
Abstract
Inhibition of glutamine synthetase (GS) is one of the most promising strategies for the discovery of novel drugs against tuberculosis. Forty-three bisphosphonic and bis-H-phosphinic acids of various scaffolds, bearing aromatic substituents, were screened against recombinant GS from Mycobacterium tuberculosis. Most of the studied compounds exhibited activities in micromolar range, with N-(3,5-dichlorophenyl)-2-aminoethylidenebisphoshonic acid, N-(3,5-difluorophenyl)-2-aminoethylidene-bisphoshonic acid and N-(3,4-dichlorophenyl)-1-hydroxy-1,1-ethanebisphosphonic acid showing the highest potency with kinetic parameters similar to the reference compound – L-methionine-S-sulfoximine. Moreover, these inhibitors were found to be much more effective against pathogen enzyme than against the human ortholog. Thus, with the bone-targeting properties of the bisphosphonate compounds in mind, this activity/selectivity profile makes these compounds attractive agents for the treatment of bone tuberculosis.
Introduction
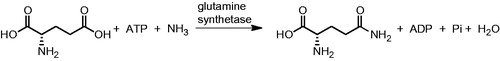
Glutamine synthetase (GS, glutamate-ammonia ligase, E.C. 6.3.1.2), one of the key enzymes in nitrogen metabolismCitation1, catalyses the formation of glutamine from glutamate and ammonia in the presence of adenosine triphosphate (ATP) (). This enzyme, together with glutamate synthase (GOGAT, E.C. 1.4.1.13), is a component of a metabolic cycle that enables the incorporation of inorganic nitrogen (in the form of ammonium ions) into the organic molecules present in living cells. Glutamate, the product of the GS-GOGAT cycle, is the primary nitrogen donor in the synthesis of purines, pyrimidines, amino acids, glucosamine-6-phosphate, carbamoyl phosphate and a range of other nitrogen-containing products.
Figure 1. Reaction catalysed by glutamine synthetase.
The numerous implications of GS in various biological processes make the enzyme indispensable for cell vitality in all living organismsCitation2. Bioinformatic studies using multiple sequence alignment showed the gene encoding GS to be one of the oldest yet foundCitation3. As the result of the long evolutionary process, three distinct classes of GSs have been developed: GS1, GS2 and GS3. GS1 is mainly expressed in prokaryotic cells and is constructed from 12 identical subunits, forming two hexameric rings connected face to faceCitation4,Citation5. GS2 is primarily found in eukaryotes, but its expression has also later been confirmed in some actinomycetes and rhizobiaCitation6,Citation7. This form of the enzyme is composed of 10 subunits forming two pentameric rings. The most recently characterised class (GS3) was first discovered in the anaerobic bacterium Bacteroides fragilis and later found in other anaerobesCitation8. Its dodecameric structure is formed by association of two hexameric rings, but on the opposite molecular surface than found in GS1 and GS2.
The active site of GS is formed of two cones connected at their narrow ends, in which two or three metal ions (Mg2+ or Mn2+) resideCitation9. The glutamate and ammonium ion enter from one side, while ATP arrives via the opposite end. In the first step of the enzymatic reaction, glutamate is phosphorylated to yield γ-glutamyl phosphate. Ammonia then replaces the phosphate to form the amide moiety of glutamine. The commonly held belief that this catalytic mechanism is the same for all the existing forms of GS is supported by the presence of highly conserved residues at the glutamate and ammonium binding sitesCitation1,Citation10.
The most numerous group of GS inhibitors is composed of glutamate analogues. The majority of these are organophosphorus and organosulfur compounds, with L-methionine-S-sulfoximine (MetSox, 1) and phosphinothricin (2) being the most representative examples ()Citation1,Citation11. These structures are phosphorylated in the enzyme active site and subsequently act as transition state analoguesCitation12,Citation13.
Figure 2. Representative examples of glutamine synthetase inhibitors.
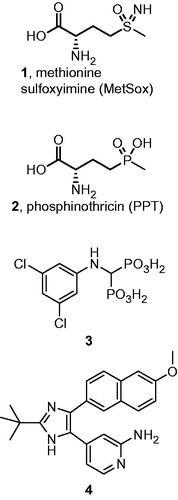
More recently, compounds that interact with the ATP binding pocket have been developed. A number of aminomethylenebisphosphonic acids (e.g. structure 3, ) were initially shown to be effective inhibitors of plant GSCitation14,Citation15. Later, aromatic derivatives of the imidazole functionality (e.g. compound 4) were discovered as bacterial GS inhibitorsCitation16,Citation17.
