
Abstract
A novel series of 3-benzyl-substituted-4(3H)-quinazolinones were designed, synthesized and evaluated for their in vitro antitumor activity. The results of this study demonstrated that 2-(3-benzyl-6-methyl-4-oxo-3,4-dihydroquinazolin-2-ylthio)-N-(3,4,5-trimethoxyphenyl)acetamide, 2-(3-benzyl-6,7-dimethoxy-4-oxo-3,4-dihydroquinazolin-2-ylthio)-N-(3,4,5-trimethoxyphenyl)acetamide and 3-(3-benzyl-6-methyl-4-oxo-3,4-dihydroquinazolin-2-ylthio)-N-(3,4,5-trimethoxyphenyl)-propanamide have shown amazing broad spectrum antitumor activity with mean GI50 (10.47, 7.24 and 14.12 µM. respectively), and are nearly 1.5–3.0-fold more potent compared with the positive control 5-FU with mean GI50, 22.60 µM. On the other hand, compounds 6 and 10 yielded selective activities toward CNS, renal and breast cancer cell lines, whereas compound 9 showed selective activities towards leukemia cell lines. Molecular docking methodology was performed for compounds 7 and 8 into ATP binding site of EGFR-TK which showed similar binding mode to erlotinib, while compound 11 into ATP binding site of B-RAF kinase inhibited the growth of melanoma cell lines through inhibition of B-RAF kinase, similar to PLX4032.
Introduction
Cancer is a disease characterized by a shift in the controlled mechanisms that govern cell proliferation and differentiationCitation1. Malignancy is caused by abnormalities in cells, which might be due to inherited genes or caused by outside exposure of the body to chemicals, radiation, or even infectious agentsCitation2,Citation3. Several techniques have been adopted for the treatment and eradication of cancerous cells. These techniques involved surgery, radiation, immunotherapy, chemotherapy and chemoprevention. Ideal anticancer drugs would eradicate cancer cells without harming normal tissues. Unfortunately, no currently available agents meet this criterion, and clinical use of drugs involves a weighing of benefits against toxicity in a search of favorable therapeutic indexCitation4. Many of chemotherapeutic agents currently used in cancer therapy are agents which inhibit tumor growth by inhibiting the replication and transcription of DNA. The wide occurrence of the heterocycles in bioactive natural products made them important synthetic targets. Quinazolines represent a class of heterocyclic compounds of great importance in biological chemistryCitation5–11.
Quinazolines are frequently used in medicine because of their wide spectrum of biological activitiesCitation5–11. In the course of identifying various chemical substances which may serve as leads for designing novel antitumor agents, we are particularly interested in the present work with quinazoline derivatives which have been identified as a new class of cancer chemotherapeutic agents with significant therapeutic efficacy against solid tumorsCitation10,Citation11. It is well known that quinazoline derivatives are potent inhibitors of epidermal growth factor receptor (EGFR)Citation12–17. The EGFR is cellular trans-membrane tyrosine kinases that are over-expressed in a significant number of human tumors (e.g., breast, ovarian, colon, renal, and prostate)Citation18,Citation19. Overexpression of EGFR family receptors have been observed in these tumors, approximately in 60% of all tumors. A number of small molecule EGFR kinase inhibitors have been evaluated in cancer clinical trialsCitation12–19. For example (), anilinoquinazoline-containing compounds gefitinib (IressaTM),Citation13c–19 erlotinib (TarcevaTM),Citation15a lapatinib (TykerbTM, also known as GW-572016) and Vandetanib (ZactimaTM) were recently approved for the treatment of breast cancer and non-small-cell lung cancerCitation15b–17. On the other hand, melanoma is the most aggressive type of skin cancer in which V600E-B-RAF kinase is over-expressed in 63% of all malignant melanomasCitation20a. Vemurafenib (PLX4032) is a drug which targets inhibition of V600E-B-RAF kinaseCitation20a. In 2011, it was approved by the U.S. Food and Drug Administration (FDA) for the treatment of late-stage melanoma, so inhibition of V600E-B-RAF kinase is a very potential avenue for the treatment of melanomaCitation20a. Moreover, a series of salicylanilides were synthesized and their inhibitory activity against tyrosine kinases was determined. Some of them indeed have proven to be potent and selective EGFR tyrosine kinase inhibitorsCitation20b (). Many more compounds are still under evaluation in clinical trials for the treatment of cancerCitation18b,Citation19.
Figure 1. Reported and designed quinazoline derivatives as antitumor.
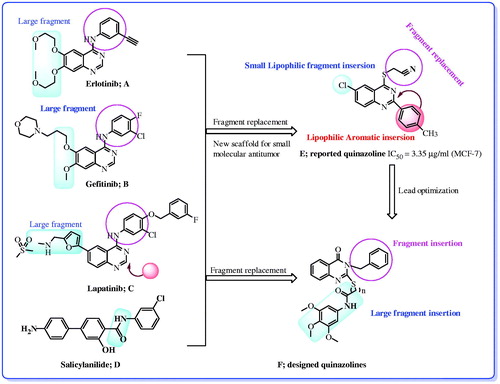
Continuing our studies on quinazoline derivatives as attractive candidates as antitumor agentsCitation10b,Citation11a,Citation20b, we have designed a number of new quinazoline derivatives containing benzyl group with different fragments and biologically evaluated the in vitro antitumor activities (). In the present study, the substitution pattern at the 6 and 7 positions of quinazoline pharmacophore was selected so as to confer different electronic environment that would affect the lipophilicity, and hence the activity of the target molecules. The objectives of forming these hybrids are an attempt to reach an active antitumor agent with potentiated activity and selectivity toward cancerous cells. Molecular docking methodology was used to identify the structural features required for the antitumor properties of these new series.
Materials and methods
Chemistry
Melting points were recorded on Barnstead 9100 Electrothermal melting apparatus at the Pharmaceutical Chemistry Department, King Saud University, Riyadh, Saudi Arabia. IR spectra (KBr) were recorded on a FT-IR Perkin-Elmer spectrometer (ν cm−1) at Research Center, King Saud University, Riyadh, Saudi Arabia. Nuclear magnetic resonance (1H and 13C NMR) spectra were recorded on Bruker 700 MHz or 500 MHz spectrometer using DMSO-d6 or CDCl3 as solvents at Research Center, King Saud University, Riyadh, Saudi Arabia. The chemical shifts are expressed in δ ppm using TMS as internal standard. Mass spectra were recorded on a Perkin-Elmer, Clarus 600T GC/MS, or Varian TQ 320 GC/MS/MS mass spectrometers at Research Center, King Saud University, Riyadh, Saudi Arabia. Single crystal X-ray crystallography was recorded on Bruker APEXII CCD diffractometer at Chemistry Department, University of Malaya, Kuala Lumpur, Malaysia and structure refinement and resolve was carried out using SHELXS97Citation21a. Solvent evaporation was performed under reduced pressure using Buchan Rotatory Evaporator at the Pharmaceutical Chemistry Department, King Saud University, Riyadh, Saudi Arabia. Thin-layer chromatography was performed on precoated (0.25 mm) silica gel GF254 plates (E. Merck, Germany); compounds were detected with 254 nm UV lamp. Silica gel (60–230 mesh) was employed for routine column chromatography separations. Compounds 1–3 and 14–15 were prepared according to reported procedureCitation11b, while compounds 13 and 21 were prepared according to the reported methodCitation7a,Citation21b respectively.
General procedure for the synthesis of 3-benzyl-2-mercapto-substitutedquinazolin-4(3H)-one (1–4)
A mixture of substituted anthranilic acid (10 mmol) and benzylisothiocyanate (10 mmol, 1.49 g) in 30 ml absolute ethanol containing triethylamine (11 mmol, 1.1 g) was heated under reflux for 2 h. The reaction mixture was filtered while hot, the solvent was removed under reduced pressure and the solid obtained was dried and recrystallized from ethanol.
