
Abstract
The interest towards coumarin-based structures stems from their polypharmacological profile. Herein, we present a series of Mannich bases and 7-azomethine-linked coumarin derivatives exhibiting antiplatelet and antithrombotic activities, in addition to the already known anti-inflammatory and antioxidant activities. Among others, compounds 15 and 16 were found to be the most potent and selective inhibitors of platelet aggregation whereas compound 3 also proved to be the most potent in the clot retraction assay. Structure–activity relationship studies were conducted to elucidate the molecular determinants responsible for the herein observed activities. The chance of inhibiting cyclooxygenase-1 was also investigated for evaluating the platelet aggregation induced by arachidonic acid. Taken together, these results suggest that the investigation of other targets connected to the antiplatelet activity, such as phosphodiesterase-3 (PDE3), could be a viable strategy to shed light on the polypharmacological profile of coumarin-based compounds. Docking simulations towards PDE3 were also carried out.
Introduction
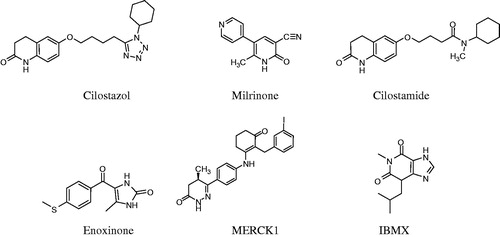
Venous and arterial thromboembolic diseases are still the most frequent causes of deathCitation1. Platelets play an important role in hemostatic and thrombotic processes, modulated by different physiological mediators. Among them, the second messenger cyclic adenosine monophosphate (cAMP) is a strong platelet activation inhibitor. The intracellular cAMP levels are controlled through its synthesis rate by adenylate cyclase and/or its hydrolysis by phosphodiesterases (PDEs)Citation2,Citation3. Three classes of PDEs that is PDE2, PDE3 and PDE5 are contained in platelets. The PDE3 isoform accounts for 80–90% of total platelet PDE and its inhibition induces strong antiplatelet effect. Antiplatelet agents have been proposed as additional therapeutic tools to prevent thromboembolism through multiple mechanisms of action (ADP receptor antagonists GP(glucoprotein)IIb/IIIa antagonists, phosphodiesterase or cyclo-oxygenase inhibitors)Citation4–6. Cilostazol, a PDE3 inhibitor has been proposed as antiplatelet drug endowed with considerable antithrombotic effects in vivoCitation7. Cilostamide, enoximone and milrinone are also known as specific inhibitors of the PDE3 isoform ().
Figure 1. Structures of some phosphodiesterase inhibitors.
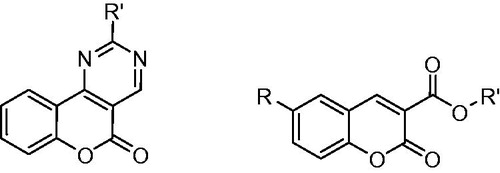
Coumarin derivatives comprise a large class of phenolic substances occurring in plantsCitation8 and presenting multiple biological activitiesCitation9–11. Both natural and synthetic derivatives were found to possess significant anti-inflammatory and antioxidant activities and might be potentially used to treat various ailments (cancer, burns and rheumatic diseases)Citation10,Citation12. Among coumarin derivatives, warfarin, a vitamin K antagonist, was also found to play significant role against thromboembolic diseasesCitation13,Citation14. Several coumarins have also been developed till now as potential antithrombotic and antiplatelet agents ()Citation15–21.
Figure 2. General structures of coumarin derivatives presenting antithrombotic or antiplatelet aggregation activities.
In the course of our research on the design and synthesis of biological active coumarin derivatives, we have already synthesized two series of weak basic coumarin derivatives: (1) coumarin Mannich bases (7-hydroxy-8-substituted coumarin derivatives; , compounds 1–11)Citation22 and (2) 7-azomethine-linked coumarin derivatives (, compounds 12–19)Citation23. Some of these compounds were found to show potent in vivo anti-inflammatory activity both in acute and chronic inflammation, presenting potential either prophylactic or even therapeutic activities without causing ulcerogenicity. Some of them significantly inhibit lipoxygenase, resulting in a potent free radical scavengingCitation22–24 and NO donating activityCitation25. The just claimed biological relevance of these series of compounds, supported by literature dataCitation12,Citation21, prompted us to further investigate their possible antiplatelet and antithrombotic activities as well as their serine protease inhibition (methods given in Supplementary material), being most of them published in a preliminary reportCitation23.
Table 1. Chemical structures and corresponding molecular descriptors of coumarin derivatives 1–19.
