH3), 7.43–7.46 (m, 3H, Ar), 7.52 (s, 1H, thiazole), 7.69–7.90 (m, 2H, Ar), 8.55–8.56 (m, 1H, Ar), 8.9 (s, 1H, Ar), 11.46 (bs, 1H, NH, D2O exch.); 13C NMR (101 MHz, DMSO-d6): δ 13.44, 100.89, 124.28, 124.98, 125.37, 127.67, 127.89, 130.34, 130.89, 134.65, 135.72, 144.01, 146.42, 148.00, 152.67, 171.01. Anal. Calcd for C16H12Cl2N4S: C, 52.90; H, 3.33; N, 15.42. Found: C, 53.05; H, 3.14; N, 15.69.
1-(4-(2,4-Dimethoxyphenyl)thiazol-2-yl)-2-(1-(pyridin-3-yl)ethylidene)hydrazine (23): m.p. 205–208 °C; 95% yield; 1H NMR (400 MHz, DMSO-d6): δ 2.36 (s, 3H, CH3), 3.87 (s, 3H, OCH3), 4.00 (s, 3H, OCH3), 7.34 (s, 1H, thiazole), 7.74–7.76 (m, 3H, Ar), 8.24–8.28 (m, 2H, Ar) 8.74–8.79 (m, 1H, Ar), 9.03 (s, 1H, Ar), 10.95 (bs, 1H, NH, D2O exch.); 13C NMR (101 MHz, DMSO-d6): δ 13.59, 57.39, 57.97, 100.53, 122.76, 122.56, 125.87, 126.84, 126.98, 129.54, 132.67, 134.64, 134.93, 141.63, 145.02, 149.43, 150.32, 170.24. Anal. Calcd for C18H18N4O2S: C, 61.00; H, 5.12; N, 15.81. Found: C, 61.19; H, 5.31; N, 15.99.
1-(4-(2-Chlorophenyl)thiazol-2-yl)-2-(1-(pyridin-4-yl)ethylidene)hydrazine (25): m.p. 222–225 °C; 87% yield; 1H NMR (400 MHz, DMSO-d6): δ 2.36 (s, 3H, CH3), 7.41–7.50 (m, 2H, Ar), 7.89 (s, 1H, thiazole), 7.56–7.57 (m, 2H, Ar), 8.38–8.40 (d, J = 8.0 Hz, 2H, Ar), 8.66–8.67 (m, 2H, Ar), 10.43 (bs, 1H, NH, D2O exch.); 13C NMR (101 MHz, DMSO-d6): δ 13.89, 100.66, 123.88, 123.99, 124.78, 128.66, 130.74, 132.66, 132.89, 136.44, 147.45, 149.54, 157.67, 171.98. Anal. Calcd for C16H13ClN4S: C, 58.44; H, 3.98; N, 17.04. Found: C, 58.62; H, 3.79; N, 17.27.
1-(4-(2-Bromophenyl)thiazol-2-yl)-2-(1-(pyridin-4-yl)ethylidene)hydrazine (26): m.p. 227–230°C; 80% yield; 1H NMR (400 MHz, DMSO-d6): δ 2.42 (s, 3H, CH3), 7,32–7.34 (m, 1H, Ar), 7.39 (s, 1H, thiazole), 7.48 (m, 1H, Ar), 7.88–7.90 (d, J = 8.0 Hz, 2H, Ar), 8.27–8.29 (d, J = 8.0 Hz, 2H, Ar), 8.90–8.91 (m, 2H, Ar), 12.50 (bs, 1H, NH, D2O exch.); 13C NMR (101 MHz, DMSO-d6): δ 13.90, 100.66, 125.12, 125.54, 125.89, 128.45, 130.48, 130.97, 133.78, 135.54, 143.54, 148.52, 157.33, 171.19. Anal. Calcd for C16H13BrN4S: C, 51.48; H, 3.51; N, 15.01. Found: C, 51.20; H, 3.24; N, 15.22.