3-Benzyl-2-mercaptoquinazolin-4(3H)-one (1)
Yield 95%, m.p. 240–242 °C, 1H NMR (700 MHz, DMSO-d6): δ 13.06 (s, 1H), 7.96 (d, 1H, J = 15.34 Hz), 7.75 (t, 1H, J = 14.0 Hz), 7.43 (d, 1H, J = 15.34 Hz), 7.34–7.29 (m, 5H), 7.24 (d, 1H, J = 12.5 Hz), 5.67 (s, 2H). 13C NMR (175 MHz, DMSO-d6): δ49.2, 115.9, 116.2, 125.0, 127.4, 127.6, 127.8, 128.7, 136.1, 137.0, 139.6, 159.8, 176.0.
3-Benzyl-6-chloro-2-mercaptoquinazolin-4(3H)-one (2)
Yield 95%, m.p. 248–251 °C; 1H NMR (700 MHz, DMSO-d6): δ 13.17 (s, 1H), 7.88 (s, 1H), 7.80 (d, 1H, J = 8.36 Hz), 7.34 (d, 1H, J = 16.73 Hz), 7.33–7.24 (m, 4H), 7.23 (s, 1H), 5.65 (s, 2H). 13C NMR (175 Hz, DMSO-d6): δ 49.3, 117.3, 118.4, 126.7, 127.4, 127.7, 128.6, 128.9, 136.0, 136.8, 138.4, 159.0, 175.9.
3-Benzyl-2-mercapto-6-methylquinazolin-4(3H)-one (3)
Yield 95%, m.p. 259–261 °C; 1H NMR (700 MHz, DMSO-d6): δ 13.01 (s, 1H), 7.75 (s, 1H), 7.58 (d, 1H, J = 15.34 Hz), 7.34–7.29 (m, 5H), 7.23 (s, 1H), 5.67 (s, 2H), 2.35 (s, 3H). 13C NMR (175 Hz, DMSO-d6): δ 20.9, 49.1, 115.7, 116.1, 127.1, 127.3, 127.5, 128.6, 134.7, 137.1, 137.2, 137.6, 159.8, 175.5.
3-Benzyl-2-mercapto-6,7-dimethoxyquinazolin-4(3H)-one (4)
Yield 82%, m.p. 268–270 °C; IR (KBr, cm−1) ν: 3180 (NH), 1684 (CO), 1251 (CS); 1H NMR (500 MHz, DMSO-d6): δ 12.86 (s, 1H), 7.30–6.94 (m, 7H), 5.66 (s, 2H), 3.85 (s, 6H). 8.04 (d, 1H, J = 1.5 Hz), 7.82 (d, 2H, J = 8.0 Hz), 7.68 (d, 1H, J = 8.5 Hz), 7.37–7.22 (m, 2H), 2.33 (s, 3H, CH3). 13C NMR (125 MHz, DMSO-d6): δ 48.6, 55.7, 56.0, 97.7, 106.7, 107.9, 126.8, 127.1, 128.1, 135.0, 136.8, 146.7, 155.3, 158.8, 174.3. MS: (M, 328).
General procedure for the synthesis of compounds 5–12
A mixture of appropriate 2-mercapto-3-benzylquinazolin-4(3H)-one (1) (2 mmol) and/or the appropriate 2-chloro-N-phenylacetamide and/or phenyl 2-chloroacetate (2 mmol) in 15 ml acetone containing anhydrous potassium carbonate (3 mmol, 415 mg) was stirred at room temperature for 10–12 h. The reaction mixture was filtered, the solvent was removed under reduced pressure and the solid obtained was dried and recrystallized from ethanol.
2-(3-Benzyl-4-oxo-3,4-dihydroquinazolin-2-ylthio)-N-(3,4,5-trimethoxyphenyl)acetamide (5)
Yield 92%, m.p; 201–203 °C; IR (KBr, cm−1) ν: 3287 (NH), 1674, 1655 (CO); 1H NMR (500 MHz, DMSO-d6): δ 9.88 (s, 1H), 8.34 (d, 1H, J = 7.5 Hz), 7.80 (t, 1H, J = 7.0 Hz), 7.70 (d, 1H, J = 8.0 Hz), 7.51 (t, 1H, J = 7.0 Hz), 7.40–7.31 (m, 5H), 6.73 (s, 2H), 5.42 (s, 2H), 3.99 (s, 2H), 3.78 (s, 3H), 3.74 (s, 6H). 13C NMR (125 MHz, DMSO-d6): δ 36.3, 47.8, 56.0, 61.0, 96.8, 119.6, 124.8, 126.7, 127.8, 128.0, 128.1, 128.8, 134.1, 134.5, 134.7, 135.0, 146.4, 153.3, 158.0, 161.3, 166.3. MS: (M, 491).
2-(3-Benzyl-6-chloro-4-oxo-3,4-dihydroquinazolin-2-ylthio)-N-(3,4,5-trimethoxyphenyl)acetamide (6)
Yield 88%, m.p. 190–192 °C; IR (KBr, cm−1) ν: 3243 (NH), 1663, 1650 (CO); 1H NMR (500 MHz, DMSO-d6): δ 9.48 (s, 1H), 8.28 (s, 1H), 7.71 (dd, 1H, J = 7.0, 1.5 Hz), 7.62 (d, 1H, J = 8.5 Hz), 7.37–7.31 (m, 5H), 6.71 (s, 2H), 5.41 (s, 2H), 3.99 (s, 2H), 3.79 (s, 3H), 3.76 (s, 6H). 13C NMR (125 MHz, DMSO-d6): δ 36.3, 48.0, 56.0, 61.0, 97.0, 120.6, 126.6, 127.2, 127.8, 128.2, 128.8, 132.4, 133.9, 134.5, 134.7, 135.3, 145.0, 153.3, 158.1, 160.4, 166.0. MS: (M, 526 and M + 2, 528).
2-(3-Benzyl-6-methyl-4-oxo-3,4-dihydroquinazolin-2-ylthio)-N-(3,4,5-trimethoxyphenyl)acetamide (7)
Yield 87%, m.p; 199–200 °C; IR (KBr, cm−1) ν: 3310 (NH), 1685, 1654 (CO); 1H NMR (500 MHz, DMSO-d6): δ 9.94 (s, 1H), 8.13 (s, 1H), 7.62 (s, 2H), 7.38–7.30 (m, 5H), 6.73 (s, 2H), 5.42 (s, 2H), 3.97 (s, 2H), 3.79 (s, 3H), 3.75 (s, 6H), 2.63 (s, 3H). 13C NMR (125 MHz, DMSO-d6): δ 21.3, 36.3, 47.8, 56.0, 61.0, 96.9, 119.3, 124.7, 127.4, 127.8, 128.1, 128.8, 134.1, 134.5, 134.9, 136.3, 137.1, 144.5, 153.3, 157.0, 161.4, 166.4. MS: (M, 505).
2-(3-Benzyl-6,7-dimethoxy-4-oxo-3,4-dihydroquinazolin-2-ylthio)-N-(3,4,5-trimethoxyphenyl)acetamide (8)
Yield 86%, m.p. 205–206 °C; IR (KBr, cm−1) ν: 3304 (NH), 1677, 1656 (CO); 1H NMR (500 MHz, DMSO-d6): δ 9.71 (s, 1H), 7.63 (s, 1H), 7.37–7.34 (m, 5H), 7.04 (s, 1H), 6.73 (s, 2H), 5.42 (s, 2H), 4.00 (s, 2H), 3.98 (s, 6H), 3.78 (s, 3H), 3.74 (s, 6H).13C NMR (125 MHz, DMSO-d6): δ 36.2, 47.8, 56.0, 56.4, 56.5, 61.0, 97.0, 105.6, 106.6, 112.7, 127.7, 128.0, 128.8, 134.1, 134.7, 135.0, 142.8, 148.9, 153.4, 155.6, 155.7, 160.9, 166.4. MS: (M, 551).
3-(3-Benzyl-4-oxo-3,4-dihydroquinazolin-2-ylthio)-N-(3,4,5-trimethoxyphenyl)propanamide (9)
Yield 90%, m.p. 195–196 °C; IR (KBr, cm−1) ν: 3292 (NH), 1685, 1662 (CO); 1H NMR (500 MHz, DMSO-d6): δ 10.40 (s, 1H), 8.12 (d, 1H, J = 7.5 Hz), 7.83 (t, 1H, J = 7.0, 7.5 Hz), 7.61 (d, 1H, J = 8.0 Hz), 7.48 (t, 1H, J = 7.5 Hz), 7.36–7.27 (m, 5H), 7.01 (s, 2H), 5.33(d, 2H, J = 7.0 Hz), 4.74 (d, 1H, J = 6.5 Hz), 3.73 (s, 6H), 3.61 (s, 3H), 1.60 (s, 3H).13C NMR (125 MHz, DMSO-d6): δ 17.7, 46.7, 47.0, 55.6, 60.0, 97.0, 118.8, 125.8, 126.2, 126.6, 126.7, 127.4, 128.6, 133.6, 134.9, 135.5, 146.7, 152.7, 156.2, 160.8, 169.0. MS: (M, 505).