Materials and methods
All the reagents and chemicals used are of analytical grade and were obtained as follows: adenosine diphosphate (ADP), bovine thrombin (T3399), acetylsalicylic acid (ASA), calf collagen type III, epinephrine bitartrate, sodium citrate, dimethylsulphoxide (DMSO) and Triton were purchased from Sigma Chemical Co. (St. Louis, MO). U46619 and SQ 29548 were acquired from Caymanchem (Ann Arbor, MI). Thrombin, 50 NIH-E/mg, Trypsin (pancreas protease) 200Fip U/g and the substrates tosyl-Gly-Pro-Arg-pNA, N-tosyl-arginine-methyl ester (TAME), N-Benzoyl-l-tyrosine ethyl ester (BTEE) were purchased from Merck KGaA, Darmstadt, Germany and α-Chymotrypsin from Sigma (51 U/mg) for in vitro evaluation of serine proteases inhibition. UV–Vis spectra were obtained on a Perkin-Elmer 554 double beam spectrophotometer (Perkin-Elmer Corporation Ltd., Lane Beaconsfield, Bucks, UK). Chlorophenol red β-d-galactopyranoside was purchased from Roche Molecular Biochemicals (Indianapolis, IN). Each in vitro experiment was performed at least in triplicate and the standard deviation of absorbance was less than 10% of the mean.
Compounds 1 and 12 are commercially available (Sigma-Aldrich). Compounds 2–11 and 13–19 were previously synthesized in our labCitation22,Citation23. All the studied compounds were dissolved in DMSO and then diluted in the appropriate assay medium up to 0.5% DMSO final concentration.
The concentration-dependent inhibition of platelet aggregation and clot retraction was studied in vitro on rat platelet rich plasma (PRP). The inhibitory potency of the compounds was presented as % activity or as IC50 that is the concentration required to inhibit the response by 50% compared to the control experiment. IC50 was calculated by regression analysis of specific concentration–response curves using least-squares method. The same procedure was also used to assess serine proteases inhibition.
Guinea pig, Fischer and male Wistar rats fed on regular diet were used for the in vivo experiments. The animals were starved at 20°C and fasted 18 h with free access to water before the experiments. All the experiments were performed according to ethical standard guidelines and were approved by Italian Ministry of Health (DL116/92). Data were taken at least from four independent experiments.
Biological studies
Blood manipulation
Blood from male Wistar rats or guinea pigs, was obtained by cardiac puncture after CO2 euthanasia; it was collected in plastic tubes and anticoagulated with sodium citrate 3.8% (1 part citrate: 9 part blood). Blood was centrifuged for 15 min at 180 × g to obtain PRP or 10 min at 2000 × g to obtain platelet poor plasma (PPP).
Platelet aggregation
PRP from guinea pig was used to perform aggregation in the PAP-4D (BioData, Horsham, PA) following Born’s turbidimetric methodCitation26. Aggregation was recorded as percent change in light transmission: the baseline value was set using PRP and maximal transmission using PPP. PRP was pre-incubated at 37°C for 5 min with solvent or compound under study, before addition of platelet agonist. Maximal aggregation was obtained stimulating platelets with 3 µM ADP, 50 µM Arachidonic Acid (AA) or 2 μM. Tests were performed within 3 h avoiding platelet inactivation. The effects of compounds were expressed as percent inhibition compared with control samples. In control samples, 0.5% DMSO did not interfere with platelet aggregation.
Platelet viability
Platelets viability was quantitatively evaluated by measuring the activity of acid phosphatase, a platelet enzyme whose activity is stable independently from platelet stimulation and is not released from vital platelets. A modification of the method described by Bellavite et al.Citation27 was used. Briefly, 150 μl of PRP (platelet-rich plasma) from guinea pig was added to each well of microtiter plate in presence of different concentrations of test compounds dissolved in DMSO or in the presence of 1% triton or 0.5% DMSO. After 30 min of incubation the acid phosphatase activity was measured using substrate p-nitrophenyl phosphate (PNPP) at final concentration of 5 mM. This reaction was allowed to proceed for 30 min at 37°C in the dark and stopped with 150 μl of 2 N NaOH per well. Optical density at 405 nm was measured. Platelet viability was calculated assuming untreated platelets as 100% viability and 1% triton treated platelets as 0% viability.
Clot retraction
PRP from rats was diluted with tyrode buffer (137 mM NaCl, 20 mM Hepes, 5.6 mM glucose, 1 mM MgCl2, 2.7 mM KCl, 3.3 mM NaH2PO4, pH = 7.4) to obtain a final concentration of 200 000 plts µl−1 and the assay was performed according to Davidson et al.Citation28 Briefly, 447.5 µl aliquots of the above platelet suspension was added to siliconize glass tubes and incubated 10 min with 2.5 µl solvent (DMSO) or the compound under study at appropriate concentrations at 37°C. Fibrin clot retraction was induced by addition of 50 µl thrombin 20 U ml−1. Pictures of clots were taken 2 and 60 min after thrombin addition using a digital camera. Quantification of retraction was performed measuring clot area by means of a free access program from Research Services Branch (RSB) of the National Institute of Mental Health (NIMH), part of the National Institutes of Health (NIH), Bethesda, MD. Data were expressed as percentage of retraction = ((area t0 − area t)/area t0) × 100, where t0 was the area of the clot 2 min after thrombin addition and t was the area at the test time.