1-(4-(3-Bromophenyl)thiazol-2-yl)-2-(1-(pyridin-4-yl)ethylidene)hydrazine (27): m.p. 260–262 °C; 89% yield; 1H NMR (400 MHz, DMSO-d6): δ 2.40 (s, 3H, CH3), 7.37–7.41 (m, 1H, Ar), 7.53–758 (m,1H, Ar), 7.66 (s, 1H, thiazole), 7.88–7.90 (d, J = 8.0 Hz, 1H, Ar), 8.09–8.10 (m, 1H, Ar), 8.21–8.22 (m, 2H, Ar), 8.86–8.87 (m, 2H, Ar), 12.18 (bs, 1H, NH, D2O exch.); Citation13C NMR (101 MHz, DMSO-d6): δ 13.79, 101.11, 125.33, 125.46, 125.67, 128.43, 130.12, 130.34, 134.34, 135.44, 144.79, 148.12, 157.20, 171.11. Anal. Calcd for C16H13BrN4S: C, 51.48; H, 3.51; N, 15.01. Found: C, 51.26; H, 3.36; N, 14.80.
1-(4-(4-Bromophenyl)thiazol-2-yl)-2-(1-(pyridin-4-yl)ethylidene)hydrazine (30): m.p. 275–277 °C; 97% yield; 1H NMR (400 MHz, DMSO-d6): δ 2.39 (s, 3H, CH3), 7.57–7.61 (m, 2H, Ar), 7.63 (s, 1H, thiazole), 7.82–7.84 (d, J = 8.0 Hz, 2H, Ar), 8.19–8.21 (d, J = 8.0 Hz, 2H, Ar), 8.85–8.86 (m, 2H, Ar), 12.04 (bs, 1H, NH, D2O exch.); Citation13C NMR (101 MHz, DMSO-d6): δ 13.80, 100.45, 123.22, 124.56, 125.78, 126.88, 131.11, 131.34, 147.58, 149.89, 153.45, 171.15. Anal. Calcd for C16H13BrN4S: C, 51.48; H, 3.51; N, 15.01. Found: C, 51.33; H, 3.25; N, 14.79.
1-(4-(4-Biphenyl)thiazol-2-yl)-2-(1-(pyridin-4-yl)ethylidene)hydrazine (33): m.p. 245–247 °C; 78% yield; 1H NMR (400 MHz, DMSO-d6): δ 2.24 (s, 3H, CH3), 7.41–7.49 (m, 5H, Ar), 7.53 (s, 1H, thiazole), 7.70–7.89 (m, 4H, Ar), 7.96–8.22 (m, 2H, Ar), 8.55–8.77 (m, 2H, Ar), 11.84 (bs, 1H, NH, D2O exch.); Citation13C NMR (101 MHz, DMSO-d6): δ 13.89, 100.55, 124.67, 125.07, 125.69, 127.41, 127.92, 128.66, 130.62, 131.50, 133.32, 135.64, 147.77, 149.06, 155.01, 171.22. Anal. Calcd for C22H18N4S: C, 71.32; H, 4.90; N, 15.12. Found: C, 71.60; H, 4.72; N, 14.98.
1-(4-(2,4-Dichlorophenyl)thiazol-2-yl)-2-(1-(pyridin-4-yl)ethylidene)hydrazine (34): m.p. 241–243 °C; 92% yield; 1H NMR (400 MHz, DMSO-d6): δ 2.43 (s, 3H, CH3), 7.43–7.46 (m, 3H, Ar), 7.55 (s, 1H, thiazole), 7.79–7.81 (m, 2H, Ar), 8.55–8.56 (m, 2H, Ar), 11.98 (bs, 1H, NH, D2O exch.); Citation13C NMR (101 MHz, DMSO-d6): δ 13.67, 100.80, 121.12, 123.54, 125.88, 127.86, 129.02, 130.44, 134.45, 135.16, 146.01, 148.55, 152.43, 171.05. Anal. Calcd for C16H12Cl2N4S: C, 52.90; H, 3.33; N, 15.42. Found: C, 52.69; H, 3.57; N, 15.20.