3-(3-Benzyl-6-chloro-4-oxo-3,4-dihydroquinazolin-2-ylthio)-N-(3,4,5-trimethoxyphenyl)propanamide (10)
Yield 89%, m.p. 196–198 °C; IR (KBr, cm−1) ν: 3307 (NH), 1683, 1663 (CO); 1H NMR (500 MHz, DMSO-d6): δ 10.41 (s, 1H), 8.04 (s, 1H), 7.88 (d, 1H, J = 7.0 Hz), 7.69 (d, 1H, J = 7.0 Hz), 7.43–7.28 (m, 5H), 7.0 (s, 2H), 5.31 (s, 2H), 4.71 (d, 1H, J = 6.5 Hz), 3.71 (s, 6H), 3.60 (s, 3H), 1.58 (d, 3H, J = 6.5 Hz). 13C NMR (125 MHz, DMSO-d6): δ 17.6, 48.8, 47.2, 55.6, 60.1, 96.8, 120.0, 125.6, 126.8, 127.5, 128.0, 128.6, 130.2, 133.5, 134.9, 135.0, 135.2, 145.4, 152.7, 157.0, 159.9, 169.0. MS: (M, 539, M + 2, 541).
3-(3-Benzyl-6-methyl-4-oxo-3,4-dihydroquinazolin-2-ylthio)-N-(3,4,5-trimethoxyphenyl)propanamide (11)
Yield 85%, m.p 217–219 °C; IR (KBr, cm−1) ν: 3293 (NH), 1686, 1662 (CO); 1H NMR (500 MHz, DMSO-d6): δ 10.42 (s, 1H), 7.91 (s, 1H), 7.62 (d, 1H, J = 7.0 Hz), 7.51 (d, 1H, J = 7.0 Hz), 7.43–7.25 (m, 5H), 7.01 (s, 2H), 5.30 (s, 2H), 4.72 (d, 1H, J = 6.5 Hz), 3.72 (s, 6H), 3.60 (s, 3H), 2.42 (s, 3H), 1.58 (d, 3H, J = 6.5 Hz). 13C NMR (125 MHz, DMSO-d6): δ 17.6, 20.7, 46.6, 46.8, 55.6, 60.0, 96.8, 118.5, 125.7, 125.9, 126.7, 127.4, 128.6, 133.5, 135.0, 135.6, 136.0, 136.2, 144.9, 152.9, 155.1, 160.7, 169.1. MS: (M, 519).
3-(3-Benzyl-6,7-dimethoxy-4-oxo-3,4-dihydroquinazolin-2-ylthio)-N-(3,4,5-trimethoxyphenyl)propanamide (12)
Yield 84%, m.p; 150–152 °C; IR (KBr, cm−1) ν: 3275 (NH), 1684, 1670 (CO); 1H NMR (500 MHz, DMSO-d6): δ 10.34 (s, 1H), 7.43–7.27 (m, 7H), 7.05 (s, 2H), 5.31 (s, 2H), 4.69 (s, 3H), 3.87 (s, 3H), 3.83 (s, 3H),3.72 (s, 3H), 3.61 (s, 1H), 3.51 (s, 3H), 1.59 (s, 3H).13C NMR (125 MHz, DMSO-d6): δ 17.6, 46.5, 46.8,55.6, 55.7, 60.1, 96.8, 105.6, 106.7, 11.6, 126.7, 127.4, 128.5, 133.6, 135.1, 135.7, 143.1, 148.2, 152.7, 154.1, 155.0, 160.1, 169.2. MS: (M, 565).
Ethyl 2-(3-benzyl-6-methyl-4-oxo-3,4-dihydroquinazolin-2-ylthio)acetate (14)
A mixture of 3-benzyl-2-mercapto-6-methylquinazolin-4(3H)-one (3) (10 mmol, 2.82 g) ethyl 2-bromoacetate (11 mmol, 1.84 g), and anhydrous potassium carbonate (12 mmol, 1.66 g) in (100 ml) dry acetone was stirred at room temperature for 6 h. The reaction mixture was filtered, the solvent was removed under reduced pressure, and the solid obtained was dried and recrystallized from ethanol. Yield 95%; m.p; 130–133 °C; 1H NMR (700 MHz, DMSO-d6), δ 7.90 (s, 1H), 7.64 (d, 1H, J = 12.55 Hz), 7.38 (d, 1H, J = 12.55 Hz), 7.34 (d, 2H, J = 9.76 Hz), 7.428 (s, 3H,), 5.33 (s, 2H), 4.13 (q, 3H, J = 9.76 Hz), 4.08 (s, 2H), 2.43 (s, 3H), 1.19 (s, 3H). 13C NMR (175 MHz, DMSO-d6): δ 14.6, 21.2, 34.6, 47.4, 61.5, 118.8, 126.2, 126.4, 127.2, 127.9, 129.0, 136.0, 136.4, 136.7, 145.2, 155.5, 161.2, 168.7. MS: (M, 368).
2-(3-Benzyl-6-methyl-4-oxo-3,4-dihydroquinazolin-2-ylthio)acetohydrazide (15)
A solution of the ethyl 2-(3-benzyl-6-methyl-4-oxo-3,4-dihydroquinazolin-2-ylthio)acetate (14) (3.68 g, 0.01 mol) and hydrazine hydrate (85 %, 3 ml) in ethanol (40 ml) was heated under reflux for 2 h. The obtained solid was filtered, dried, and recrystallized from ethanol. Yield 95%, m.p. 207–210 °C; 1H NMR (700 MHz, DMSO-d6): δ 9.36 (s, 1H), 7.91 (s, 1H), 7.66 (d, 1H, J = 13.94 Hz), 7.50 (d, 1H, J = 15.34 Hz), 7.38–7.27 (m, 5H), 5.34 (s, 2H), 4.35 (s, 2H), 3.93 (s, 2H), 2.44 (s, 3H).13C NMR (175 MHz, DMSO-d6) δ21.2, 35.0, 47.3, 118.9, 126.3, 126.4, 127.2, 127.9, 129.0, 136.1, 136.3, 136.6, 145.2, 155.5, 161.3, 166.6. MS: (M, 354).
2-(3-Benzyl-6-methyl-4-oxo-3,4-dihydroquinazolin-2-ylthio)-N′-substituted benzylideneacetohydrazide (16–18)
A solution of acid hydrazide 15 (1 mmol, 354 mg) in methanol (10 mL) and the appropriate aldehyde (1 mmol) was stirred at room temperature for 12–15 h. The reaction mixture was washed with hexane, filtered and dried.
2-(3-benzyl-6-methyl-4-oxo-3,4-dihydroquinazolin-2-ylthio)-N'-benzylideneacetohydrazide (16)
Yield 92%; m.p. 225–227 °C; 1H NMR (700 MHz, DMSO-d6): δ 11.81 (s, 0.37 H), 11.65 (s, 0.63 H), 8.27 (s, 0.37 H), 8.07 (s, 0.63 H), 7.90 (s, 1H), 7.70 (s, 2H), 7.63–7.59 (m, 1H), 7.45–7.41 (m, 3H), 7.35–7.32 (m, 6H), 5.36 (s, 2H), 4.56 (s, 1.2 H), 4.09 (s, 0.8 H), 2.41 (s, 3H). 13C NMR (175 MHz, DMSO-d6) δ21.2, 34.3, 47.3, 118.9, 126.3, 126.4, 127.2, 127.3, 127.5, 127.9, 129.0, 129.3, 130.5, 134.6, 136.1, 136.3, 136.6, 143.8, 145.3, 147.1, 155.7, 161.1, 164.0, 169.0, MS: (M, 422).