Enhancement of cAMP level in SK–N–MC cells
SK–N–MC cells were grown in 75 cm2 culture flasks at 37°C in a humidified atmosphere with 5% CO2 in Eagle's minimal essential medium (EMEM), supplemented with 10% (v/v) fetal calf serum, 1% non-essential amino acids, 1% penicillin–streptomycin, 1% l-glutamine and 1% disodium–pyruvate in the presence of 500 µg ml−1 Hygromicine. Cells were used for experiments when they reached about 70–80% confluence and transferred in 96-well plates before the assay. The cells express the reporter gene for β-galactosidase under the control of multiple cAMP responsive elements. To investigate the variation of cAMP levels, the compounds or the vehicle were added at proper concentrations to the cell media followed 5 min later by addition of forskolin (1 µM) or saline (3 µl). After a 6 h incubation at 37°C, media were aspirated and the cells were lysed with 25 µl of 0.1 × assay buffer (composition mM: 10 NaH2PO4, 10 Na2HPO4, pH 8, 0.2 MgSO4, 0.01 MnCl2) and after 10 min with 100 µl of 1 × assay buffer (composition mM: 100 NaH2PO4, 100 Na2HPO4, pH = 8, 2 MgSO4, 0.1 MnCl2) containing 0.5% Triton and 40 mM mercaptoethanol. Color was developed using 25 µl of 1 mg ml−1 substrate solution and quantitated on microplate reader at absorbance 550 nm after overnight incubation at 20°CCitation29. In another pool of experiments forskolin was not added to evaluate the ability of substances to directly stimulate cAMP production. Data are expressed as % of maximal response produced by IBMX (3-isobutyl-1-methylxanthine) 3 µM.
Cell viability
Cell viability was determined through colorimetric quantification of formazan derived from thiazolyl blue tetrazolium bromide (MTT) metabolic reductionCitation30. SK–N–MC cells were suspended at final concentration of 105 cells/ml and plated in 96 well plates. The following day cells were incubated with the compounds under study or the vehicle (0.5% DMSO) for 6 h at proper concentrations. At the end of the incubation, 10 µl of 5 mg/ml MTT solution was added to each well and the plate returned in incubator for 3 h. Afterwards, the culture medium was removed, the cells washed with PBS and 200 µl of formazan solubilization solution (0.1 N HCl in anhydrous isopropanol) added. Culture medium absorbance was spectrophotometrically read at 570 nm (Biorad microplate reader 550, Segrate, MI, Italy). Cells viability was expressed as relative viability compared to control.
2D-QSAR studies: descriptors, regression analysis and statistical parameters
Quantitative structure–activity relationship (QSAR) is expected to uncover how biological activity is dependent on molecular structure modificationsCitation31. Such a methodology, today applied also for regulatory purposesCitation32, is often based on the application of a multivariate equation that relates molecular features to the analyzed activityCitation33. Herein, the multi-regression analyses (MRA) were used in the QSAR model derivation. They were obtained using the C-QSAR programCitation34,Citation35. The resulting best fit models with the statistical parameters of the regressions are given in Equation (Equation1(1) ). In each regression, the 99% confidence limits for each term are indicated in parentheses, n represents the number of data points, r: correlation coefficient between observed and calculated values, s: standard deviation, q2: square of cross-validated correlation coefficient (calculated through leave-one-out procedureCitation36) and F-values for individual term. Clog P is a calculated partition coefficient in octanol/water and is a measure of hydrophobicity calculated with C-QSAR ProgramCitation34,Citation35.
Structural geometry optimization and calculation of 3D descriptors
The molecules were constructed using SPARTANCitation37. Semi-empirical energy minimization (AM1 method)Citation38 along with Monte Carlo (MMFF94)Citation39 conformation analysis were applied to identify the lowest energy conformations of all the synthesized compounds. The energies of most stable conformers were calculated at HF/6-31G* level of theory. The electrostatic potential and the HOMO and LUMO energy values were also determinedCitation40.
Docking studies
Binding studies were performed on human PDE3 to elucidate main binding interactions of coumarin compounds with PDE3B. The X-ray structure of human PDE3B complexed with inhibitor MERCK1 (PDB: 1SO2)Citation41 was used in the docking calculations after removing the inhibitor from the PDB file, obtained from the RCSB Protein Data Bank (PDB)Citation42. Energy minimizations and protein setup was performed using the USCF CHIMERA (University of California, San Francisco, CA)Citation43. UCSF Chimera implements the following force fields: AMBER FF99 for standard residuesCitation44 and GAFF to associate non-standard residues with parameters other than charges. Charges are computed using Antechamber programCitation45.