1-(4-(2,4-Dimethoxyphenyl)thiazol-2-yl)-2-(1-(pyridin-4-yl)ethylidene)hydrazine (35): m.p. 230–233 °C; 93% yield; 1H NMR (400 MHz, DMSO-d6): δ 2.56 (s, 3H, CH3), 3.83 (s, 3H, OCH3), 3.87 (s, 3H, OCH3), 7.17–7.277 (m, 1H, Ar), 7.32 (s, 1H, thiazole), 7.47–7.54 (m, 1H, Ar), 7.68–7.70 (m, 1H, Ar), 8.00–8.10 (m, 2H, Ar), 8.57–8.66 (m, 2H, Ar) 12.58 (bs, 1H, NH, D2O exch.); Citation13C NMR (101 MHz, DMSO-d6): δ 13.67, 57.88, 58.12, 100.11, 122.59, 126.17, 126.44, 126.94, 128.88, 133.12, 134.33, 134.77, 145.13, 149.77, 150.78, 170.06. Anal. Calcd for C18H18N4O2S: C, 61.00; H, 5.12; N, 15.81. Found: C, 60.78; H, 4.96; N, 15.54.
Determination of hMAO isoform activity
The effects of the test compounds on the enzymatic activity of hMAO isoform were evaluated by a fluorimetric method following the experimental protocol previously described by usCitation33. 0.1 mL of sodium phosphate buffer (0.05 M, pH 7.4) containing different concentrations of the test drugs and adequate amounts of recombinant hMAO-A or hMAO-B required and adjusted to obtain the same reaction velocity in our experimental conditions, i.e. to oxidize in the control group 165 pmol of p-tyramine/min, were incubated for 15 min at 37 °C in a flat-black-bottom 96-well microtest™ plate (BD Biosciences, Franklin Lakes, NJ) placed in a dark fluorimetric chamber. The reaction was started by adding 200 µM Amplex® Red reagent, 1 U/mL horseradish peroxidase and 1 mM p-tyramine. The production of H2O2 and, consequently, of resorufin was quantified at 37 °C in a multidetection microplate fluorescence reader (Fluostar Optima, BMG Labtech GmbH, Offenburg, Germany) based on the fluorescence generated (excitation, 545 nm, emission, 590 nm) over a 15 min period, in which the fluorescence increased linearly. Control experiments were carried out simultaneously by replacing the test compounds and reference inhibitors with appropriate dilutions of the vehicles. In addition, the potential ability of the above test drugs to modify the fluorescence generated in the reaction mixture due to non-enzymatic inhibition (e.g. for directly reacting with Amplex® Red reagent) was determined by adding these drugs to solutions containing only the Amplex® Red reagent in a sodium phosphate buffer. The drugs, vehicle and chemicals used in the experiments were the new compounds, moclobemide (F. Hoffmann-La Roche Ltd., Basel, Switzerland), dimethyl sulfoxide (DMSO), R-(−)-deprenyl hydrochloride, iproniazid phosphate, isatin (Sigma-Aldrich, Spain), resorufin sodium salt, clorgyline hydrochloride, p-tyramine hydrochloride, sodium phosphate and horseradish peroxidase (supplied in the Amplex® Red MAO assay kit from Molecular Probes). Unless otherwise specified, results shown in the text and tables are expressed as mean ± standard error of the mean (S.E.M.) from n experiments. Significant differences between two means were determined by one-way analysis of variance (ANOVA) followed by the Dunnett’s post-hoc test. Graph Pad Prism Software (GraphPad Software, San Diego, CA) was used to perform statistical analyses and to calculate IC50 values and kinetic parameters.
Study of AChE inhibition
Percent inhibition of AChE: The spectrophotometric method of EllmanCitation34 was used to assess the inhibitory activity of the selected compounds 26–29 and 34 toward AChE. 3 mL of 0.1 M (pH 7.4) phosphate buffer containing DTNB (0.25 mM) and AChE (0.083 U mL−1) was mixed in a polystyrene cuvette of 1 cm path length with 1 µL of the studied compounds, previously solubilized in DMSO in order to obtain a 9 mM solution. The reaction was started by adding 30 μL of an aqueous solution (pH = 7.4, phosphate buffer) of acetylthiocoline (10 mM) to the enzyme-inhibitor mixture. The production of the yellow anion was recorded at 412 nm and 25 °C between 0.5 and 1.5 min. As a control, an identical solution of the enzyme without the inhibitor is processed following the same protocol to determine the 100% of enzyme activity.