2-(3-benzyl-6-methyl-4-oxo-3,4-dihydroquinazolin-2-ylthio)-N′-(2-fluorobenzylidene)acetohydrazide (17)
Yield 92%; m.p. 219–221 °C; 1H NMR (700 MHz, DMSO-d6): δ 11.95 (s, 0.33 H), 11.76 (s, 0.67 H), 8.50 (s, 0.33 H), 8.28 (s, 0.67 H), 7.90 (s, 2H), 7.63–7.59 (m, 1H), 7.47–7.45 (m, 1H), 7.35–7.20 (m, 8H), 5.36 (s, 2H), 4.56 (s, 1.2 H), 4.08 (s, 0.8 H), 2.41 (s, 3H). 13C NMR (175 MHz, DMSO-d6) δ21.2, 34.2, 37.3, 116.5, 118.9, 122.1, 125.3, 125.4, 126.4, 126.7, 126.8, 127.2, 127.8, 127.9, 129.1, 132.2, 136.1, 136.2, 136.6, 155.8, 160.5, 161.9, 164.1, 169.3. MS: (M, 460).
2-(3-Benzyl-6-methyl-4-oxo-3,4-dihydroquinazolin-2-ylthio)-N′-(2-methoxybenzylidene)acetohydrazide (18)
Yield 90%; m.p. 203–205 °C; 1H NMR (700 MHz, DMSO-d6), 11.79 (s, 0.36 H), 11.60 (s, 0.64 H), 8.61 (s, 0.36 H), 8.40 (s, 0.64 H), 7.90 (s, 1H), 7.82 (d, 0.64 H, J = 11.15 Hz), 7.77 (d, 0.36, J = 12.55 Hz), 7.63 (d, 0.36 H, J = 13.94 Hz), 7.60 (d, 0.64 H, J = 13.94 Hz), 7.45 (d, .036, J = 13.94 Hz), 7.40 (d, 0.64 H, J = 13.94 Hz), 7.35–7.34 (m, 6H), 7.31 (d, 1H, J = 11.15 Hz), 7.30 (d, 0.36 H, J = 5.76 Hz), 7.29 (d, 0.64, J = 25.1 Hz), 5.36 (s, 2H), 4.55 (s, 1.2 H), 4.06 (s, 0.8 H), 3.86 (s, 3H), 2.42 (s, 3H). 13C NMR (175 MHz, DMSO-d6): δ 21.2, 34.3, 47.3, 56.2, 112.3, 118.9, 121.1, 122.5, 125.9, 126.3, 127.2, 127.8, 129.0, 131.8, 132.0, 136.1, 136.6, 139.5, 142.6, 145.3, 155.7, 155.9, 158.1, 161.2, 163.8, 169.1.
2-(3-Benzyl-6-methyl-4-oxo-3,4-dihydroquinazolin-2-ylthio)-N-(4-oxo-2-phenylthiazolidin-3-yl)acetamide (19)
A solution of 2-(3-benzyl-6-methyl-4-oxo-3,4-dihydroquinazolin-2-ylthio)-N′-benzylideneacetohydrazide (16) (0.5 mmol, 221 mg) in acetic acid (10 ml) was refluxed for 12 h with thiglycolic acid (3 mmol, 271 mg). The reaction mixture was cooled, the solvent was removed under reduced pressure; the residue was neutralized with 10% sodium carbonate solution. The solid obtained was filtered, dried and recrystallized from acetic acid. Yield 61%; m.p. 207–209 °C; 1H NMR (700 MHz, DMSO-d6), 11.44 (s, 1H), 7.95 (d, 1H, J = 8.36 Hz), 7.64 (d, 1H, J = 13.95 Hz), 7.52–7.11 (m, 11H), 5.34 (s, 2H), 5.09 (s, 2 H), 4.38–4.30 (m, 1H), 3.94–3.88 (m, 1H), 3.76–3.59 (m, 1H), 2.46 (s, 3H). 13C NMR (175 MHz, DMSO-d6): 20.7, 21.2, 25.3, 43.6, 46.7, 114.0, 115.6, 120.0, 126.1, 126.7, 126.9, 127.1, 127.3, 127.4, 127.5, 127.7 127.9, 128.7, 128.8, 129.4, 136.2, 136.6, 137.0, 137.7, 137.9, 145.6, 150.6, 154.6, 161.9, 162.4. MS: (M, 516).
2-Mercapto-5H-[1,3,4]thiadiazolo[2,3-b]quinazolin-5-one (20)
A solution of 3-benzyl-2-mercaptoquinazolin-4(3H)-one (13) (354 mg, 1 mmol), carbon disulphide (760 mg, 10 mmol) and potassium hydroxide (170 mg, 3 mol) in ethanol (10 ml) was stirred at room temperature for 4 h the reaction mixture was heated under reflux for 6 h. The reaction mixture was cooled and the solvent was evaporated under reduced pressure. The solid obtained was dissolved in water, acidified with concentrated hydrochloric acid; the solid obtained was filtered, washed with water, dry and crystallized from ethanol. Yield 90%; m.p. 198–200 °C; 1H NMR (500 MHz, DMSO-d6): δ 14.72 (s, 1H), 7.55 9 (d, 1H, J = 7.5 Hz), 7.28 (t, 1H, J = 7.0 Hz), 6.90 (d, 1H, J = 8.0 Hz), 6.68 (t, 1H, J = 7.5 Hz), 13C NMR (125 MHz, DMSO-d6): δ 101.1, 113.7, 114.0, 125.0, 130.7, 145.1, 158.6, 173.8. MS: (M, 235).
2-(Methylthio)-5H-[1,3,4]thiadiazolo[2,3-b]quinazolin-5-one (21)
A solution of 2-mercapto-5H-[1,3,4]thiadiazolo[2,3-b]quinazolin-5-one (20) (470 mg, 2 mmol), in 10 ml acetone containing anhydrous potassium carbonate (3 mmol, 415 mg) was stirred with iodomethane (426 mg, 3 mmol) at room temperature for 6 h. The reaction mixture was filtered, the solvent was removed under reduced pressure, and the solid obtained was washed with water, dried and recrystallized from ethanol. Yield 76%; m.p. 96–98 °C; 1H NMR (500 MHz, CDCl3): δ 8.42 (d, 1H, J = 7.5 Hz), 7.79 (t, 1H, J = 7.0 Hz), 7.63 (d, 1H, J = 8.0 Hz), 7.49 (t, 1H, J = 7.0 Hz), 2.84 (s, 3H), 13C NMR (125 MHz, CDCl3): δ = 15.3, 118.9, 126.2, 127.6, 134.8, 147.2, 156.2, 157.1, 158.5. MS: (M, 249).
Antitumor screening
A primary anticancer assay was performed for an approximately 60 human tumor cell line panel derived from nine neoplastic diseases, in accordance with the protocol of the Drug Evaluation Branch, National Cancer Institute, Bethesda, MDCitation22–26.
Docking methodology
Docking studies have been performed using MOE 2008.10 (2008). Docking procedure was followed using the standard protocol implemented in MOE 2008.10 and the geometry of resulting complexes was studied using the MOE’s Pose Viewer utilityCitation27.
Results and discussion
Chemistry
Synthesis of 2-mercapto-3-benzylquinazolin-4(3H)-one derivatives (1–4) as key intermediates was achieved by the reaction of various anthranilic acid with benzylisothiocyanate in absolute ethanol in 82–88% yield. Compounds 1–4 were treated with 2-chloro-N-(3,4,5-trimethoxyphenyl)acetamide or 3-chloro-N-(3,4,5-trimethoxyphenyl)propanamide in anhydrous acetone in the presence of potassium carbonate to give the corresponding 2-(quinazolin-2-ylthio)-N-(3,4,5-trimethoxyphenyl)acetamide or 2-(quinazolin-2-ylthio)-N-(3,4,5-trimethoxyphenyl)propanamide 5–12 in 84–92% yield (Scheme 1).
Scheme 1. Reactions of 2-mercapto-3-benzylquinazolin-4(3H)-one (1) with 2-chloro-N-(3,4,5-trimethoxyphenyl)acetamide and 3-chloro-N-(3,4,5-trimethoxyphenyl)propanamide.
