
Abstract
Membrane-type I matrix metalloproteinases (MT1-MMP) is an enzyme critical to the remodeling and homeostasis of extracellular matrix, and when over expressed it contributes to metastasis and cancer cell progression. Because of its role and implication as a biomarker that is upregulated in various cancers, MT1-MMP has become an attractive target for drug discovery. A small pilot library of peptidomimetics containing a phosphoramidate core as a zinc-binding group was synthesized and tested for inhibitory potency against MT1-MMP. From this library, a novel two residue peptidomimetic scaffold was identified that confers potency against MT1-MMP at submicromolar concentrations. The results of this study confirm that for this scaffold, valine is favored as a P1 residue and leucine in the P1′ position. Furthermore, steric tolerance was observed for the N-terminus, thus implicating that a second-generation library could be constructed to extend the scaffold to P2 without concomitant loss of affinity within the MT1-MMP catalytic domain.
Introduction
Matrix metalloproteinases play a key role in extracellular matrix remodeling, metastasis, and angiogenesis in cancerCitation1–3. Membrane-type I matrix metalloproteinase (MT1-MMP) or MMP-14 is a transmembrane protein that catalyzes the hydrolysis of collagen, the main component of extracellular matrix. Unlike secreted MMPs, pro-MT1-MMP is processed within the cell then transported to the plasma membrane through trans-Golgi trafficking or invadopodia by exocytosis, respectivelyCitation4,Citation5. MT1-MMP is upregulated in metastatic cancer cells and, therefore, an attractive target for both diagnostic and therapeutic applicationsCitation6. The clinical significance of MT1-MMP expression in aggressive cancer phenotypes emphasizes the need for the design of novel inhibitors for diagnostic and therapeutic applications.
There has been some success with the development of selective inhibitors against several MMPs, and there are a handful of peptide and antibody inhibitors of MT1-MMPCitation7–12. However, there are fewer efforts dedicated toward the development of small moleculesCitation13–15, with some recent progress in allosteric inhibitionCitation16. A challenge in designing inhibitors for MT1-MMP is that although a few sites for hydrophobic interactions exist, the catalytic site is shallow and mostly solvent exposed with a pocket of moderate depth in S1′. This explains MT1-MMP versatility for processing a variety of substrates from type-I collagen to large biomolecules such as pro-MMP2 and CD44Citation17. The most potent small-molecule inhibitors of MT1-MMP contain a hydroxamic acid as a strong zinc-binding motif. A disadvantage of employing a hydroxamic acid for zinc coordination is broad spectrum inhibition. A general limitation in hydroxamate-based inhibitors developed against MT1-MMP is that interactions within the active site are restricted to only the prime subsites. The structural nature of phosphoramidates enables the probing of both the prime and non-prime catalytic subsites as well as coordination to the active-site zinc. Phosphoramidate inhibitors focusing exclusively on the prime side of the scaffold have been developed for MMP-1 with fair potency in the low micromolar rangeCitation18. It is envisaged that selective inhibition of MT1-MMP could be achieved by capitalizing on interactions of peptide residues in the phosphoramidate scaffold with both prime and non-prime subsites of MT1-MMP and not through dependence on a strong zinc binding motifCitation19.
Our group has developed potent and selective inhibitors for the Zn-dependent folate hydrolase known as prostate-specific membrane antigen (PSMA or GCPII). We therefore expected that the phosphoramidate core could be applied to other Zn-dependent enzymes such as matrix metalloproteinases. The focus of this study involved the rational design of the phosphoramidate peptidomimetics through extensive virtual screening of amino acids (phenylalanine, methionine, threonine, leucine, valine, histidine, isoleucine, aspartic acid, and glutamic acid) at the P1 and P1′ positions. A subset of amino acids was selected from the screening study for the development of a small library of peptidomimetics, which were evaluated for potency against MT1-MMP. Herein, we report the identification of a two-residue peptidomimetic phosphoramidate scaffold with promising potency against MT1-MMP.