All the molecules were constructed with ChemDraw program (ChemDraw Ultra© v.12, CambridgeSoft, Cambridge, MA, 1986–2009. http://www.cambridgesoft.com/software/chemDraw/) and converted in 3D-Structures with the OpenBabel (version 2.2.3, OELib Cheminformatics Company)Citation46.
Docking calculations were performed with the Autodock Vina software (Autodock Vina version 1.1.1. Copyright© 1989–2010, The Scripps Research Institute, San Diego, CA, 2010)Citation47. PyRx program (PyRx-Python Prescription version 0.5, 2008–2010 Sargis Pallakyan, The Scripps Research Institute, San Diego, CA, 2010; http://pyrx.scripps.edu/) was employed to generate the docking input files and to analyze the results. The protein was kept rigid, while ligands were allowed to experience full conformational freedom. A grid box of size x = 52.95, y = 42.56 and z = 53.43 points was applied. The blind docking performed on MERCK1 and on all the inhibitors described in this study revealed that all the poses were properly aligned in the active site of PDE3B. Docking was carried out with exhaustiveness value of 64 and a maximum output of 100 binding modes. With this setting, the program Autodock Vina returned for the co-crystallized (i.e. ligand MERCK1) a good match between docking and crystal pose with the root mean square deviation (RMSD) value of 2.40 Å. The final output of docking procedure is a set of solutions ranked according to corresponding scoring function values, each defined by the 3D coordinates of its atoms and expressed as PDB file.
Results
Study of antiplatelet activity
The potential role of coumarin derivatives as antiplatelet agents has been tested using two different platelet aggregation inducers: (a) 3 µM ADP and (b) 50 µM AA ().
Table 2. Activity of studied compounds in ADP (3 µM) or arachidonic acid (50 µM)-induced platelet aggregation (guinea pig) and in clot retraction assay (rat).
During the stimulation with ADP, platelets undergo shape change, release granule contents and start to aggregateCitation48. From the group of coumarin Mannich bases (1–11), compounds 3 and 5 were found slightly active with IC50 values equal to 390 and 385 μM, respectively. It may be inferred that a moderate lipophilicity (Clog P nearly 2.5) may play a role in this activity. On the other hand, the second group of compounds, including 12–19, was found to significantly reduce platelet aggregation induced by ADP. Among them, compound 14 the most potent anti-inflammatory agentCitation23 was found to be also the most potent antiplatelet agent (IC50 = 135 μM), thus suggesting that the antiplatelet activity is improved by the presence of an oxime group at 7-position.
Using AA as inducer, among the compounds 1–11, compounds 3, 5 and 11 were found to be the most actives. Among them, compound 3 is the most active and this could likely be attributed to its molar refractivity value, the highest in the panel. More in general, the group of 7-azomethine linked to coumarin derivatives shows the highest activities. Compound 16, that is the one having the higher lipophilic content, was found to be the most active (IC50 = 19.9 μM) and, most importantly, even more active than acetyl salicylic acid (IC50 = 55 μM), normally used as reference. Although without the selectivity of 16, also compounds 14 and 15 were found to be active.
2D-QSAR studies of coumarin antiplatelet activity induced by arachidonic acid indicated that lipophilicity and ELUMO were the most significant descriptors and played a relevant role for modeling the biological data. The data from both groups in were used to derive the following equation:
(1)
n = 9, r = 0.948, r2 = 0.898, q2 = 0.750, s = 0.152, Q = 6.24, F2,9 = 26.374, α = 0.01 range in log 1/IC50: 3.34–4.7
Clog P expresses the lipophilicity of the whole molecule and ELUMO is an electrophilicity parameter. It appears to be directly proportional to the electronic affinity of the compounds. The equation coefficient with ELUMO has a negative sign. Thus, the increase of ELUMO decreases the biological response. No intercorrelation exists between lipophilicity and ELUMO. Compound 19 was outlier of this equation, since it is simultaneously characterized by a low lipophilicity value and by the lowest ELUMO value. It was therefore omitted in the derivation of Equation (Equation1(1) ).
The coumarin derivatives were unable to prevent platelet aggregation induced by U-46619 (2 μM) up to 500 µM (data not shown). Moreover, none of the tested analogues, at 500 μM concentration, showed platelet toxicity in rat platelet viability using the PNPP assay. Only compounds 3 and 5 were found to slightly reduce platelet viability (74 and 87%) at 500 μM ().