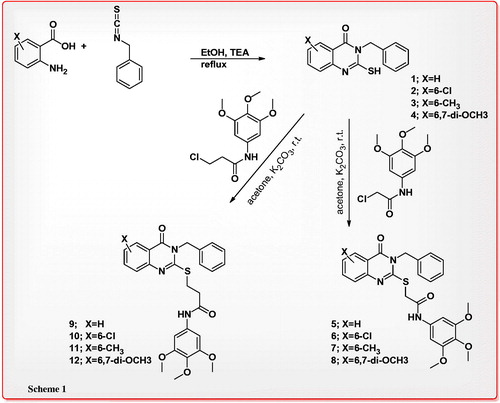
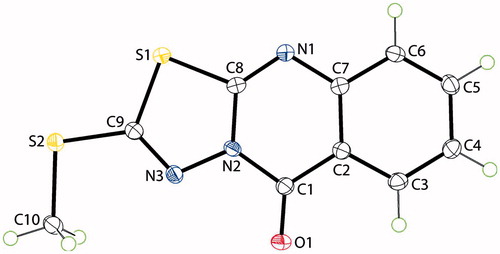
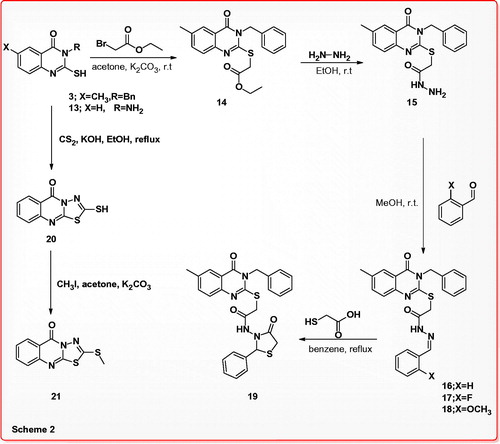
On the other hand, 3-benzyl-2-mercapto-6-methylquinazolin-4(3H)-one (3) was reacted with ethyl 2-bromoacetate in dry acetone in the presence of potassium carbonate at room temperature to afford ethyl 2-(3-benzyl-6-methyl-4-oxo-3,4-dihydroquinazolin-2-ylthio)acetate (14), which was treated with hydrazine hydrate in ethanol at room temperature to furnish 2-(3-benzyl-6-methyl-4-oxo-3,4-dihydroquinazolin-2-ylthio)acetohydrazide (15) as key intermediate in 90% yield. Schiff’s bases 16–18 were obtained in 90–92% yields, by reaction of the acid hydrazide 15 with various aldehydes in methanol at room temperature, compound 16 was subjected cyclization via boiling with acetic acid containing thioglycolic acid to give 2-phenylthiazolidin-4-one 19 in 61% yield. Reaction of 3-amino-2-mercaptoquinazolin-4(3H)-one (13) with carbon disulfide in ethanol containing potassium hydroxide gave 2-mercapto-5H-[1,3,4]thiadiazolo[2,3-b]quinazolin-5-one (20) in 90% yield, the last compound was alkylated by stirring with iodomethane in acetone containing potassium carbonate to yield 2-(methylthio)-5H-[1,3,4]thiadiazolo[2,3-b]quinazolin-5-one (21) in 76% yield. The structure of compound 21 was also confirmed by X-ray crystallography analysisCitation21b (Scheme 2) and the molecular solid state structure and numbering system are indicated in .
Figure 2. X-ray crystal structure and numbering system of compound 21.
Scheme 2. Synthesis, reactions of 3-benzyl-2-mercaptoquinazolin-4(3H)-one and 2-(3-benzyl-6-methyl-4-oxo-3,4-dihydroquinazolin-2-ylthio)acetohydrazide.
Antitumor activity
In vitro antitumor evaluation of eight compounds indicated in was selected by National Cancer Institute, Bethesda, Maryland, USA, on the basis of degree of the structure variation and computer modeling techniques for evaluation of their anticancer activity.
Table 1. Percentage growth inhibition (GI %) of in vitro subpanel tumor cell lines at 10 µM concentration.
A single dose (10 µM) of the test compounds 5–11 and 21 was used in the full NCI 60 cell lines panel assayCitation22–26. The data reported as mean-graph of the percent growth of the treated cells, and presented as percentage growth inhibition (GI %) caused by the test compounds ().
The compounds 5–11 displayed significant activities in the in vitro screening on the tested cell lines in 10 µM concentration with positive cytotoxic effect (PCE) of 23/53, 52/53, 52/53, 49/49, 41/57, 46/57 and 46/58, respectively, whereas 7/58 for compound 21 (). Compounds 5–11 showed cytotoxic effects on the most of the cancer cell lines with mean growth inhibition percentages (MGI %) of 10%, 35%, 44%, 50%, 19%, 29% and 34%, respectively (). Compound 21 showed slight selective activity against HOP-62, NCI-H226, NCI-H522, SNB-75, OVCAR-4, PC-3 and MCF7 cancer cell lines with GI values of 13%, 20%, 11%, 28%, 11%, 11% and 21%, respectively ().
Table 2. Mean growth inhibition percentage and positive cytotoxic effect of quinazoline derivatives.
With regard to broad spectrum antitumor activity, close examination of the data presented in , revealed that compounds 7, 8 and 11 are the most active members of this study, showing effectiveness toward numerous cell lines belong to different tumor subpanels. Consequently, compounds 7, 8 and 11 were selected and tested against a panel of 60 different tumor cell lines at a 5-log dose rangeCitation22–26. Three response parameters, GI50, TGI, and LC50 were calculated for each cell line, using the known drug 5-Fluorouracil (5-FU) as a positive control. Compounds 7, 8 and 11 exhibited remarkable growth inhibitory activity pattern against renal cancer (GI50 = 4.26, 3.47 and 12.30, µM), non-small cell lung cancer (GI50 = 5.65, 3.92 and 7.83 µM), breast cancer (GI50 = 4.16, 4.16 and 6.16 µM), ovarian cancer (GI50 = 9.54, 6.30 and 10.23 µM) and melanoma cancer (GI50 = 6.58, 4.24 and 3.70 µM), CNS (GI50 = 5.43, 2.50 and 3.38 µM), prostate cancer (GI50 = 27.22, 4.89 and 18.8 µM), respectively. Compounds 7, 8 and 11 are almost 1.5–3.0 fold more active than the positive control 5-FU, with GI50, (10.47, 7.24 and 14.12 µM), TGI (58.8, 36.30 and 60.25 µM) and LC50 (>100, 87.09 and 95.49 µM) values, respectively (). Comparison of the antitumor activities of compounds 7, 8 and 11 with the activities of Gefitinib and Erlotinib showed that compounds 7, 8 and 11 () possess activities almost equal to or higher than those of gefitinib and erlotinib against most cell lines except non-small lung (EKVX and NCI-H522), melanoma (SK-MEL-28), ovarian cancer (IGROV1 and SK-OV-3) renal cancer (ACHN and TK-10) and breast cancer (MDA-MB-468). On the other hand, compounds 6 and 10 yielded selective activities toward CNS, renal and breast cancer cell lines, compound 9 showed selective activities toward leukemia cell lines, whereas compound 5 possessed moderate antitumor activity.
Table 3. Compounds 7, 8 and 11 median growth inhibitory (GI50, µM), total growth inhibitory (TGI, µM) and median lethal (LC50, µM) concentration of in vitro subpanel tumor cell lines.
Table 4. GI50 values (µM) of compounds 7, 8, 11, Gefitinib and Erlotinib over the most cell lines of non-small lung cancer, colon cancer, CNS cancer, melanoma, Ovarian Cancer, Renal Cancer and breast cancer subpanels.
Regarding the activity toward individual cell lines, compounds 9 and 10 showed selective activity against leukemia cell lines CCRF-CEM, K-562, MOLT-4 and PRMI-8226 with GI values of 26%, 22%, 19%, 23%, 31%, 40% and 44%, 58% respectively. Compound 10 revealed selective activities towards SR leukemia cell lines with GI values of 34%, whereas compound 11 disclosed weak activity against K-562 and PRMI-8226 cell lines with GI values of 23% and 17%.