Kinetic analysis of AChE inhibition: 3 mL of 0.1 M (pH 7.4) phosphate buffer containing DTNB (0.25 mM) and AChE (0.083 U mL−1) was mixed with the opportune volume of compound 26, solubilized in H2O/DMSO (10:1) in order to obtain the final solutions of 26 between 0.5 and 2.5 μM and the final DMSO content of the assay mixture <0.05%. The reaction was started by adding the proper volume of an aqueous solution (pH 7.4, phosphate buffer) of acetylthiocoline (10 mM) to the enzyme–inhibitor mixture. The production of the yellow anion was recorded at 412 nm and 25 °C between 0.5 and 1.5 min. To obtain estimates of the mechanism of action of compound 26, the plot of 1/v versus [I] was constructed at different concentrations of the inhibitor (0.5, 1.0, 1.5, 2.0 and 2.5 µM) and three concentrations of substrate (75, 100 and 150 µM). Each determination was performed in triplicate. The recorded data were analyzed with the enzyme kinetic module of SigmaPlot, version 8.02 a.
Molecular modeling studies
The hMAO-A and hMAO-B enzyme models 2Z5XCitation49 and 2V5ZCitation40 were downloaded from the Protein Data Bank (PDB)Citation39 site, while the crystallographic structure with PDB code 1EVECitation42 was used for the AChE target. Both E and Z isomers of the 3D compounds 26, 14 and 2 were built by means Maestro GUI 9.7 in the Scrodinger Suite 2014Citation50. Protonated and tautomeric forms at pH 7.4 for each molecule were calculated using the “LigPrep” moduleCitation51 and the lowest energy conformations, generated in the absence of the receptor with the OPLS_2005 force field, were used as starting points for the docking simulations. To study all interactions of each target with the ligands, water molecules were removed and all hydrogen atoms were added during the preparation procedure of the enzymes carried out by “Protein Preparation Wizard” function. Docking simulations were performed with the software Glide 6.2Citation36 on the hMAO-A, hMAO-B and AChE crystal structures. Semi-flexible approach was considered: the flexible ligands were sequentially docked in the binding pocket of the rigid targets recording 10 poses per ligand. A cube of length 25 Å was defined as binding region, using the “Receptor Grid Generation” panel with the default options, centered on the former positions of the removed co-crystallized inhibitors with the proteins, into each catalytic site. The accuracy of GlideSP has been shown to successfully reproduce experimentally observed binding modes of co-crystallized inhibitors, in terms of root-mean squared deviation (RMSD) that were respectively: 2Z5X PBD entry: 0.2875 Å; 2V5Z PDB entry: 1.1359 Å; 1EVE PBD entry: 2.9617 Å.
The ligand binding affinity was estimated by means of Glide SP scoring function (Gscore) and binding energies (dG Bind) were calculated by “Prime MM-GBSA” utilityCitation37. The most stable conformations of compounds 26 and 2, complexed with the hMAO-B crystal model, were submitted to a dynamic simulation of 5.0 ns carried out by Desmond packageCitation41: “Simulation Event Analysis” and “Simulation Interactions Diagram” panels were used to display the results.
As regards the AChE target, the best docking Gscore complexes were also minimized by “Embrace” method, as implemented in the Schrödinger Software MacroModelCitation43, performed using an Interaction Energy Mode, to estimate the embrace free energy (EFE) in kcal/mol. Finally, the Pymol 1.7.0.0Citation52 molecular graphics system was used to visualize the molecules, the results of the docking and dynamic experiments and to generate all figures.