Methods
Inhibition assay of cdMT1-MMP
The methods for inhibitor synthetic procedures and docking studies are located in the Supplemental material file. Enzyme was purchased as cdMMP-14 recombinant from Enzo Life Sciences (Plymouth Meeting, PA). Enzyme inhibition assays were carried out according to R&D Systems protocol (Minneapolis, MN). Inhibitors were dissolved in DMSO to prepare 10 mM stock solutions. Dilutions of the inhibitors were made using 50 mM TRIS pH = 8.5. The recombinant cdMT1-MMP was diluted to obtain 1 ng/µL in assay buffer containing 50 mM TRIS pH = 8.5, 3 mM CaCl2, and 1 µM of ZnCl2. The FRET substrate QXL-520 was diluted to obtain 2 µM in assay buffer. To a black 96-well plate, 50 μL of enzyme (50 ng/assay) was added followed by 10 μL of inhibitor and finally 40 μL of substrate. The wells were measured using kinetic analysis in a FLUOstar Omega microplate reader over a 30 min time interval.
Results
While previous inhibitors have been designed to interact with the prime side of the catalytic domain, we sought to probe both prime and non-prime sides with a phosphoramidate-based inhibitor scaffold. A Glide-based docking study was carried out to select for P1 and P1′ residues in the model scaffold () using Maestro 9.8 Glide docking workflow. The inhibitors were prepared for rigid docking using ligand preparation without constraints and subsequently docked into a receptor grid generated from the 3MA2Citation20 crystal structure available for the MT1-MMP catalytic domain. The structures for which the Glide potential energies were in the range of −7 to −10 kcal/mol were selected as synthetic candidates for the first-generation library. The residues in both the P1 and P1′ positions that resulted in favorable docking scores were valine, phenylalanine, and leucine. We also explored the effect of the N-terminus residue on inhibitor potency and whether planar or tetrahedral geometry was significant for P2 residue tolerance. This was achieved with Boc and benzamide derivatives of the N-terminal amine.
Table 1. Library of P1/P1′ residue-containing phosphoramidate inhibitors.
The peptidomimetic phosphoramidate-based inhibitors were synthesized beginning with a Boc-protected amino-alcohol analog (detailed procedures are available in the Supplementary material) PCl3 (Scheme 1). The intermediate phosphite 6 was generated by sequential reaction with benzyloxydichlorophosphine and water. The P1′ residue was installed as the C-terminal amide using Atherton–Todd chemistry to provide protected phosphoramidates 7. In some cases, N-deprotection was followed by derivatization to benzamides 8. Ultimately, hydrogenolysis provided the desired inhibitors 1–5 in quantitative yield and was characterized by 1H, 31P NMR, and high-resolution mass spectrometry.
Scheme 1. Synthesis of peptidomimetic phosphoramidate-based inhibitors of MT1-MMP.
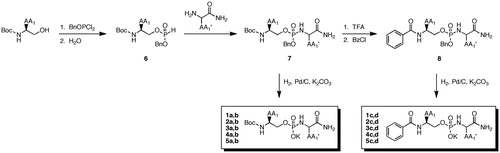
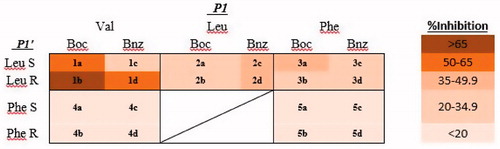
Based on the ClogP values, all phosphoramidate inhibitors were relatively lipophilic (). The docking scores in general did not appear to correlate with inhibitor potency, however, in silico inspection of the molecular bonding interactions between the inhibitors and the enzymatic residues in the catalytic site were consistent with respect to the coordination of the phosphoryl core of the inhibitor to Zn294 and interactions of P1 and P1′ in register with either S1 or S1′ subsites. To assess inhibitory potency against MT1-MMP, a preliminary screening of the library was carried out by incubating the catalytic domain of MT1-MMP with at 300 nM for each inhibitor. The relative extent of inhibition () was determined using a FRET-based assay. Upon cursory inspection of the P1′ residue, leucine promoted greater potency at 300 nM than phenylalanine (). No consistent preference for an N-terminal benzamide or the Boc group was observed for the compounds examined, indicating reasonable steric tolerance for a P2 residue if the scaffold was to be expanded in a subsequent generation.
