
Abstract
5α-R isozymes (types 1 and 2) play an important role in prostate gland development because they are responsible for intraprostatic dihydrotestosterone (DHT) levels when the physiological serum testosterone (T) concentration is low. In this study, we synthesized seven novel dehydroepiandrosterone derivatives with benzimidazol moiety at C-17, and determined their effect on the activity of 5α-reductase types 1 and 2. The derivatives with an aliphatic ester at C-3 of the dehydroepiandrosterone scaffold induced specific inhibition of 5α-R1 activity, whereas those with a cycloaliphatic ester (cyclopropyl, cyclobutyl, or cyclopentyl ring) or an alcohol group at C-3 inhibited the activity of both isozymes. Derivatives with a cyclohexyl or cycloheptyl ester at C-3 showed no inhibitory activity. In pharmacological experiments, derivatives with esters having an alcohol or the aliphatic group or one of the three smaller cycloaliphatic rings at C-3 decreased the diameter of male hamster flank organs, with the cyclobutyl and cyclopentyl esters exhibiting higher effect. With exception of the cyclobutyl and cyclopentyl esters, these compounds reduced the weight of the prostate and seminal vesicles.
Introduction
In male tissues, 5α-reductase (5α-R) (Enzyme Commission (EC) 1.3.99.5) converts testosterone (T) to 5α-dihydrotestosterone (DHT). The 5α-R isoenzymes (type 1 and type 2) of different species have been characterized: each isoenzyme is encoded by a different geneCitation1,Citation2. Although these isozymes have similar substrates, the amino acid sequences of these isozymes have ∼50% identity, thus showing different sensitivity for the same inhibitorCitation1. Comparison of the amino acid sequences of 5α-R types 1 and 2 has shown 60% identity for the human isozymes and 77% identity for those of the ratCitation1. When the two classes of 5α-R isozymes are compared between human and rat, they average 47% identity, thus indicating that both isozymes are true homologuesCitation1. Because, when isolated in pure crystalline form, the activities of these enzymes are lost, a study of structure–activity relationship cannot be performedCitation1.
The binding of T or DHT to the androgen receptor (AR) induces a change in its configuration, which leads to transcription (activation or inhibition) of specific genes. AR activation permits transmission of extracellular signals into the cells to produce intracellular responses through the targeting of response elements and recruitment of cofactorsCitation3. AR is present in androgen-dependent tissues, such as hamster flank organsCitation4. pilosebaceous complexes, occurring as two pigmented spots located in the dorsal skin of male and female hamstersCitation5.
After castration, the diameters of the pigmented spots in the male hamster are reduced, thus resembling the nodules of the female hamster; however, with androgen treatment, these can return to their original sizeCitation5.
In this study, we synthesized seven novel dehydroepiandrosterone derivatives having a benzimidazol moiety at C-17 or each of these new derivatives, we evaluated its biological activity to act as inhibitor of 5α-R types 1 and 2 isoenzymes.
Finasteride (F) is a specific inhibitor of type 2 5α-R; It is used clinically to treat benign prostatic hyperplasia and male pattern baldness. In this study, finasteride was used as a reference compound for in vivo testing. We evaluated the activity of these seven steroid derivatives on androgen-dependent tissues – flank organs, prostate, and seminal vesicles – from gonadectomized male hamsters treated with T. These novel steroids may have potential for the treatment of androgen-dependent illnesses and conditions.
Materials and methods
Chemical reagents and steroid derivatives
Reagents
Solvents were laboratory grade. Melting points (uncorrected) were determined on a melting point apparatus (Fisher Johns, México, D. F., México); 1H-NMR and 13C-NMR were taken on Varian Gemini 200 and VXR-300 spectrometer, respectively (MR Resources, Fitchburg, MA). Chemical shifts are given in ppm relative to that of Me4Si (δ = 0) in CDCl3 (the abbreviations of signal patterns are as follows: s, singlet; d, doublet; t, triplet; m, multiplet). Mass spectra were obtained with an HP5985-B spectrometer (Avantes, Apeldoorn, the Netherlands). IR spectra were recorded on a Perkin-Elmer 200 s spectrometer (Perkin Elmer Life and Analytical Science, Shelton, CT). UV lamp (254 nm) was from UVP (Upland, CA).
(1, 2, 6, 7-3H) Testosterone ([3H] T; specific activity: 95 Ci/mmol) was purchased from Perkin-Elmer Analytical Sciences (Boston, MA). This labeled steroid was purified by thin layer chromatography (TLC) on HPTLC Keiselgel 60 F254 plates (Merck, Darmstadt, Germany); the solvent system recommended by the manufacturer was used.
Radioinert T and 5α-DHT were purchased from Steraloids (Wilton, NH), Lubrol PX, DL-dithiothreitol (DTT). F was obtained by extraction from Proscar® (Merck, Sharp & Dohm, Mexico City, Mexico) as follows: 180 Proscar tablets were crushed and extracted with 200 mL chloroform; after the solvent was removed under vacuum, the crude product was purified by silica gel 60 (63–200 µm) column chromatography (Sigma-Aldrich, Mexico City, Mexico) with ethyl acetate as eluantCitation6.