Conclusion
Starting from our recent discoveries in the field of new hMAO inhibitors, we synthesized and tested 36 hydrazothiazole derivatives of 2-, 3- and 4-acetylpyridine bearing different aromatic substituents at C4 of the thiazole. Most of the products were active in the low micromolar/high nanomolar range, and some of them were effective at low nanomolar concentrations towards MAO-B isoform. While the coumarin derivatives 12 and 24 were more active and more selective on hMAO-A enzyme with respect to the reference drug Moclobemide, compounds 26, 27 and 28 showed an inhibitory effect on hMAO-B at lower concentrations than that required by R-(−)-Deprenyl. Moreover, compound 28 displayed a selectivity of inhibition that was very similar to that of the above mentioned reference drug. As demonstrated by molecular modeling studies, the presence of an acetylpyridine linked at the hydrazonic nitrogen together with an aryl moiety at C4 position of the pharmacophoric nucleus could be crucial for the inhibitory effect of this scaffold of molecules on monoamine oxidase enzymes. Furthermore, 4-acetylpyridine nucleus conferred to the hydrazothiazole derivatives the best hMAO-B inhibitory activity. We have also investigated the anticholinesterase activity of hydrazones 26–29 and 34 selected from the most active and selective hMAO-B inhibitors. The percentage of inhibition of compound 26 prompted us to determine its type of inhibition and the Ki value. Our results show that 26 inhibits AChE in the low micromolar range with a competitive mechanism, in agreement with the molecular docking study; therefore, the dual effect of derivate 26 against MAO-B and AChE makes it a good candidate for further study in the effort at developing new anti-neurodegenerative agents.
Supplementary material available online
Supplementary Figures S1 and S2.
Supplemental Material.pdf
Download PDF (59 KB)Declaration of interest
This work was supported by “Progetto di Ateneo Ricerca 2013” (P. Chimenti) and Interregional Research Center for Food Safety and Health at the Magna Græcia University of Catanzaro (MIUR PON a3_00359).
The authors state no conflict of interest and have received no payment in preparation of this manuscript.
References
- Bach AW, Lan NC, Johnson DL, et al. cDNA cloning of human liver monoamine oxidase A and B: molecular basis of differences in enzymatic properties. Proc Natl Acad Sci USA 1988;85:4934–8
- Bortolato M, Chen K, Shih JC. Monoamine oxidase inactivation: from pathophysiology to therapeutics. Adv Drug Deliv Rev 2008;60:1527–33
- Murphy DL. Substrate-selective monoamine oxidases-inhibitor, tissue, species and functional differences. Biochem Pharmacol 1978;27:1889–93
- Strolin Benedetti M, Dostert P. Monoamine oxidase, brain ageing and degenerative diseases. Biochem Pharmacol 1989;38:555–61
- Trillo L, Das D, Hsieh W, et al. Ascending monoaminergic systems alterations in Alzheimer’s disease. Translating basic science into clinical care. Neurosci Biobehav Rev 2013;37:1363–79
- Davie CA. A review of Parkinson’s disease. Br Med Bull 2008;86:109–27
- Willner P, Scheel-Krüger J, Belzung C. The neurobiology of depression and antidepressant action. Neurosci Biobehav Rev 2013;37:2331–71
- Carradori S, D’Ascenzio M, Chimenti P, et al. Selective MAO-B inhibitors: a lesson from natural products. Mol Divers 2014;18:219–43
- Secci D, Carradori S, Bolasco A, et al. Discovery and optimization of pyrazoline derivatives as promising monoamine oxidase inhibitors. Curr Top Med Chem 2012;12:2240–57