Figure 1. Heat map of inhibitor potency at 300 nM based on percent inhibition of the catalytic domain of human recombinant MT1-MMP.
Discussion
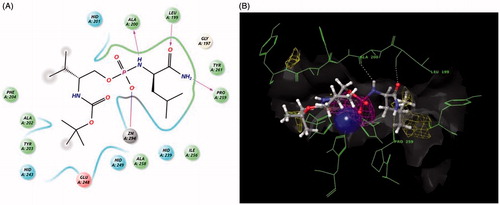
In general, stereochemistry of the P1′ residue did not appear to have a large impact on inhibitor potency. Upon closer inspection, compound 1b exhibited the greatest potency with 67% inhibition at 300 nM. When this inhibitor was docked into the enzyme, the phosphoryl oxygen was coordinated to the catalytic Zn294 (). The orientation of 1b was aligned with substrate placement into the P1 and P1′ subsites of cdMT1-MMP. Compound 1b makes three key hydrogen–bond interactions within S1′ of the active site: the carbonyl of P1′ leucine with NH of Leu199, the P1′ carbamate-NH2 to the carbonyl of Pro259 in the outer wall of S1′, and the P1′ phosphoramidate NH with the carbonyl of Ala200Citation21. Examination of the docking results for 1a revealed that hydrogen bonds were made with Leu199 and Pro259 in the active site (see Supplemental information for docking results of other compounds) while the docking of 1d revealed a hydrogen bond between the P1′ residue and the Pro259. These results suggest that a hydrogen bond to Pro259 may be a key interaction for this scaffold. In general, the docking of weaker inhibitors (inhibition <20% at 300 nM), revealed two or fewer hydrogen bonds within the MT1-MMP active site further supporting the importance of hydrogen bonds to Leu199, Pro259, and Ala200.
Figure 2. Figures exported from Maestro 9.8 (2014). (A) Two-dimensional ligand representation of 1b in the catalytic domain coordinating to Zn294 and making three hydrogen bonds to Ala200, Leu199, and Pro259. (B) Three-dimensional view with metal binding (purple) and hydrophobic (yellow) mesh density maps as well as surface. (See color rendition in the online journal article).
Conclusion
We have identified a phosphoramidate-based peptidomimetic scaffold in which a P1 valine and a P1′ leucine contribute to submicromolar inhibition of MT1-MMP. Inhibitory potency was not dependent on the structures of N-terminus Boc or benzamide groups suggesting tolerance for extending the scaffold farther with a P2 residue. The introduction of a phosphoramidate core, which as a weak zinc-binding group compared to hydroxamic acids, does not dominate binding to the active site, and could thus allow for the selective tuning of side chain interactions for both prime and non-prime subsites to increase potency. The identification of the Val-PO2-Leu phosphoramidate peptidomimetic scaffold is expected to inspire the design of a second-generation library that spans both the P2 and P2′ positions in subsequent studies for enhancing potency of peptidomimetic phosphoramidate inhibitors toward MT1-MMP.
Supplementary material available online
Supplemental Material.pdf
Download PDF (1.2 MB)Acknowledgements
The authors extend appreciation to Jeffrey P. Jones for his guidance and generous support in the use of Schrodinger Maestro software as well as Gerhard Munske for the acquisition of high resolution mass spectrometry data.
Declaration of interest
The authors declare that they have no conflicts of interest. This work was supported in part by the National Institutes of Health (R01CA140617) including support for D.E.M. by a National Institutes of Health Biotechnology Training Grant (T32GM008336).