In non-small cell lung, A549/ATCC cell line proved to be selectively sensitive to 6, 7, 8, 9, 10 and 11 with GI values of 33%, 34%, 72%, 28%, 41% and 58%, respectively. In addition, compounds 5, 10 and 11 proved to susceptible to the HOP-62, NCI-H226 and NCI-H522 cell lines with GI values of 35%, 43%, 91%, 45%, 69%, 73%, 30%, lethal and 35%, respectively. Compounds 6, 7 and 8 have dedicated activity against NCI-H226, NCI-H23, NCI-H322M and NCI-H460 cell lines with GI values of 54%, lethal, 55%, 24%, 26%, 31%, 18%, 37%, 35%, 36%, 43% and 32% respectively. Compounds 7 and 9 showed strong activity against HOP-92 and NCI-H522 cell lines in 76%, 46%, 69% and 96% respectively, Meanwhile, compounds 10 and 11 showed certain activity against NCI-H322M and NCI-H460 cell lines in 17%, 29%, 19% and 52% respectively, whereas compounds 7, 8 and 9 showed activity in 55%, 25% and 25% against HOP-62, EKVX and NCI-H322M cancer cells respectively.
In activity against colon cancer, compounds 6, 7, 8, 10 and 11 showed GI values of 49%, 57%, 48%, 48%, 66%, 44%, 30%, 39%, 24% and 22% with colon HCT-116 and HT29 cell lines, respectively, while compounds 7 and 8 demonstrated moderate activities against HCC-2998 cancer cell line with GI value 25% and 23%, respectively. On the other hand, compounds 10 and 11 verified sensitivity in 28% and 24% to colon HCT-15 cancer cells, compounds 6, 7 and 8 displayed modest activities against KM12 cancer cell with GI value 49%, 43% and 23% respectively.
In investigation of activity toward CNS cancer, compounds 5, 6, 7, 8, 9, 10 and 11 showed strong potency against CNS cancer SF-539, SNB-75 and U251 cell lines with GI values of 46%, lethal, 89%, 94%, 28%, 74%, 67%, 23%, 47%, lethal, lethal, 45%, 74%, lethal, 25%, 45%, 43%, 94%, 20%, 30 and 56%, respectively. Compounds 7, 8, 9 and 11 showed GI values of 31%, 61%, 19%, 60%, 21%, 55%, 25% and 41% to CNS cancer SF-268 and SF-295 cell lines, respectively. CNS cancer SNB-19 proved to be sensitive to compounds 8 and 11 with GI values of 37% and 27%, while compounds 6 and 10 showed certain potency to SF-268, and SF-295 cancer cells with GI values of 41% and 34% respectively.
In activity against melanoma, compounds 6, 7 and 8 are active against LOX IMVI, MALME-3M, M14, MDA-MB-435, MDA-MB-435, SK-MEL-2, SK-MEL-28, SK-MEL-5, UACC-257 and UACC-62 cell lines with GI values from 22% to 80%. Compound 5 showed moderate activities against M14 cell line in 22% and 25%, whilst compounds 9 and 10 possessed activity against UACC-62 cell line with GI values of 34% and 47%. Melanoma SK-MEL-2, SK-MEL-28 and UACC-257 cell lines were sensitive to compounds 10 and 11 in 27%, 48%, 30%, 27%, 39% and 43%, respectively, at the same time as compound 11 was active to LOX IMVI and MALME-3M cell lines in 29% and 67%, while compound 10 had slight activity against SK-MEL-5 cell line with GI value of 26%.
In investigation of activity toward ovarian cancer, compounds 5, 6 and 7 showed moderate activities against ovarian OVCAR-8 and SK-OV-3 cell lines with GI values of 55%, 32%, 59%, 28%, 28% and 61% respectively. Ovarian OVCAR-3 and OVCAR-4 cell lines were responsive to compounds 6, 7 and 8 with GI values of 23%, 57%, 31%, 19%, 77% and 52% respectively, while compounds 9 and 10 were active against OVCAR-8 and OVCAR-4 cell lines with GI values of 21%, 27%, 37% and 39%, respectively. Ovarian NCI/ADR-RES and IGROV1 cell lines were receptive to compounds 6, 7 and 7, 8 with GI values of 33%, 34% and 26%, 25%, respectively, additionally compounds 8 and 11 demonstrated lethal activities to OVCAR-8 and OVCAR-4 cell lines, respectively. OVCAR-3, OVCAR-8, SK-OV-3 cell lines were sensitive to compound 11 with GI values of 33%, 71% and 70%, respectively, just as SK-OV-3 cell line was perceptive to compound 8 with GI values of 53%.
In renal cancer, compounds 6, 7, 8 and 11 were active against 786-0, A498, ACHN, CAKI-1, RXF 393, SN12C, TK-10 and UO-31cell lines with GI values from 24%-lethal. Renal A498, ACHN, CAKI-1, SN12C and UO-31 cell lines were sensitive to compounds 9 and 10 with GI values of 36%, 65%, 22%, 55%, 28%, 37%, 18%, 21%, 55% and 26%, respectively, whereas RXF 393 cell line was responsive to compounds 5 and 10 with GI values of 29% and 43%, respectively.
Prostate PC-3 and DU-145 cell lines proved to be selectively sensitive to compounds 6, 7 and 8 with GI value of 29%, 26%, 65%, 28%, 20% and 46%, respectively, while compounds 9 and 10 possessed selective activity towards PC-3 cell line with GI value of 34% and 57%, respectively.
Activity against breast cancer; breast MCF7, MDA-MB-231/ATCC, HS 578T, BT-549, T-47D cell lines possessed convinced response to compounds 6, 7, 8, 9, 10 and 11 with GI values from 23% to 94%. Compound 5 showed GI effectiveness against breast HS 578T and BT-549 cell lines with values of 45% and 28%, additionally compound 11 had selective activity against breast MDA-MB-468 cell line with GI value of 32%.
Structure activity relationship
The antitumor activity correlation of the newly synthesized compounds () revealed that compound 5 showed moderate antitumor potency. The introduction of 6-Cl, 6-CH3 or 6,7-dimethoxy groups at quinazoline moiety of compound 5 produced compounds 6–8 with dramatically increased the antitumor activity, also the induction of chloro or methyl function at position 6 of quinazoline nucleus of compound 9 produced compounds 10, 11 with remarkable improved antitumor potency. Additionally, replacement of the 6-Cl function at quinazoline ring of compounds 6 and 10 by 6-CH3 group produced compounds 7 and 11 with significant enhanced antitumor activity, as well substitution of the 6-Cl-function at quinazoline nucleus of compound 6 with 6,7-dimethoxy functions gave compound 8 with advanced antitumor achievement, which implied that electron donating functions favor the activity rather than electron withdrawing moieties. Moreover, changeover of the 6-CH3 group of quinazoline ring of compound 7 with 6,7-dimethoxy group enhanced the antitumor activity of compound 8. On the other hand, replacement of acetamide function of compounds 6–8 by propanamide function produced compounds 9–11 with reduction of the antitumor activity, which indicated that the length of the carbon chain linking is a vital factor for the antitumor activity. Accordingly, the N-substituted phenylacetamides with one carbon length favor the activity (compounds 6–8) rather than the propanamides with two carbon lengths (compounds 9–11).
Figure 3. The antitumor activity correlation of the newly synthesized compounds.