Syntheses of steroid derivatives–overview
Steroids 2, 3a–3g and 4a–4g () were prepared as have been reported previouslyCitation7,Citation8.
Figure 1. Synthetic route for the preparation of six novel steroidal derivatives, 5a–5f and 6. (a) NaOH 10%, MeOH; (b) RCOOH, 4-dimethylaminopyridine (DMAP), N,N′-dicyclohexyl carbodiimide (DCC); (c) phosphorus oxychloride (POCl3), dimethyl formamide (DMF), CHCl3; reflux, 5 h; (d) Benzimidazole, Na2CO3, DMF; (e) Formic acid, reflux, 5 h; (f) 1. Benzimidazole, Cs2CO3, DMF, 60 °C, 2 h; 2. CHCl3/MeOH, HCl, 40 °C, 3 h.
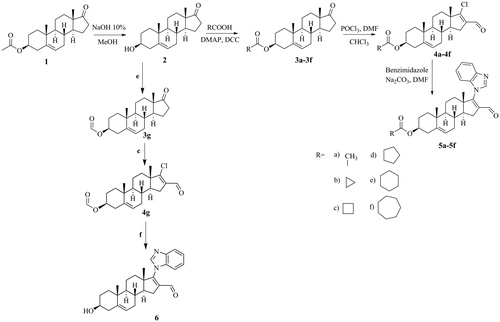
Derivatives 5a–5f
β-Acetoxy-17-(benzimidazol-1H-yl)-16-formylandrosta-5,16-diene (5a in ). Yield: 63 mg (25.93%) white solid; m.p.: 230–231 ˚C; IR (KBr) cm−1: 1725, 1670, 1248; UV (nm): 243; 1H-NMR (400 MHz, CDCl3): δ 1.1 (s, H-18, 3H), 1.1 (s, H-19, 3H), 2.1 (s, 3β-acetoxy, 3H), 4.7 (m, H-3, 1H), 5.5 (d, J = 5.4 Hz, H-6, 1H), 7.4 (m, aromatic, 3H), 7.9 (s, aromatic, 1H), 8.3 (s, H-aromatic, 1H), 9.6 (s, formyl, 1H); 13C-NMR (300 MHz, CDCl3): δ 73.5 (C3), 120.2 (C16), 120.2 (C-aromatic), 121.7 (C6), 123.8 (C-aromatic), 124.9 (C-aromatic), 140.0 (C-aromatic), 141.4 (C5), 170.5 (C=O ester), 187.5 (C=O formyl); and HRMS calculated for C29H34N2O3 (458.5919), found 459.5918.
3β-Cyclopropanoyloxy-17-(1H-benzimidazol-1-yl)-16-formylandrosta-5,16-diene (5b in ). Yield: 51 mg (21.18%) white solid; m.p.: 206–207 ˚C; IR (KBr) cm−1: 1714, 1670, 1184; UV (nm): 244, 273, 280; 1H-NMR (400 MHz, CDCl3): δ 1.1 (s, H-18, 3H), 1.1 (s, H-19, 3H), 4.6 (m, H-3, 1H), 5.4 (d, J = 5.2 Hz, H-6, 1H), 7.4 (m, aromatic, 3H), 7.9 (s, aromatic, 1H), 7.9 (s, aromatic, 1H), 9.6 (s, formyl, 1H); 13C-NMR (300 MHz, CDCl3): δ 73.5 (C3), 120.7 (C16), 120.7 (C-aromatic), 121.7(C6), 123.4 (C-aromatic), 124.0 (C-aromatic), 140.1 (C-aromatic), 141.4 (C5), 174.3 (C=O ester), 187.9 (C=O formyl); and HRMS calculated for C31H36N2O3 (484.6291), found 485.6290.
3β-Cyclobutanoyloxy-17-(1H-bezimidazol-1-yl)-16-formylandrosta-5,16-diene (5c in ). Yield: 57 mg (23.81%) white solid; m.p.: 138–139 °C; IR (KBr) cm–1: 1727, 1673, 1175; UV (nm): 245, 273, 280; 1H-NMR (400 MHz, CDCl3): δ 1.1 (s, H-18, 3H,), 1.1 (s, H-19, 3H), 4.6 (m, H-3, 1H), 5.4 (d, J = 5.2 Hz, H-6, 1H), 7.4 (m, aromatic, 3H), 7.9 (s, aromatic, 1H), 7.9 (s, aromatic, 1H), 9.6 (s, formyl, 1H); 13C-NMR (300 MHz, CDCl3): δ 73.3 (C3), 120.7 (C16), 120.6 (C-aromatic), 121.7 (C6), 123.4 (C-aromatic), 124.2 (C-aromatic), 140.1 (C-aromatic), 141.4 (C5), 174.9 (C=O ester), 187.8 (C=O formyl); and HRMS calculated for C33H38N2O3 (498.6557), found 499.6556.