Discussion
Further investigation of the antiplatelet activity of coumarin compounds
It is well known that platelets are activated by thromboxane A2 (TxA2), an AA metabolite acting as endogenous platelet activator. Cyclooxygenase-1 (COX-1) inhibitors like acetyl salicylic acid greatly reduce the production of TxA2 and subsequently inhibit platelet aggregation and formation of the platelet plugCitation49. Thus, we further explored the effect of our compounds against COX-1 (the method is given in Supplementary material). In this respect, compound 16 was tested since it demonstrated selective antiplatelet activity on aggregation induced only by AA. According to the in vitro COX-1 inhibitory activity, compound 16 was found to weakly inhibit COX-1 (33% at 10−5 M and 13% at 10−6 M). No selectivity was found against COX-2 (28% at 10−5 M and 7% at 10−6 M).
Compound 9, a coumarin Mannich base with potent anti-inflammatory activityCitation22,Citation24 and with an interesting anti-aggregatory activity was also tested. Compound 9 was found to slightly inhibit COX-1 (22% at 10−4 M) and COX-2 (21% at 10−4 M), respectively. These results pointed out that the anti-aggregatory activity is not significantly related to the COX-1 inhibitory activity of the considered coumarins.
Study of antithrombotic activity
All the coumarin derivatives were further tested in the clot retraction assay for their possible antithrombotic activity. PRP was used and clot retraction was induced by the addition of thrombin (20 U/ml). From the first group (1–11), compounds 2, 3, 5, 6 and 8 were found to be active. Compound 3 resulted again the most active; it incorporated a bulky substituent at position 8 and exhibited moderate lipophilicity (Clog P = 2.60, from the range 0.42–3.06).
From the other group of derivatives (12–19), compounds 13, 18 and 19 were found to inhibit in vitro the thrombus stability. Moderate lipophilicity seemed to strengthen antithrombotic activity (Clog P: 1.13, 1.00 and 0.69 for compounds 13, 18 and 19, respectively), while a decrease of lipophilicity led to the opposite result. Pyrrolidinyl group seems to be an important structural moiety to bias this biological activity. Unfortunately, the 2D-QSAR analysis did not lead to any significant correlation for this type of activity.
Study on possible serine protease inhibitor activity
The compounds herein reported have been already tested as antiproteolytic agents and the results have been partially reported in a previous workCitation23 (Methods are given in Supplementary material). Most of the compounds have been found to be active (data not shown). Within the examined series (1–11), compound 7, the most lipophilic compound, was the most active (IC50 = 49.5 μM) compared to the others showing only moderate activity (IC50 = 49.5–450 μM). Also, the second group of compounds (12–19) shows a strong inhibitory activity. Compound 13 (IC50: 28.8 μM) was found to be the most active, followed by compound 15 (IC50: 32 μM). The activity of the remaining compounds is within a high range of IC50 values (50–345 μM).
Building on these results, all the compounds were also evaluated as possible in vitro thrombin inhibitors. They were tested on bovine thrombin, slightly inhibited only by compounds 4 and 18 (IC50 of 380 and 330 μM respectively). The derived results do not support any kind of selectivity on thrombin.
Enhancement of c-AMP levels in SK-N-MC cells
A well-known modulator is the second messenger cAMP which acts as a strong inhibitor of platelet activation. The intracellular cAMP levels are controlled through its synthesis rate by adenylate cyclase and/or its hydrolysis by PDEsCitation50. Coumarin derivatives were tested for the enhancement of cAMP in forskolin-stimulated SK–N–MC cells after 6 h incubation (). Results from stimulated and unstimulated cells are given. The compounds presented low enhancement of cAMP intracellular levels at the unstimulated cells compared to the selective phosphodiesterase inhibitor IBMX (3-isobutyl-1-methylxanthine), which is inactive towards platelet aggregation. Among the studied compounds, only compounds 9 and 15 increase cAMP levels in the forskolin stimulated cells (EC50 of 47.9 and 43.1 μM respectively, ).
Table 3. Biological activity of coumarin derivatives on enhancement of cAMP level in SK-N-MC cells and cell viability.
The maximum tolerated concentration in SK–N–MC cell was also determined for all the analogues. Only compounds 3, 5 and 8 presented low tolerated concentration thus indicating that most of the coumarin compounds present no toxicity against platelets.
Molecular docking studies on PDEs
Taking under consideration the influence of some of our compounds on cAMP enhancement, we planned to investigate their behavior as possible phosphodiesterase inhibitors. The PDE3 isoform, which is the most abundant, exhibits low Km for cAMP, accounts for 80–90% of the total platelet PDE and is localized in the cytosolic fraction.
To analyze the binding interactions of the coumarin derivatives in the active site of PDE, docking simulations were performed taking into account all the molecules herein investigated. The human phosphodiesterase complexed with the inhibitor MERCK1 (PDB:1SO2) was chosenCitation41 as the reference model, since it was determined at the highest resolution (2.4 Å) among all the available structures. Docking was carried out using the automated docking program Autodock Vina.