- Carradori S, Secci D, Bolasco A, et al. Patent-related survey on new monoamine oxidase inhibitors and their therapeutic potential. Expert Opin Ther Pat 2012;22:759–801
- Secci D, Bolasco A, Chimenti P, Carradori S. The state of the art of pyrazole derivatives as monoamine oxidase inhibitors and antidepressant/anticonvulsant agents. Curr Med Chem 2011;18:5114–44
- Bolasco A, Carradori S, Fioravanti R. Focusing on new monoamine oxidase inhibitors. Expert Opin Ther Pat 2010;20:909–39
- Bolasco A, Fioravanti R, Carradori S. Recent development of monoamine oxidase inhibitors. Expert Opin Ther Pat 2005;15:1763–82
- Ramsay RR. Inhibitor design for monoamine oxidases. Curr Pharm Des 2013;19:2529–39
- Carreiras MC, Mendes E, Perry MJ, et al. The multifactorial nature of Alzheimer's disease for developing potential therapeutics. Curr Top Med Chem 2013;13:1745–70
- Bolognesi ML, Rosini M, Andrisano V, et al. MTDL design strategy in the contextof Alzheimeir’s disease: from lipocrine to memoquin and beyond. Curr Pharm Des 2009;15:601–13
- Cavalli A, Bolognesi ML, Minarini A, et al. Multi-target-directed ligands to combat neurodegenerative diseases. J Med Chem 2008;51:347–72
- Bautista-Aguilera OM, Esteban G, Bolea I, et al. Design, synthesis, pharmacological evaluation, QSAR analysis, molecular modeling and ADMET of novel donezepil-indolyl hybrids as multipotent cholinesterase/monoamine oxidase inhibitors for the potential treatment of Alzheimer’s disease. Eur J Med Chem 2014;75:82–95
- Di Giovanni G, García I, Colangeli R, et al. N-(furan-2-ylmethyl)-N-methylprop-2-yn-1-amine (F2MPA): a potential cognitive enhancer with MAO inhibitor properties. CNS Neurosci Ther 2014;20:633–40
- Cappelli A, Gallelli A, Manini M, et al. Further studies on the interaction of the 5-HT3 receptor with arylpiperazine ligands. Development of a new 5-HT3 receptor ligand showing potent acetylcholinesterase inhibitory properties. J Med Chem 2005;48:3564–75
- Colovic MB, Krstic DZ, Lazarevic-Pasti TD, et al. Acetylcholinesterase inhibitors: pharmacology and toxicology. Curr Neuropharmacol 2013;11:315–35
- Mehta M, Adem A, Sabbagh M. New acetylcholinesterase inhibitors for Alzheimer’s disease. Int J Alzheimers Dis 2012;2012:728983
- Chimenti P, Petzer A, Carradori S, et al. Exploring 4-substituted-2-thiazolylhydrazones from 2-, 3-, and 4-acetylpyridine as selective and reversible hMAO-B inhibitors. Eur J Med Chem 2013;66:221–7
- Secci D, Bolasco A, Carradori S, et al. Recent advances in the development of selective human MAO-B inhibitors: (hetero)arylidene-(4-substituted-thiazol-2-yl)hydrazines. Eur J Med Chem 2012;58:405–17
- Secci D, Carradori S, Bolasco A, et al. Synthesis and selective human monoamine oxidase inhibition of 3-carbonyl, 3-acyl, and 3-carboxyhydrazido coumarin derivatives. Eur J Med Chem 2011;46:4846–52
- Chimenti F, Carradori S, Secci D, et al. Synthesis and biological evaluation of novel conjugated coumarin-thiazole systems. J Heterocyclic Chem 2009;46:575–8
- Raza R, Saeed A, Arif M, et al. Synthesis and biological evaluation of 3-thiazolocoumarinyl Schiff-base derivatives as cholinesterase inhibitors. Chem Biol Drug Des 2012;80:605–15
- Chimenti F, Secci D, Bolasco A, et al. Synthesis and selective inhibition of human monoamine oxidases of a large scaffold of (4,5-substituted-thiazol-2-yl)hydrazones. Med Chem Commun 2010;1:61–72
- Carradori S, D’Ascenzio M, De Monte C, et al. Synthesis and selective human monoamine oxidase B inhibition of heterocyclic hybrids based on hydrazine and thiazole scaffolds. Arch Pharm 2013;346:17–22