3β-Cyclopentanoyloxy-17-(1H-bezimidazol-1-yl)-16-formylandrosta-5,16-diene (5d in ). Yield: 97 mg (40.74%) white solid; m.p.: 165–166 °C; IR (KBr) cm−1: 1728, 1672, 1188; UV (nm): 244, 281; 1H-NMR (400 MHz, CDCl3): δ 1.1 (s, H-18, 3H), 1.1 (s, H-19, 3H), 4.6 (m, H-3, 1H), 5.4 (d, J = 5.2 Hz, H-6, 1H), 7.4 (m, aromatic, 3H), 7.9 (s, aromatic, 1H), 7.9 (1H, s, aromatic), 9.6 (s, formyl 1H); 13C-NMR (300 MHz, CDCl3): δ 73.2 (C3), 120.6 (C16), 120.6 (C-aromatic), 121.6 (C6), 123.4 (C-aromatic), 124.0 (C-aromatic), 140.2 (C-aromatic), 141.4 (C5), 176.2 (C=O ester),187.8 (C=O formyl); and HRMS calculated for C35H40N2O3 (512.6823), found 513.6822.
3β-Cyclohexanoyloxy-17-(1H-bezimidazol-1-yl)-16-formylandrosta-5,16-diene (5e in ). Yield: 61.5 mg (25.96%) white solid; m.p.: 130–131 °C; IR (KBr) cm−1: 1725, 1674, 1195; UV (nm): 244, 273, 280; 1H-NMR: (400 MHz, CDCl3): δ 1.1 (s, H-18, 3H), 1.1 (s, H-19, 3H), 4.6 (m, H-3, 1H), 5.4 (d, J = 5.2 Hz, H-6, 1H), 7.4 (m, aromatic, 3H), 7.8 (s, aromatic, 1H), 7.9 (s, aromatic, 1H), 9.6 (s, formyl, 1H); 13C-NMR: (300 MHz, CDCl3): δ 73.0 (C3), 120.6 (C16), 120.6 (C-aromatic), 121.7 (C6), 123.4 (C-aromatic), 124.0 (C-aromatic), 140.2 (C-aromatic), 141.4 (C5), 175.6 (C=O ester), 187.9 (C=O formyl); and HRMS calculated for C37H42N2O3 (526.7089), found 527.7088.
3β-Cycloheptanoyloxy-17-(1H-bezimidazol-1-yl)-16-formylandrosta-5,16-diene (5f in ). Yield: 75 mg (31.67%) white solid; m.p.: 156–157 °C; IR (KBr) cm–1: 1726, 1673, 1186.; UV (nm): 244, 273, 280; 1H-NMR: (400 MHz, CDCl3): δ 1.1 (s, H-18, 3H), 1.1 (s, H-19, 3H), 4.6 (m, H-3, 1H), 5.4 (d, J = 4.8 Hz, H-6, 1H), 7.4 (m, aromatic, 3H), 7.8 (s, aromatic, 1H), 7.9 (s, aromatic, 1H), 9.6 (s, formyl, 1H); 13C-NMR: (300 MHz, CDCl3):δ 73.1 (C3), 120.6 (C16), 120.6 (C-aromatic), 121.6 (C6), 123.4 (C-aromatic), 124.0 (C-aromatic), 140.2 (C-aromatic), 141.4 (C5), 176.5 (C=O ester), 187.8 (C=O formyl); and HRMS calculated for C39H44N2O3 (540.7355), found 541.7354.
Derivative 6
Compound 6 () was prepared from 4 g as following:
A mixture of 3β-hydroxy-17-chloro-16-formylandrosta-5,16-diene, 4 g (0.1 mg, 0.3 mmol), benzimidazole (0.071 mg, 0.6 mmol) and Cs2CO3 (0.195 mg, 0.6 mmol) in dry DMF was stirred at 60 °C under 2 h. After, the mixture was poured onto ice-cold water (50 mL) and the obtained precipitate was filtered, washed with water and dried in vacuum. The dirty yellow solid was purified by column chromatography on florisil (Hexane:EtOAc 50:50) to give the compound 6.
3β-Hydrloxy-17-(1H-benzimidazole-1-yl)-16-formylandrosta-5,16-diene (6 in )
Yield 52%, white solid; m.p.: 203–204 °C mp; IR (FTIR-ATR): 3334, 3087, 2958, 1671, 1618, 737 cm−1; 1H-NMR (CDCl3) δ: 9.59 (s, 1H, CHO); 7.98 (s, 1H, imidazole-H); 7.85 (dd, J = 7.0, 2.8 Hz, 2H, aromatic-Hs); 7.35 (dd, J = 6.0, 2.9 Hz, 2H, aromatic-Hs); 5.40 (s, 1H, H-6); 3.55 (m, 1H, H-3); 1.05 (s, 6H, angular-Hs); 13C-NMR (CDCl3) δ: (16-CHO); 141.25 (imidazole-C); 124.21 (aromatic-C); 123.66 (aromatic-C); 122.68 (aromatic-C); 120.92 (C-6); 120.63 (aromatic-C); 71.62 (C-3); 20.57 (C-19); 19.45 (C-18). FAB-MS calcd for C27H32N2O2 (amu) 416, found 417 [M + 1]+.
Human an animal tissues and procedures
Four hours after a 57-year-old man had died from myocardial infarction; his normal prostate was extirpated in the Pathology Department of the General Hospital in Mexico City. The Ethical Committee of the General Hospital in Mexico City approved this protocol.