In the catalytic site of PDE3, the interactions of the inhibitor MERCK1 are mostly polar (). The MERCK1 inhibitor is bound in an extended conformation. The dihydropyridazinone moiety fits tightly the binding pocket formed by the backbone of the residues Ile938–Pro941 (from H13) and Lys950–Thr952 (from H14) and the side chain of His948. The nitrogen atoms of dihydropyridazinone engage hydrogen bonds with the side chain of Gln315Citation15,Citation16. The exocyclic oxygen interacts with the side chains of His275. The exocyclic methyl binds a hydrophobic pocket formed by residues Ile938–Pro941. The benzyl ring is stacked between the side chains of Phe991 and Ile955. The 2-aryl substituent extends into a nearby hydrophobic pocket formed by the side chains of Phe959, Pro975, Phe976, Met977, Leu987, Ser990 and Phe991. One end of this pocket is exposed to the solvent in the current structure, and several ordered water molecules are bound there.
Figure 3. (A) Superposition of compounds MERCK1 (cyan), 9 (purple), 15 (magenta) and 16 (green). Docked poses of (B) the ligand MERCK1 (cyan), (C) compound 9 (purple), (D) compound 15 (magenta) and (E) compound 16 (green) in the PDE binding site. A gray scale was instead used for printed version.
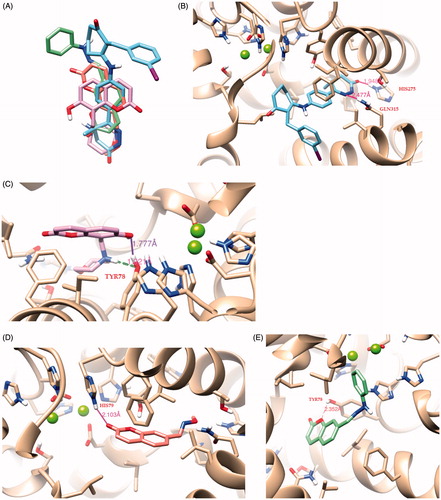
The compounds with the best scoring and poses as well as the highest biological activity are the compounds 9, 15 and 16 in . Interestingly, a nice overlap was observed among the docking poses of the here considered coumarin derivatives and MERCK1 (), thus providing interesting clues for shedding light on the binding mode in the active site of PDE.
Compound 9 returns high docking energy score (−8.9 kcal/mol). The hydroxyl group from the coumarin ring is closed to the active center and presents hydrogen bonds with Tyr78 as well as the nitrogen atom of the amine group.
Compound 15 presents also strong binding affinity with PDE (−8.6 kcal/mol). The carbonyl group from the coumarin ring presents strong hydrogen interaction with His79.
Finally, it is observed that the best compound in terms of biological activity, namely, 16, presents a very high binding energy score (−9.8 kcal/mol) with strong hydrogen interactions with the active site as depicted in . As the inhibitor is anchored in the pocket, the oxygen atom of the coumarin ring is bound to Tyr78.
Conclusions
A series of coumarin derivatives with structural modifications at positions 7- and 8-, already reported as potent in vivo anti-inflammatory agents, both in acute and chronic inflammation, was tested for their possible antithrombotic and antiplatelet activity. Most of the coumarin derivatives presented anti-aggregatory activity, while none of them showed significant platelet toxicity. Compound 3 was found with high antithrombotic activity, although it was slightly more toxic for platelets, giving the less viability to platelets. Compound 16 emerged for its selective antiplatelet activity on aggregation induced only by AA. Lipophilicity and ELUMO primarily affected anti-aggregatory activity. Compounds 9 and 16, further tested as possible COX-1 inhibitors, presented low inhibitory activity, while compounds 4 and 18 were found to slightly inhibit thrombin.
The antiplatelet and antithrombotic effect of coumarin derivatives was investigated through docking studies as PDE3 inhibitors. Compounds 9, 15 and 16 were found to be the most promising PDE3 inhibitors comparable to the selective MERCK1 inhibitor.
The above results lead us to conclude that compounds 15 and 16 are the most promising leading compounds. The multi-target potencyCitation51 of these compounds could be further used to design new series of promising antiplatelet and antithrombotic agents.
Supplementary material available online.