Molecular docking studies
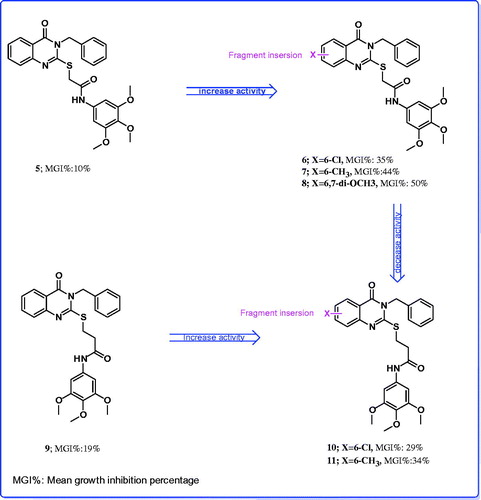
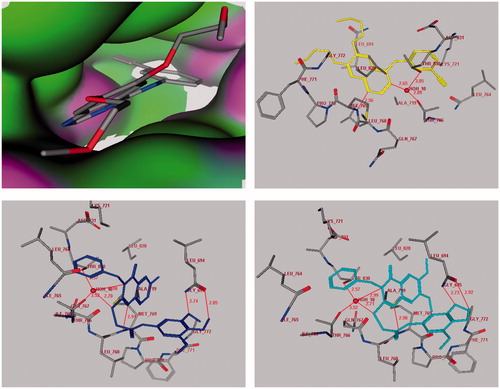
The level of antitumor activities of the compounds 7 and 8 over breast cancer, colon, renal and small lung cancer cells, in which EGFR kinase is highly expressedCitation18,Citation19,Citation27, prompted us to perform molecular docking into the ATP binding site of EGFR kinases to predict if these compounds had analogous binding mode to the EGFR kinase inhibitor. We assumed that the active target compounds 7 and 8 might demonstrate antiproliferative activity against breast cancer, colon, renal and small lung cell lines through inhibition of EGFR. Compounds 7 and 8 were docked into receptor active site of EGFR along with their inhibitor. All calculations were performed using MOE 2008.10 softwareCitation28 installed on 2.0G Core 2 Duo. The crystal structure of EGFR with erlotinib (Tarceva™) (PDB code: 1M17)Citation18,Citation19,Citation29–31 was obtained from protein data bank (PDB). The automated docking program of MOE 2008.10 was used to dock compounds 7 and 8 along with the inhibitor erlotinib into ATP binding site of EGFR. The complexes were energy-minimized with a MMFF94 force fieldCitation31 till the gradient convergence 0.01 kcal/mol was reached. The binding energies of compounds 7, 8 and erlotinib docked into the active site of EGFR were −21.04, −22.56 and −24.00 kcal/mol, respectively (). These docking studies have revealed that the quinazoline ring binds to a narrow hydrophobic pocket in the N-terminal domain of EGFR-TK where N-1 of the quinazoline ring interacts with the backbone NH of Met-769 via a hydrogen bond, and similarly, a water (HOH-10) molecule-mediated hydrogen bonding interaction is observed among the N-3 and S-atom of the quinazoline ring and the Thr-830 and Thr-766 side chain. These interactions revealed the importance of quinazoline ring for binding and the subsequent inhibitory capacity. Compounds 7, 8 complexed with EGFR-TK in a fashion similar to erlotinib and showed the occurrence of three hydrogen bonds with Met-769- (2.94–2.98Å) and HOH-10 (2.70–2.71Å and 2.98–2.99Å)-mediated hydrogen bonding interaction with Thr-830 and Thr-766 side chain. Moreover, the anilide trimethoxyphenyl fragment showed two additional hydrogen bonds with Gly-695 (2.74–2.73Å and 2.85–2.92Å).
Figure 4. Docking of the erlotinib inhibitor (Upper panel in yellow), compounds 7 (Lower left panel in blue) and 8 (Lower right panel in cyan) into the active site of epidermal growth factor receptor. Hydrogen bonds are shown in red.
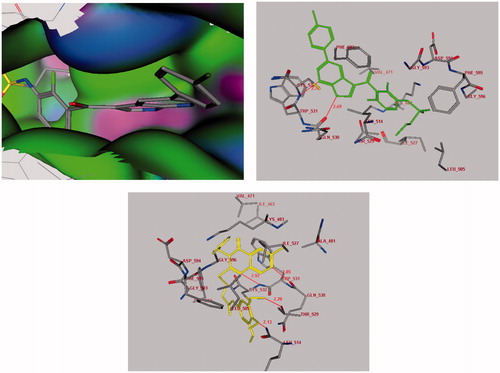
On the other hand, the level of antitumor activities of the compound 11 over melanoma cancer cells, in which V600E-B-RAF kinase is highly expressedCitation18,Citation19,Citation28, prompted us to perform molecular docking into the ATP binding site of V600E-B-RAF kinase to predict if this compound 11 has analogous binding mode to the V600E-B-RAF kinase inhibitors. The crystal structure of V600E-BRAF kinase in complex with PLX4032 (PDB code: 3OG7)Citation28,Citation32,Citation33 was obtained from protein data bank (PDB). The binding energies of compound 11 and PLX4032 docked into the active site of V600E-BRAF kinase were −30.65 and −36.11 kcal/mol, respectively (). The docking study has revealed that the ligand 11 has bound in the active site of one of the protomers in the protein dimer through the formation of four hydrogen bonds with Gln-530 (2.85Å), Cys-532 (2.92Å), Leu-505 (2.13Å) and Thr-529 (2.20Å). Moreover, there was one arene π–π interaction between the binding site and the ligand which occurred between quinazoline fragment and Trp-531. The results of this molecular docking study can support the postulation that our active compound may inhibit the growth of melanoma cell lines through inhibition of B-RAF kinase, similar to PLX4032Citation33.
Figure 5. Docking of the PLX4032 inhibitor (Upper panel in green) and compound 11 (Lower panel in yellow) into the V600E-B-RAF kinase domain. Hydrogen bonds are shown in red.
Conclusion
A new derivatives of 3-benzyl-4(3H)-quinazolinones were synthesized and evaluated for their in vitro antitumor activity. The results of this study indicated that compounds 7, 8 and 11 possess amazing broad spectrum antitumor activity with mean GI50 (10.47, 7.24 and 14.12 µM respectively) and are nearly 1.5–3.0 fold more potent compared with the positive control 5-FU with mean GI50 22.60 µM.
Supplementary material available online
Supplemental Material.pdf
Download PDF (4.5 MB)Declaration of interest
The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding the work through the research group project No. RGP-VPP-163. The authors would like to express their gratitude and thanks to the National Cancer Institute (NCI), Bethesda Maryland, USA, for doing the antitumor testing of the new compounds.