The tissue was rinsed and immediately chilled in ice-cold 150 mM NaCl and stored at −20 °C. The frozen human prostate was thawed on ice, rinsed, and minced in buffer A (20 mM sodium phosphate, pH 6.5, containing 0.32 M sucrose, 0.1 mM DTT (Sigma-Aldrich, Mexico City, Mexico) with an IKA® A11 basic tissue mill (IKA Laboratory Equipment, Mexico City, Mexico). Unless otherwise specified, the following procedures were carried out at 4 °C.
5α-R type 2 isozyme isolated from human prostate
Human prostate was used in this experiment because this tissue is an abundant source of 5α-R2 for the study of the effect(s) of 5a–5f and 6, which were designed for inhibition of the activity of this enzyme in humans. In this tissue, 5α-R type 1 is not as abundant as is type 2.
Human prostate tissue was homogenized in two volumes of buffer A with a tissue homogenizer Ultra-Turrax IKA, T18 basic (Wilmington, NC). Homogenates were centrifuged (1500 × g; 60 min)Citation9 in a SW 60 Ti rotor (Beckman Instruments, Palo Alto, CA). The pellets were re-suspended in buffer A, and stored at −70 °C. This suspension had a final concentration of 5 mg protein/mL, as determined by the Bradford methodCitation10 and was used as source of 5α-R type 2 isozyme.
5α-R type 1 isozyme isolated from rat liver
All procedures involving animals were approved by the Institutional Care and Use Committee of the Metropolitan University of Mexico (UAM; Xochimilco, Mexico). All animals used in this study were obtained from the Animal Care Facility at UAM.
Two adult (8-month-old) had been fasted overnight to decrease glycogen levels before their livers were extirpated for use as a source of 5α-R1, as recommend Levy et al.Citation11 To prepare microsomes, the livers (30 g) were minced in one volume of buffer A, by using the tissue mill. Unless otherwise specified, the following procedures were carried out at 4 °C. The tissue was homogenized; the suspension was centrifuged (10 000 × g; 30 min; 0 °C) (Beckman L70K ultracentrifuge, Mexico City, Mexico); and the pellet was discarded. The supernatant was filtered through a nylon mesh filter (pore size 11 µm, distributed by “OEM-Membrane Solution”, Dallas, TX) and centrifuged (100 000 × g; 60 min). The microsomal pellet was re-suspended in five volumes of buffer A with a homogenizer, and the protein concentration was determined by the Bradford method. The suspension was re-centrifuged (100 000 × g; 30 min) and the pellet was re-suspended in buffer A to give a final concentration of 20 mg protein/mL. The microsomal suspension was stored at −80 °C prior to the preparation of the solubilized steroid 5α-R type 1.
The solubilization of 5α-R type 1 isozyme from the rat liver microsomes was carried out according to Levy et al.Citation11
Gonadectomized male hamsters
For the experiments in vivo, 88 adult male golden hamsters (2.5-month-old; 150–200 g) were used. After gonadectomies had been performed on 80 hamsters under isoflurane anesthesia, the castrated hamsters were allowed to recover for 30 d prior to experimentationCitation5,Citation12. The castrated hamsters and the remaining eight intact hamsters were housed in a room with controlled temperature (22 °C) and light–dark periods of 12 h; the hamsters were fed with food and water ad libitum. Thirty days post-gonadectomy; the hamsters were separated in 10 groups consisting of four animals per group. Hamsters were treated for 6 d as described in section “Effect of steroid derivatives on flank organs... ” and thereafter sacrificed with CO2. This experiment was carried out twice under the same conditions.
Biological activity of steroidal derivatives
Experiments in vitro
The activity of the 5α-R type 1and 2 were determined by following the conversion of T to DHT, as previously describedCitation9,Citation11. The reagent mixture (final volume, 1 mL) contained 1 mM of DTT in 40 mM sodium phosphate buffer (pH 8.0 for type 1; pH 6.5 for type 2), 2 nM [1,2,6,7 3H] T, 6.31 µM T (for type 1), and 2 mM NADPH. The reaction, carried out in duplicate, was started when this mixture was added to the enzymatic fraction (for type 1, 90 µg protein/6.7 µL from the solubilized microsomes; for type 2, 50 µg/80 µL from the membrane fraction of human prostate). After incubation (37.5 °C; 60 min), the reaction was stopped by adding dichloromethane (1 mL) and mixing; this was considered as the end point. As control, this procedure was carried out without the addition of the enzyme fraction.
In order to extract and purify DHT formed by the activities of 5α-R types 1 and 2, after the individual reaction mixtures had been agitated (1 min) by using a Type 16700 mixer (Barnstead Thermoline, Proveedor Científico, México, D. F.), the dichloromethane phase of each was placed into individual tubes. This extraction was repeated four additional times. Each pooled dichloromethane extract was evaporated to dryness under a nitrogen stream and then suspended in methanol (50 µL); each preparation was spotted onto HPTLC Keiselgel 60 F254 plates. T, DHT, and a mixture of both standards were applied to the plate in distinct lanes on either side of the spotted preparation samples. The plates were developed in chloroform–acetone (9:1) and were air-dried; the chromatography was repeated two additional times. DHT was detected by using phosphomolybdic acid reagent (Sigma-Aldrich), and T, by fluorescence (UV lamp; 254 nm). The radioactivity on the plate was scanned by using a Bioscanner AR2000 (Bioscan, Washington, DC). The zones that showed chromatographic behavior identical to that of the standards (retention factor, Rf) were quantified as T or DHT. The DHT transformation yields were calculated from the lanes, taking into account the entire radioactivity in the TLC, and were plotted by using SigmaPlot 12 software (Systat Software, Inc., San Jose, CA). For the control incubations, the chromatographic separations and identifications were carried out in the same manner.