Supplemental Material.pdf
Download PDF (32.3 KB)Acknowledgements
Dr Christos Kontogiorgis and Dr Dimitra Hadjipavlou-Litina are grateful to Biobyte Corp. and Dr Hansch (deceased in 2011) and Dr Leo for their support and free access to the C-QSAR program. This part of research has been done using the above program via Internet. Biobyte Corp., 201 West 4th Street, Suite 204, Claremont, CA 91711, USA. Dr Christos Kontogiorgis is grateful to the Foundation of Education and European Culture, established in Athens, for the financial support for the postgraduate research. Dr Christos Kontogiorgis kindly thanks Professor J. Stürzebecher, Jena, Germany, for his help in the thrombin inhibition test and Fragos A., Pharmacist, Abbott Laboratories, Thessaloniki, Greece, for the kind gift of warfarin.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Sattari M, Lowenthal DT. Novel oral anticoagulants in development: Dabigatran, Rivaroxaban, and Apixaban. Am J Ther 2011;18:332–8
- Alvarez R, Taylor A, Fazzari JJ, Jacobs JR. Regulation of cyclic AMP metabolism in human platelets. Sequential activation of adenylate cyclase and cyclic AMP phosphodiesterase by prostaglandins. Mol Pharmacol 1981;20:302–9
- Tani T, Sakurai K, Kimura Y, et al. Pharmacological manipulation of tissue cyclic AMP by inhibitors. Effects of phosphodiesterase inhibitors on the functions of platelets and vascular endothelial cells. Adv Second Messenger Phosphoprotein Res 1992;25:215–27
- Angiolillo DJ. The evolution of antiplatelet therapy in the treatment of acute coronary syndromes: from aspirin to the present day. Drugs 2012;72:2087–116
- Wijeyeratne YD, Heptinstall S. Anti-platelet therapy: ADP receptor antagonists. Br J Clin Pharmacol 2011;72:647–57
- Schneider DJ. Anti-platelet therapy: glycoprotein IIb-IIIa antagonists. Br J Clin Pharmacol 2011;72:672–82
- Rondina MT, Weyrich AS. Targeting phosphodiesterases in anti-platelet therapy. Handb Exp Pharmacol 2012;210:225–38
- Egan D, O'Kennedy R, Moran E, et al. The pharmacology, metabolism, analysis, and applications of coumarin and coumarin-related compounds. Drug Metab Rev 1990;22:503–29
- Kimura Y, Tani T, Kanbe T, Watanabe K. Effect of cilostazol on platelet aggregation and experimental thrombosis. Arzneimittelforschung 1985;35:1144–9
- Murray RDH, Mendez I, Brawn SA. The natural coumarins. New York: Wiley; 1982
- Catto M, Pisani L, Leonetti F, et al. Design, synthesis and biological evaluation of coumarin alkylamines as potent and selective dual binding site inhibitors of acetylcholinesterase. Bioorg Med Chem 2013;21:146–52
- Fylaktakidou KC, Hadjipavlou-Litina DJ, Litinas KE, Nicolaides DN. Natural and synthetic coumarin derivatives with anti-inflammatory/antioxidant activities. Curr Pharm Des 2004;10:3813–33
- Wilkinson P. New oral anticoagulants: an alternative to warfarin. Br J Hosp Med (Lond) 2012;7:498–501
- Deitelzweig S, Jaff MR. Medical management of venous thromboembolic disease. Tech Vasc Interv Radiol 2004;7:63–7
- Pochet L, Doucet C, Schynts M, et al. Esters and amides of 6-(chloromethyl)-2-oxo-2H-1-benzopyran-3-carboxylic acid as inhibitors of alpha-chymotrypsin: significance of the “aromatic” nature of the novel ester-type coumarin for strong inhibitory activity. J Med Chem 1996;39:2579–85
- Pochet L, Doucet C, Dive G, et al. Coumarinic derivatives as mechanism-based inhibitors of alpha-chymotrypsin and human leukocyte elastase. Bioorg Med Chem 2000;8:1489–501
- Wouters J, Huygens M, Pochet L, et al. Structural approach of the mechanism of inhibition of alpha-chymotrypsin by coumarins. Bioorg Med Chem Lett 2002;12:1109–12
- Frédérick R, Robert S, Charlier C, et al. 3,6-Disubstituted coumarins as mechanism-based inhibitors of thrombin and factor Xa. J Med Chem 2005;48:7592–603
- Bruno O, Brullo C, Schenone S, et al. Synthesis, antiplatelet and antithrombotic activities of new 2-substituted benzopyrano[4,3-d]pyrimidin-4-cycloamines and 4-amino/cycloamino-benzopyrano[4,3-d]pyrimidin-5-ones. Bioorg Med Chem 2006;14:121–30
- Bruno O, Brullo C, Schenone S, et al. Synthesis and pharmacological evaluation of 5H-[1]benzopyrano[4,3-d]pyrimidines effective as antiplatelet/analgesic agents. Bioorg Med Chem 2004;12:553–61