- D’Ascenzio M, Carradori S, Secci D, et al. Identification of the stereochemical requirements in the 4-aryl-2-cycloalkylidenhydrazinylthiazole scaffold for the design of selective human monoamine oxidase B inhibitors. Bioorg Med Chem 2014;22:2887–95
- Chimenti F, Secci D, Bolasco A, et al. Synthesis, semipreparative HPLC separation, biological evaluation, and 3D-QSAR of hydrazothiazole derivatives as human monoamine oxidase B inhibitors. Bioorg Med Chem 2010;18:5063–70
- Chimenti F, Secci D, Bolasco A, et al. Investigations on the 2-thiazolylhydrazyne scaffold: synthesis and molecular modeling of potent and selective human monoamine oxidase inhibitors. Bioorg Med Chem 2010;18:5715–23
- Yáñez M, Fraiz N, Cano E, Orallo F. Inhibitory effects of cis- and trans-resveratrol on noradrenaline and 5-hydroxytryptamine uptake and on monoamine oxidase activity. Biochem Biophys Res Commun 2006;344:688–95
- Ellman GL, Courtney KD, Andres V, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 1961;7:88–95
- Friggeri L, De Vita D, Pandolfi F, et al. Design, synthesis and evaluation of 3,4-dihydroxybenzoic acid derivatives as antioxidants, bio-metal chelating agents and acetylcholinesterase inhibitors. J Enzyme Inhib Med Chem 2014. doi:10.3109/14756366.2013.866657
- Glide, version 6.2. New York (NY): Schrödinger, LLC; 2014
- Prime version 3.4. New York (NY): Schrödinger, LLC; 2014
- De Colibus L, Li M, Binda C, et al. Three-dimensional structure of human monoamine oxidase A (MAO A): relation to the structures of rat MAO A and human MAO B. Proc Natl Acad Sci USA 2005;36:12684–9
- Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res 2000;28:235–42
- Binda C, Wang J, Pisani L, et al. Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: safinamide and coumarin analogs. J Med Chem 2007;50:5848–52
- Desmond Molecular Dynamics System, version 3.7, D. E. Shaw Research, New York, NY, 2014. Maestro-Desmond Interoperability Tools, version 3.7. New York (NY): Schrödinger; 2014
- Kryger G, Silman L, Sussman JL. Structure of acetylcholinesterase complexed with E2020 (Aricept): implications for the design of new anti-Alzheimer drugs. Structure 1999;7:297–307
- MacroModel, version 10.3, Schrödinger, LLC, New York, NY, 2014
- Chimenti F, Bizzarri B, Bolasco A, et al. Synthesis and biological evaluation of novel 2,4-disubstituted-1,3-thiazoles as anti-Candida spp. agents. Eur J Med Chem 2011;46:378–82
- Secci D, Bizzarri B, Bolasco A, et al. Synthesis, anti-Candida activity, and cytotoxicity of new (4-(4-iodophenyl)thiazol-2-yl)hydrazine derivatives. Eur J Med Chem 2012;53:246–53
- Chimenti F, Bizzarri B, Bolasco A, et al. Synthesis and anti-Helicobacter pylori activity of 4-(coumarin-3-yl)thiazol-2-ylhydrazone derivatives. J Heterocyclic Chem 2010;47:1269–74
- Hassan AA, Ibrahim YR, El-Sheref EM, et al. Synthesis and antibacterial activity of 4-aryl-2-(1-substituted ethylidene)thiazoles. Arch Pharm 2013;346:562–70
- Carradori S, Rotili D, De Monte C, et al. Evaluation of a large library of (thiazol-2-yl)hydrazones and analogues as histone acetyltransferase inhibitors: enzyme and cellular studies. Eur J Med Chem 2014;8:569–78
- Son SY, Ma J, Kondou Y, et al. Structure of human monoamine oxidase A at 2.2-A resolution: the control of opening the entry for substrates/inhibitors. Proc Natl Acad Sci USA 2008;105:5739–44
- Maestro, version 9.7. New York (NY): Schrödinger, LLC; 2014
- LigPrep, version 2.9. New York (NY): Schrödinger, LLC; 2014
- The PyMOL Molecular Graphics System, version 1.7.0.0 Schrödinger, LLC