The 5α-R type 1 activity was calculated from the percentage of the labeled DHT that had been formed, taking into consideration the recovery, blank values, specific activity of [3H] T, and the ratio of added [3H] T to unlabeled T. The 5α-R type 2 activity was calculated, taking into consideration the recovery, blank values, and the specific activity of [3H] TCitation13.
The Rf of the T standard was 0.56. The radioactive zone that had chromatographic behavior identical to that of the standard T corresponded to 70% of the accounted radioactivity in the plate. The radioactivity contained in the zone of the experimental chromatogram, which had an Rf identical to that of the DHT standard (Rf: 0.67), was identified as the transformed DHT; it corresponded to 20–27% of the total radioactivity accounted for in the TLC. This result was considered to be 100% of the activity of 5α-R (types 1 or 2) for the development of inhibition plots. Unmodified [3H]T, identified (Rf: 0.56) from control incubations (i.e. those that had not contained tissue), had identical chromatographic behavior as did the non-labeled standard T.
The radioactivity contained in the zone corresponding to DHT standard (Rf: 0.67) of the control chromatogram was 1% of the total radioactivity accounted in the plate and was considered as an error; it was subtracted from the experimental chromatograms.
Effect of steroids derivatives on activity of 5α-reductase types 1 and 2
The effect of steroids 5a–5f and 6 on the activity of 5α-R type 1 or 2 was determined in the same conditions in vitro as described in section “Experiments in vitro”, but in presence of six different concentrations ([1 × 10−11, 1 × 10−10 M, …1 × 10−3 M]) of each one of 5a–5f and 6 derivatives. Extraction and purification of the DHT formed from these incubations were carried out as described in section “Experiments in vitro”.
Control tubes (without inhibitors) were prepared with the same incubating medium and labeled T plus either type 1 or type 2 5α-R, under the same conditions (section “Experiments in vitro”). DHT transformations in presence of 5a–5f and 6 were calculated from the lanes, taking into account the entire radioactivity in the plate, and were plotted by using SigmaPlot 12 software for inhibition curves.
Determination of the 50% inhibitory concentration of steroids 5a–5f and 6 on the activities of 5α-reductase types 1 and 2
To determine the 50% inhibitory concentration (IC50) of steroids 5a–5f and 6 on the activities of 5α-R types 1 and 2, the inhibition plots obtained in section “Effect of steroids derivatives... ” were analyzed with the SigmaPlot 12.5 software.
Experiment in vivo
Effect of steroid derivatives on flank organs, prostate, and seminal vesicles of castrated hamsters
For six consecutive days, each of the steroid derivatives 5a–5f and 6 (2 mg/kg body weight [BW]) dissolved in 200 µL sesame oil, together with 1 mg T/kg (BW), was administered by sc injection to a group of gonadectomized hamsters (four animals per derivative). Three groups of gonadectomized animals were used as controls: the first group was injected sc with 200 µL sesame oil; the second group, with 1 mg T/kg (BW); and the third group, with 1 mg T plus 1 mg F/kg (BW) prepared also in sesame oil. Additionally, one group of four intact hamsters was used as intact control. On the seventh day, the animals were sacrificed with CO2. The diameters of the flank organs were measured by using a vernier caliper; the prostate and seminal vesicles of each hamster were dissected and weighedCitation5,Citation12.
Two separate experiments were performed for each group of steroid-treated hamsters. The results were analyzed by using one-way analysis of variance and Dunnett’s Method to compare means, by using JMP IN 5.1 software (JMP, Statistical Discovery, Cary, NC).
Results
Chemistry
The synthetic strategy toward compounds 5a–5f and 6 is summarized in . The dehydroepiandrosterone derivatives 5a–5f and 6 were synthesized using a previously published procedure of Njar et al.Citation7 and Garrido et al. with slight modificationsCitation8. The new compounds 5a–5f and 6 were obtained with moderate yields and its structure and purity were confirmed by spectroscopic and spectrometric methods and melting point (see experimental section).
The yields of the products after the various steps in the syntheses were the following: 98.54% for the free alcohol, 2, resulting from the reaction of the acetoxydehydroepiandrosterone with NaOH in methanol; 83.2% for esters 3a–3f resulting from 2 being treated with the corresponding acid, dicyclohexylcarbodiimide, and 4-dimethylaminopyridine; 55–70% for 4a–4f, the 16-formyl-17-chloro-3β-acyloxy compounds formed when 3a–3f were reacted with phosphorus oxychloride in dimethylformamide and chloroform; and 20–40% for 5a–5f, the derivatives having aliphatic esters at C-3, a formyl group at C-16, and a double bond and benzimidazole ring at C-17.