- Roma G, Cinone N, Di Braccio M, et al. Synthesis, antiplatelet activity and comparative molecular field analysis of substituted 2-amino-4H-pyrido[1,2-a]pyrimidin-4-ones, their congeners and isosteric analogues. Bioorg Med Chem 2000;8:751–68
- Kontogiorgis CA, Hadjipavlou-Litina DJ. Synthesis and antiinflammatory activity of coumarin derivatives. J Med Chem 2005;48:6400–8
- Kontogiorgis CA, Hadjipavlou-Litina DJ. Synthesis and biological evaluation of novel coumarin derivatives with a 7-azomethine linkage. Bioorg Med Chem Lett 2004;14:611–14
- Kontogiorgis CA, Savvoglou K, Hadjipavlou-Litina DJ. Antiinflammatory and antioxidant evaluation of novel coumarin derivatives. J Enzyme Inhib Med Chem 2006;21:21–9
- Kontogiorgis CA, Hadjipavlou Litina D. Nitric oxide release from coumarin-7-azomethine derivatives in the presence of thiol. Arzneimittelforschung 2007;57:143–6
- Born GV. Aggregation of blood platelets by adenosine diphosphate and its reversal. Nature 1962;194:927–9
- Bellavite P, Andrioli G, Guzzo P, et al. A colorimetric method for the measurement of platelet adhesion in microtiter plates. Anal Biochem 1994;216:444–50
- Davidson I, Henry JB. Clinical diagnosis of laboratory methods. 15th ed. Philadelphia: WB Sanders Company;1974:438–9
- Apodaca R, Dvorak CA, Xiao W, et al. A new class of diamine-based human histamine H3 receptor antagonists: 4-(aminoalkoxy)benzylamines. J Med Chem 2003;46:3938–44
- Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods 1986;89:271–7
- Nicolotti O, Carotti A. QSAR and QSPR studies of a highly structured physicochemical domain. J Chem Inf Model 2006;46:264–76
- Nicolotti O, Benfenati E, Carotti A, et al. REACH and in silico methods: an attractive opportunity for medicinal chemists. Drug Disc Today 2014;19:1757–68
- Gissi A, Gadaleta D, Floris M, et al. An alternative QSAR-based approach for predicting the bioconcentration factor for regulatory purposes. Altex 2014;31:23–36
- C-QSAR, version 4 31. Claremont (CA): Biobyte Corp.; 2003. Available from: www.biobyte.com [last accessed Nov 2013]
- Hansch C, Leo AJ. Exploring QSAR fundamentals and applications in chemistry and biology. In: Heller SR, ed. ACS Professional Reference Book. Washington DC: American Chemical Society. 1995:279–80
- Cramer RD, Patterson DE, Bunce JD. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J Am Chem Soc 1988;110:5959–67
- SPARTAN SGI, version 5.1.3 OpenGL. Irvine (CA): Wavefunction, Inc
- Dewar MJS, Zoebisch EG, Healy EF, Stewart JJP. Development and use of quantum mechanical molecular models. 76.AM1: a new general purpose quantum mechanical molecular model. J Am Chem Soc 1985;107:3902–9
- Halggren TA. Merck molecular force field I. Basis, form, scope, parameterization and performance of MMFF94. J Comput Chem 1996;17:490–519
- Stewart JJP. Optimization of parameters for semi-empirical methods I-Method. J Comp Chem 1989;10:209–20
- Scapin G, Patel SB, Chung C, et al. Crystal structure of human phosphodiesterase 3B: atomic basis for substrate and inhibitor specificity. Biochemistry 2004;43:6091–100
- http://www.pdb.org
- USCF CHIMERA. University of California, San Francisco, CA, USA; 1994. Available from: http://www.cgl.ucsf.edu/chimera [last accessed 10 Feb 2010]
- Lindorff Larsen K, Piana S, et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010;78:1950–8
- Wang J, Wolf RM, Caldwell JW, et al. Development and testing of a general amber force field. J Comput Chem 2004;25:1157–74
- OpenBabel, version 2.2.3, Open Source Chemistry Toolbox. OELib Cheminformatics Company, USA; 2006. Available from: http://sourceforge.net/projects/openbabel [last accessed 10 Feb 2010]
- Autodock Vina v. 1.1.1., The Scripps Research Institute, San Diego, CA, USA; 2010. Copyright© 1989–2010
- Jin J, Kunapuli SP. Coactivation of two different G protein-coupled receptors is essential for ADP-induced platelet aggregation. Proc Natl Acad Sci USA 1998;95:8070–4
- Nurtjahja-Tjendraputra E, Ammit AJ, Roufogalis BD, et al. Effective anti-platelet and COX-1 enzyme inhibitors from pungent constituents of ginger. Thromb Res 2003;111:259–65
- Eisert WG. Near-field amplification of antithrombotic effects of dipyridamole through vessel wall cells. Neurology 2001;57:S20–3
- Nicolotti O, Giangreco I, Miscioscia TF, Carotti A. Improving quantitative structure-activity relationships through multiobjective optimization. J Chem Inf Model 2009;49:2290–302