References
- Harris CC, Hollstein M. Clinical implications of the P53 tumor suppressor gene. N Engl J Med 1993;329:1318–27
- Liott LA, Steeg PS, Steller-Stevenson WG. Cancer metastasis and angiogenesis: an imbalance of positive and negative regulation. Cell 1991;64:327–36
- Mignatti P. Rifkin DB. Biology and biochemistry of proteinases in tumor invasion. Physiol Rev 1993;73:161–5
- Auerbuch D. Alkylating agents. In: Chabner BA. Collins JM, eds. Cancer chemotherapy: principles and practice. Philadelphia: Lippincott; 1990:314–28
- (a) El-Azab AS, Kamal EH. Synthesis and anticonvulsant evaluation of some new 2,3,8-trisubstituted-4(3H)-quinazoline derivatives. Bioorg Med Chem Lett 2012;22:327–33. (b) Alanazi AM, Abdel-Aziz A-MA, Al-Suwaidan I, et al. Design, synthesis and biological evaluation of some novel substituted quinazolines as antitumor agents. Eur J Med Chem 2014;79:446–54. (c) Aziza MA, Nassar MW, Abdel-Hamide SG, et al. Synthesis and antimicrobial activities of some new 3-heteroaryl- quinazolin-4-ones. Indian J Heterocycl Chem 1996;6:25–30. (d) El-Azab AS. Synthesis of some new substituted 2-mercaptoquinazoline analogs as potential antimicrobial agents. Phosphorus Sulfur Silicon Relat Elem 2007;183:333–48
- (a) Habib NS, Ismail KA, El-Tombary AA, Abdel-Aziem T. Antilipidemic agents, Part. IV: synthesis and antilipidemic testing of some heterocyclic derivatives of hexadecyl and cyclohexyl hemisuccinate esters. Pharmazie 2000;55:495–9. (b) Al-Omar MA, El-Azab AS, El-Obeid HA, Abdel Hamide SG. Synthesis of some new 4(3H) quinazoline analogs as potential antioxidant agents. J Saudi Chem Soc 2006;10:113–28. (c) Alafeefy AM, Kadi AA, El-Azab AS, et al. Synthesis, analgesic and anti-inflammatory evaluation of some new 3H-quinazolin-4-one derivatives. Arch Pharm 2008;341:377–85. (d) Kumar A, Sharma S, Archana A, et al. Some new 2,3,6-trisubstituted quinazolinones as potent anti-inflammatory, analgesic and COX-II inhibitors. Bioorg Med Chem 2003;11:5293–9
- (a) Alagarsamy V, Solomon VR, Dhanabal K. Synthesis and pharmacological evaluation of some 3-phenyl-2-substituted-3Hquinazolin-4-one as analgesic, anti-inflammatory agents. Bioorg Med Chem 2007;15:235–41. (b) El-Azab AS, Kamal EH, Attia SM. Synthesis and anticonvulsant evaluation of some novel 4(3H)-quinazolinones. Monatsh Chem 2011;142:837–48
- (a) El-Azab AS, Kamal EH. Design and synthesis of novel 7-aminoquinazoline derivatives: antitumor and anticonvulsant activities. Bioorg Med Chem Lett 2012;22:1879–85. (b) Kashaw SK, Kashaw V, Mishra P, et al. Synthesis, anticonvulsant and CNS depressant activity of some new bioactive 1-(4-substituted-phenyl)-3-(4-oxo-2-phenyl/ethyl-4H-quinazolin-3-yl)-urea. Eur J Med Chem 2009;44:4335–43
- (a) Archana V, Srivastava K, Kumar A. Synthesis of some newer derivatives of substituted quinazolinonyl-2-oxo/thiobarbituric acid as potent anticonvulsant agents. Bioorg Med Chem 2004;12:1257–64. (b) Alafeefy A, El-Azab AS, Mohamed MA, et al. Synthesis of some new substituted iodoquinazolinederivatives and their antimicrobial screening. J Saudi Chem Soc 2011;15:319–25
- (a) Al-Omary FA, Abou-Zeid LA, Nagi MN, et al. Non-classical antifolates. Part 2: synthesis, biological evaluation, and molecular modeling study of some new 2,6-substituted-quinazolin-4-ones. Bioorg Med Chem 2010;18:2849–63. (b) Al-Obaid AM, Abdel-Hamide SG, El-Kashef HA, et al. Synthesis, in vitro antitumor activity and molecular modeling study of certain 2-thieno-4(3H)-quinazolinone analogs. Eur J Med Chem 2009;44:2379–91
- (a) El-Azab AS, Al-Omar MA, Abdel-Aziz AA, et al. Design, synthesis and biological evaluation of novel quinazoline derivatives as potential antitumor agents: molecular docking study. Eur J Med Chem 2010;45:4188–98. (b) El-Azab AS, Abdel-Hamide SG, Sayed-Ahmed MM, et al. Novel 4(3H)-quinazolinone analogs: synthesis and anticonvulsant activity. Med Chem Res 2013;22:2815–27. (c) Abdel Gawad NM, Georgey HH, Youssef RM, El-Sayed NA. Synthesis and antitumor activity of some 2, 3-disubstituted quinazolin-4(3H)-ones and 4,6-disubstituted-1,2,3,4-tetrahydroquinazolin-2H-ones. Eur J Med Chem 2010;45:6058–67
- (a) Fry DW, Kraker AJ, McMichael A, et al. A specific inhibitor of the epidermal growth factor receptor tyrosine kinase. Science 1994;265:1093–95. (b) Traxler PM, Furet P, Mett H, et al. 4-(Phenyl amino)pyrrolopyrimidines: potent and selective, ATP site directed inhibitors of the EGF-receptor protein tyrosine kinase. J Med Chem 1996;39:2285–92
- (a) Hynes JH, Tomazic A, Kumar A, et al. Inhibition of human dihydrofolate reductase by 2,4-diaminoquinazolines bearing simple substituents on the aromatic ring. J Heterocyclic Chem 1991;28:1981–6. (b) Harris NV, Smith C, Bowden K. Antifolate and antibacterial activities of 5-substituted 2,4-diaminoquinazolines. J Med Chem 1990;33:434–44. (c) Barlési F, Tchouhadjian C, Doddoli C, et al. Gefitinib (ZD1839, Iressa) in non-small-cell lung cancer: a review of clinical trials from a daily practice perspective. Fundam Clin Pharmacol 2005;3:385–93
- (a) Arteaga CL, Johnson DH. Tyrosine kinase inhibitors-ZD1839 (Iressa). Curr Opin Oncol 2001;13:491–8. (b) Barker AJ, Gibson KH, Grundy W, et al. Studies leading to the identification of ZD1839 (IRESSA): an orally active, selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to the treatment of cancer. Bioorg Med Chem Lett 2001;11:1911–14
- (a) Ganjoo KN, Wakelee H. Review of erlotinib in the treatment of advanced non-small cell lung cancer. Biologics 2007;1:335–46. (b) Kopper L. Lapatinib: a sword with two edges. Pathol Oncol Res 2008;14:1–8
- (a) Dhillon S, Wagstaff AJ. Lapatinib. Drugs 2007;67:2101–8. (b) Burris HA, Hurwitz HI, Dees EC, et al. Phase I safety, pharmacokinetics, and clinical activity study of lapatinib (GW572016), a reversible dual inhibitor of epidermal growth factor receptor tyrosine kinases, in heavily pretreated patients with metastatic carcinomas. J Clin Oncol 2005;23:5305–13
- (a) Wood ER, Truesdale AT, McDonald OB, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res 2004;64: 6652–59. (b) Hennequin LF, Stokes ES, Thomas AP, et al. Novel 4-anilinoquinazolines with C-7 basic side chains: design and structure activity relationship of a series of potent, orally active, VEGF receptor tyrosine kinase inhibitors. J Med Chem 2002;45:1300–12
- (a) Fricker J. Tyrosine kinase inhibitors: the next generation. Lancet Oncol 2006;7:621. (b) Garofalo S, Rosa R, Bianco R, Tortora G. EGFR-targeting agents in oncology. Expert Opin Therap Patents 2008;18:889–901
- (a) Madhusudan S, Ganesan TS. Tyrosine kinase inhibitors in cancer therapy. Clin Biochem 2004;37:618–35. (b) Cockerill GS, Lackey KE. Small molecule inhibitors of the class 1 receptor tyrosine kinase family. Curr Top Med Chem 2002;2:1001–10
- (a) Harris PA. Cancer drug design and discovery. 2nd ed. New York: Academic Press; 2014. (b) Liechti C, Séquin U, Bold G, et al. Salicylanilides as inhibitors of the protein tyrosine kinase epidermal growth factor receptor. Eur J Med Chem 2004;39:11–26
- (a) Sheldrick GM. A short history of SHELX. Acta Crystallogr Sect A 2008;64:112–22. (b) El-Azab AS, Abdel-Aziz AA, Al-Swaidan IA, et al. 2-Methyl-sulfanyl-9H-1,3,4-thia-diazolo[2,3-b]quinazolin-9-one. Acta Crystallogr Sect E Struct Rep Online 2012;68:o2134
- Grever MR, Schepartz SA, Chabner BA. The National Cancer Institute: cancer drug discovery and development program. Semin Oncol 1992;19:622–38
- Monks A, Scudiero D, Skehan P, et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J Natl Cancer Inst 1991;83:757–66
- Boyd MR, Paull K. Some practical considerations and applications of the national cancer institute in vitro anticancer drug discovery screen. Drug Dev Res 1995;34:91–109
- Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer 2006;6:813−23
- Alley MC, Scudiero DA, Monks PA, et al. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res 1988;48:589–601
- MOE 2008.10 of Chemical Computing Group. Inc. Available from: https://doi.org/http://www.chemcomp.com/press_releases/2008-11-04.htm
- Brose MS, Volpe P, Feldman M, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res 2002;62:6997–7000
- Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem 2002;277:46265–72
- Bridges AJ. The rationale and strategy used to develop a series of highly potent, irreversible, inhibitors of the epidermal growth factor receptor family of tyrosine kinases. Curr Med Chem 1999;6:825–43
- Lv PC, Wang KR, Li QS, et al. Design, synthesis and biological evaluation of chrysin long-chain derivatives as potential anticancer agents. Bioorg Med Chem 2010;18:1117–23
- Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010;467:596–9
- Choi WK, El-Gamal MI, Choi HS, et al. New diarylureas and diarylamides containing 1,3,4-triarylpyrazole scaffold: synthesis, antiproliferative evaluation against melanoma cell lines, ERK kinase inhibition, and molecular docking studies. Eur J Med Chem 2011;46:5754–62