Compound 6 was obtained in a yield of 52%, forming an alcohol group at C-3 and one benzimidazole group at C-17; maintaining the formyl group and the double bond at C-16 of the dehydroepiandrosterone.
All compounds were isolated in crystalline form and the melting points showed a maximum of one degree difference.
Activity of compounds 5a–5f and 6 as inhibitors of 5α-R 1 and 2
shows the IC50 values of 5a–5f and 6 required to inhibit 50% of the activity of 5α-R isoenzymes 1 and 2. Compared to 5b–5d and 6, steroid 5a exhibited the highest inhibitory activity (IC50 = 1.4 µM) for isoenzyme type 1. For the 5α-R type 2 isoenzyme, derivative 5 b displayed the highest inhibitory activity (IC50 = 0.004 µM). Steroids 5e and 5f failed to show an inhibitory activity for either isoenzyme.
Figure 2. Biological activities of finasteride and the six novel steroidal derivatives 5a–5f and 6. IC50: concentration of compound required to inhibit 50% of the activity of 5α-reductase isoenzymes 1 and 2.
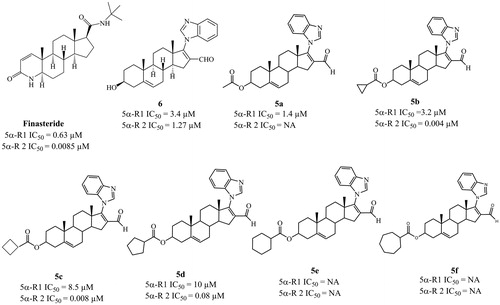
Flank organ test
Castration decreased (p < 0.005) the diameter of the male hamster flank organs, as compared to those of the normal animals (C). Treatment with vehicle alone had no effect, whereas sc injections of 1 mg/kg of T for 6 d significantly increased (p < 0.005) the diameters of the pigmented spots of the flank organs in the castrated male hamsters ().
Figure 3. Diameters of the pigmented spot (±standard deviation) of flank organs of castrated and intact (C) hamsters. For 6 d, the castrated hamsters received subcutaneous injections of the various test materials, with the animals treated with vehicle only (V). The experiment was carried out in duplicate. The asterisks show the instances of statistically significant difference (p < 0.05) between the group of hamsters treated with testosterone (T) and those treated with T and with either finasteride (F) or with the synthesized steroids (5a–5f, and 6 see for structures).
Compared to T (1 mg/kg (BW), steroids 5a–5f and 6 (2 mg/Kg (BW)) were able to counteract the effect of T, thus resulting in significantly reduced (p < 0.005) diameters of the flank organs, with structures 5a–5f and 6 showing the greatest anti-androgenic activity. F produced a statistically significant decrease (p < 0.005) in the diameters of the flank organ spots, as compared to those in treated only with T.
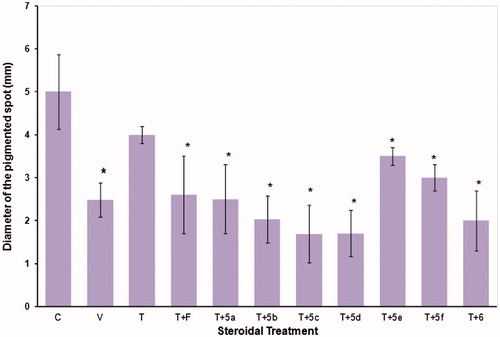
Weight of the prostate gland and seminal vesicles of hamsters
At sacrifice, there were statistically significant (p < 0.05) reductions in the weights of the prostates and seminal vesicles, as compared to glands from intact hamsters (data not shown). However, treatment with 1 mg/kg (BW) of T significantly reversed the effect of castration on these glands ().
Figure 4. Weight of prostate and seminal vesicles glands (± standard deviation) from castrated and intact animals (C) hamsters. For 6 d, the castrated hamsters received different subcutaneous injections of the various test materials, with the castrated control animals (V) treated with vehicle only. The experiment was carried out in duplicate. The asterisks show the instances of statistically significant difference (p < 0.05) between the group of hamsters treated with testosterone (T) and those treated with T and with either finasteride (F) or with the synthesized steroids (5a–5f, and 6 see for structures).
F and the steroids 5a, 5e, 5f, and 6 when administrated together with T, significantly decreased the weight of the prostate and seminal vesicles (p < 0.05); however, compared to T, compound 5b reduced the weight only of the seminal vesicles and steroids 5c and 5d did not display any statistically significant effect ().