References
- Friedl P, Wolf K. Proteolytic interstitial cell migration: a five-step process. Cancer Metastasis Rev 2009;28:129–35
- Wolf K, Wu YI, Liu Y, et al. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat Cell Biol 2007;9:893–904
- Galvez BG, Matias-Roman S, Albar JP, et al. Membrane type 1-matrix metalloproteinase is activated during migration of human endothelial cells and modulates endothelial motility and matrix remodeling. J Biol Chem 2001;276:37491–500
- Wang X, Ma D, Keski-Oja J, Pei D. Co-recycling of MT1-MMP and MT3-MMP through the Trans-Golgi Network. J Biol Chem 2004;279:9331–6
- Monteiro P, Rossé C, Castro-Castro A, et al. Endosomal WASH and exocyst complexes control exocytosis of MT1-MMP at invadopodia. J Cell Biol 2013;203:1063–79
- Nagase H, Woessner Jr JF. Matrix metalloproteinases. J Biol Chem 1999;274:21491–4
- Ottl J, Gabriel D, Murphy G, et al. Recognition and catabolism of synthetic heterotrimeric collagen peptides by matrix metalloproteinases. Chem Biol 2000;7:119–32
- Suojanen J, Salo T, Koivunen E, et al. A novel and selective membrane type-1 matrix metalloproteinase (MT1-MMP) inhibitor reduces cancer cell motility and tumor growth. Cancer Biol Ther 2009;8:2362–70
- Devy L, Huang L, Naa L, et al. Selective inhibition of matrix metalloproteinase-14 blocks tumor growth, invasion, and angiogenesis. Cancer Res 2009;69:1517–26
- Kaimal R, Aljumaily R, Tressel SL, et al. Selective blockade of matrix metalloprotease-14 with a monoclonal antibody abrogates invasion, angiogenesis, and tumor growth in ovarian cancer. Cancer Res 2013;73:2457–67
- Zhu L, Wang H, Wang L, et al. High-affinity peptide against MT1-MMP for in vivo tumor imaging. J Control Release 2011;150:248–55
- Zarrabi K, Dufour A, Li J, et al. Inhibition of matrix metalloproteinase 14 (MMP-14)-mediated cancer cell migration. J Biol Chem 2011;286:33167–77
- Jani M, Tordai H, Trexler M, et al. Hydroxamate-based peptide inhibitors of matrix metalloprotease 2. Biochimie 2005;87:385–92
- Ikejiri M, Bernardo MM, Bonfil RD, et al. Potent mechanism-based inhibitors for matrix metalloproteinases. J Biol Chem 2005;280:33992–4002
- Hurst DR, Schwartz MA, Jin Y, et al. Inhibition of enzyme activity of and cell-mediated substrate cleavage by membrane type 1 matrix metalloproteinase by newly developed mercaptosulphide inhibitors. Biochem J 2005;392:527–36
- Remacle AG, Golubkov VS, Shiryaev SA, et al. Novel MT1-MMP small-molecule inhibitors based on insights into hemopexin domain function in tumor growth. Cancer Res 2012;72:2339–49
- Itoh Y, Seiki M. MT1-MMP: a potent modifier of pericellular microenvironment. J Cell Physiol 2006;206:1–8
- Kortylewicz Z, Galardy RE. Phosphoramidate peptide inhibitors of human skin fibroblast collagenase. J Med Chem 1990;33:263–73
- Rao BG. Recent developments in the design of specific matrix metalloproteinase inhibitors aided by structural and computational studies. Curr Pharm Des 2005;11:295–322
- Grossman M, Tworowski D, Dym O, et al. The intrinsic protein flexibility of endogenous protease inhibitor TIMP-1 controls its binding interface and affects its function. Biochemistry 2010;49:6184–92
- Fernandez-Catalan C, Bode W, Huber R, et al. Crystal structure of the complex formed by the membrane type 1-matrix metalloproteinase with the tissue inhibitor of metalloproteinases-2, the soluble progelatinase A receptor. EMBO J 1998;17:5238–48