Discussion
We investigated the effect of seven novel dehydroepiandrosterone derivatives with a benzimidazol moiety at C-17, 5a–5f and 6, on the activity of 5α-R types 1 and 2 isozymes. The aliphatic ester moiety at C-3 in steroid 5a induced specific inhibition of the activity of 5α-R1, because no effect of 5a on 5α-R2 activity was detected. However, having a cyclopropyl, cyclobutyl, or cyclopentyl ring (5b–5d) as the cycloaliphatic ester moiety at C-3 of the dehydroepiandrosterone scaffold inhibited the activity of both isozymes, with the potency of these steroids decreasing according to the size of the cycloaliphatic ring. Further, a cyclohexyl or cycloheptyl ring at C-3 of these derivatives failed to have an inhibitory effect on either isozyme. F is considered as a weak inhibitor of 5α-R1Citation14. Although these derivatives showed less potency than did F in inhibiting 5α-R1, this study contributes to the understanding of the effect that different moieties at C-3 of the dehydroepiandrosterone skeleton have on the activity of 5α-R1. Steroids 5b and 5c showed higher potency than F, which is considered to be a potent inhibitor of 5α-R2Citation14, with IC50 values 0.004, 0.008, and 0.0085, respectively, thus indicating that 5b and 5c could have therapeutic potency.
5α-R types 1 and 2 play an important role in the pathologies of prostate cancer and benign prostatic hyperplasia because they are present in this gland and show different locations and biochemical characteristicsCitation1,Citation2. Type 1 has been identified in prostate epithelial cells, and type 2 is principally found in the stromal compartmentCitation15. 5α-R1 has also been identified in the liver and skin; it is active at neutral or basic pHCitation1. A different pH of activity has been reported for the type 2 enzyme because it is active in acidic media. Weisser and Krieg reported that stromal cells from benign prostatic hyperplasia produce more 5α-reduced metabolites (DHT and 5α-dione) than normal cellsCitation16. Furthermore, the development and progression of prostate cancer (PCa) are also related to alterations in the 5α-R enzymeCitation15. Immunostaining techniques for 5α-R1 and 5α-R2 using specific antibodies on different human PCa specimens have demonstrated that 5α-R1 is increased and 5α-R2 is decreased during the course of PCaCitation15. However, the expression of both 5α-R isozymes increases in recurrent and metastatic cancers, thus suggesting that both isozymes may be important in the development and progression of PCaCitation15. Moreover, the fact that finasteride, a 5α-R2 inhibitor, reduced the prevalence of PCaCitation15 also suggests that both isozymes could be required for the development and progression of PCa. Therefore, type 1 and 2 5α-R isozymes could be important therapeutic targets for this disease.
Various 5α-R inhibitors have been developed for relieving the symptoms produced in benign prostatic hyperplasia. These 5α-R inhibitors reduce bladder obstruction present in this disease by inhibiting the conversion of T in its more active form DHT. Moreover, these inhibitors eliminate the need for surgery and prevent further progression of the disease. The most extensively studied 5α-R inhibitors are finasteride® and dutasteride®Citation14. Finasteride and dutasteride are 4-azasteroids that have demonstrated low affinity for ARs and thus were not expected to produce undesirable anti-androgen effects such as impotence, muscle growth impairment or gynecomastiaCitation17. However, when these drugs are used for long periods, they could induce hepatoxicityCitation18,Citation19; thus, synthesis of new 5α-R inhibitors with low side effects has been a goal for researchers.
In this study, we used hamsters for experiments in vivo because they have two androgen-dependent pilosebaceous complexes (flank organs) in the dorsal skin, which had been used to measure the anti-androgenic effect of different drugsCitation5,Citation18. In the current model, 5a–5d and 6 induced a visible pharmacological effect on the diameter of flank organs, due to the inhibition of 5α-R types 1 and 2Citation5,Citation20–22. Steroid 5c–d and 6 was very active in decreasing the size of the flank organs.
The lipophilicity of steroids 5a–5f could be advantageous for improving the ability of these molecules to cross through cell membranes. However, in the bloodstream, these esters will be hydrolysed by the esterases present in the blood, and the resultant hydrolysed molecules may contain a carboxyl moiety at C-3, similar to that present in 5a and finally hydrolysed to an alcohol moiety at C-3 as in compound 6. This possibility was considered in this study, and we demonstrated that steroid 6 is also an inhibitor of 5α-R2. Because these esters could be considered pro-drugs, they could be used to mask the side effects and toxicity produced by the formyl group present at C-16 of these derivatives to different organs and systems of the bodyCitation23. Thus, when these ester moieties are hydrolysed by plasma esterases, the active drug will be released.
The pharmacological effect showed 5e and 5f compounds may be because they were hydrolyzed by the esterases present in the blood stream. As a result, a carbonyl group or alcohol may have formed in the C-3 of these steroids, inducing the formation of 5a or 6 which were active in vivo. However, we cannot explain the deficient pharmacological activity exhibited by 5c–5d in prostate and seminal vesicles by this hydrolysis mechanism. Thus, it could be possible 5c–5d enter to the prostate and seminal vesicles, where the presence of P450 enzymes could deactivated them. This hypothesis will be tested in a near future.
Conclusion
To varied degrees, the seven novel steroidal derivatives acted as inhibitors of the activity of 5α-R types 1 or 2, or both, and bound to androgen and progesterone receptors. In particular, compounds 5b, 5c, and 5d and 6 inhibited 5α-R isoenzymes and reduced the diameter of the androgen-dependent flank organs. From these results, we infer (1) that the inhibitory activity for the 5α-R types 1 and 2 isoenzymes decreased with an increase of the ring size in the ester moiety at C-3, and (2) that this inhibitory effect apparently was reflected in the reduction in the diameter of the flank organs. On the basis of these results, we conclude that 5b–5f and 6 may have therapeutic potential for the treatment of some androgen-dependent diseases such as prostate cancer and benign prostatic hyperplasia and, therefore, that they should be further tested in other animal models.
Acknowledgements
The authors thank Veronica Yakoleff for editing the article.
Declaration of interest
This study was supported by grants from Consejo Nacional de Ciencia y Tecnología (CONACYT project number 165049) and from Dirección General de Asuntos del Personal Académico (DGAPA project number IN 211312). The authors declare that that are no real or perceived conflicts of interest arising from intellectual, personal, or financial circumstances of the research. Additionally, all authors are aware, and approve, of the contents and order of authorship of the article.
References
- Russell DW, Wilson JD. Steroid 5alpha-reductase: two genes/two enzymes. Ann Rev Biochem 1994;63:25–61
- Liang TEHM, Cascieri MA, Cheung AH, et al. Species differences in prostatic steroid 5α-reductases of rat, dog, and human. Endocrinology 1985;117:571–9
- Lee HJ, Chang C. Recent advances in androgen receptor action. Cell Mol Life Sci CMLS 2003;60:1613–22
- Shiba K, Hamaguchi T, Kataoka K, et al. Cloning of the hamster androgen receptor gene. J Dermatol Sci 2001;26:163–8
- Cabeza M, Heuze Y, Quintana H, Bratoeff E. Comparison between two different hamster models used for the determination of testosterone and finasteride activity. Asian J Anim Veter Adv 2010;5:202–9
- Trapani G, Dazzi L, Pisu MG, et al. A rapid method for obtaining finasteride, a 5α-reductase inhibitor, from commercial tablets. Brain Res Protocol 2002;9:130–4
- Njar VCO, Kato K, Nnane IP, et al. Novel 17-azolyl steroids, potent inhibitors of human cytochrome 17α-hydroxylase-C17,20-lyase (P45017α): potential agents for the treatment of prostate cancer. J Med Chem 1998;41:902–12
- Garrido M, Cabeza M, Cortés F, et al. Cytotoxic effect of novel dehydroepiandrosterone derivatives on different cancer cell lines. Eur J Med Chem 2013;68:301–11
- Hirosumi J, Nakayama O, Fagan T, et al. FK143, a novel nonsteroidal inhibitor of steroid 5α-reductase: (1) in vitro effects on human and animal prostatic enzymes. J Steroid Biochem Mol Biol 1995;52:357–63
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–54
- Levy MA, Brandt M, Greway AT. Mechanistic studies with solubilized rat liver steroid 5.alpha.-reductase: elucidation of the kinetic mechanism. Biochemistry 1990;29:2808–15
- Wright AS, Douglas RC, Thomas LN, et al. Androgen-induced regrowth in the castrated rat ventral prostate: role of 5α-reductase. Endocrinology 1999;140:4509–15
- Bratoeff E, Ramírez E, Flores E, et al. Molecular interactions of new pregnenedione derivatives. Chem Pharmaceut Bull 2003;51:1132–6
- Machetti F, Guarna A. Novel inhibitors of 5α-reductase. Expert Opin Therapeut Patents 2002;12:201–15
- Thomas LN, Douglas RC, Lazier CB, et al. Type 1 and Type 2 5α-reductase expression in the development and progression of prostate cancer. Eur Urol 2008;53:244–52
- Weisser H, Krieg M. In vitro inhibition of androstenedione 5α-reduction by finasteride in epithelium and stroma of human benign prostatic hyperplasia. J Steroid Biochem Mol Biol 1998;67:49–55
- Liang T, Heiss CE, Cheung AH, et al. 4-Azasteroidal 5 alpha-reductase inhibitors without affinity for the androgen receptor. J Biol Chem 1984;259:734–9
- Dadras SS, Cai X, Abasolo I, Wang Z. Inhibition of 5alpha-reductase in rat prostate reveals differential regulation of androgen-response gene expression by testosterone and dihydrotestosterone. Gene Expr 2001;9:183–94
- Raynaud JP. Prostate cancer risk in testosterone-treated men. Proceedings of the 17th international symposium of the Journal of Steroid Biochemistry and Molecular Biology. Recent advances in steroid biochemistry and molecular biology; 2006 May 31–Jun 3; Seefeld, Tyrol, Austria; vol. 102
- Vermorken AJM, Goos CMAA, Wirtz P. Evaluation of the hamster flank organ test for the screening of anti-androgens. Br J Dermatol 1982;106:99–101
- Takayasu S, Adachi K. The in vivo and in vitro conversion of testosterone to 17β-hydroxy-5α-androstan-3-one (dihydrotestosterone) by the sebaceous gland of hamsters. Endocrinology 1972;90:73–80
- Takayasu S, Adachi K. The intranuclear binding of 17/3-hydroxy-5α-androstan-3-one and testosterone by hamster sebaceous glands. Endocrinology 1975;96:525–9
- Heck dHA, Casanova M, Starr TB. Formaldehyde toxicit. New understanding. Crit Rev Toxicol 1990;20:397–426