Currently, the most important application of GS inhibitors is in the development of antibiotic strategies for use against the human pathogen Mycobacterium tuberculosis. GS inhibitors were shown to selectively block the formation of the poly(glutamine-glutamate) cell walls of these bacteriaCitation18,Citation19. The efficacy of MetSox was proven in vivoCitation20. M. tuberculosis infections are known to frequently spread outside the lungs, reaching various parts of the human body. One major site of extrapulmonary tuberculosis is the bonesCitation21. Importantly in this respect, pharmacokinetic studies of bisphosphonates indicate that, regardless their structure, the overwhelming fraction of the absorbed active substance is accumulated in the bones due to the presence of the bisphosphonate moietyCitation22. Thus, the application of bone-targeting bisphosphonate M. tuberculosis GS (MtGS) inhibitors may prove an excellent strategy for treating osseous tuberculosis.
Herein, we present the results of an extensive screening of compounds with various scaffolds containing the bisphosphonic acid group, each designed to inhibit MtGS. Detailed kinetic studies of the most active structures provided insight into their molecular mode of action. Their selectivity towards MtGS was estimated by comparison with the activities demonstrated against recombinant human glutamine synthetase (HsGS).
Materials and methods
General
All used chemicals were obtained from commercial suppliers (Sigma-Aldrich, Poznan, Poland; Merck, Warsaw, Poland; Fermentas, Warsaw, Poland; POCh, Gliwice, Poland) as reagents of analytical grade. Deionized ultra-pure water was used in all biochemical experiments. Escherichia coli GJ4547 (kindly provided by Dr. Gowrishankar, Centre for DNA Fingerprinting and Diagnostics, Hyderabad, India) and BL21(DE3) CodonPlus-RIL (Stratagene, La Jolla, CA) strains were used for recombinant M. tuberculosis and human GS expression, respectively. The recombinant plasmids (Vector pTrc99c, Vector pNIC-BSA4) used in GS studies were kindly provided by Prof. Sherry L. Mowbray from Uppsala University. During the enzyme preparation, purity of the obtained proteins was analyzed by means of SDS-PAGE electrophoresis, while protein concentration was determined according to Bradford protein assay with bovine serum albumin used as standard.
Synthetic procedures
Compounds 5b, 5c, 5e–5w, 6m, 6k, 6r, 6t, 6w, 7c, 7h, 7w, 8c, 8g, 8h, 8k and 9a–w were described in preceding studiesCitation15,Citation23–25.
Aminomethylenebisphosphonic acids (5). The mixture of amine (25 mmol), triethylortoformate (30 mmol) and diethyl phosphite (100 mmol) was heated at 100–110 °C for 6 h. The volatiles were removed under reduced pressure and crystallized compound was filtered and washed with hexane. Obtained crude tetra-ester was dissolved in methylene chloride and TMSBr (10 eq) was added. The mixture was stirred overnight. Subsequently, methanol (10 mL) was added and stirring was continued for 2 h. The reaction mixture was evaporated under reduced pressure, and crude product was purified using flash chromatography (C18 column, water/acetonitrile).
N-phenylaminomethylenebisphosphonic acid (5a). Yield 56%. 1H NMR D2O δ 3.97 (t, 1H, 2JPH = 20.4, PCHP), 6.86–6.95 (m, 5H, Ph), 31P NMR D2O δ 13.98, 13C NMR D2O δ 53.92 (t, 1JPC = 132.4, PCHP), 117.03, 122.37, 129.48, 142.87 (4 × s, Ph), ESI-MS m/z: calc. 265.9984 [M–H+]−, found 265.9973 [M–H+]−.
N-(4-isopropylphenyl)aminomethylenebisphosphonic acid (5d). Yield 63.5%. 1H NMR D2O δ 1.07 and 1.02 (d each, 3H, 3JHH = 6.9 and 7.0), 2.78 (m, 1H, CH), 3.84 (t, 1H, 2JHP = 19.1, CHP2), 7.06 (d, 2H, 3JHH = 8.7, ArH), 7.20 (m, 2H, ArH), 31P NMR D2O 11.97 δ 13C NMR D2O δ 23.54 (s, CH3), 32.46 (s, CH(CH3)2), 54.22 (t, 1JPC = 130.0, PCHP), 112.92, 126.95, 136.05, 147.85 (4 × s, Ar), ESI-MS m/z: calc. 308.0453 [M–H+]−, found 308.0450 [M–H+]−.
N-(3-trifluoromethylphenyl)aminomethylenebisphosphonic acid (5p). Yield 29.9%. 1H NMR D2O δ 4.15 (t, 1H, 2JPH = 21.3, PCHP), 6.93 (d, 1H, J = 8.3, Ar), 6.96 (d, 1H, J = 7.6, Ar), 7.00 (s, 1H, Ar), 7.26 (t, 1H, J = 8.0, Ar); 31P NMR D2O 16.64 δ 13C NMR D2O δ 50.64 (t, 1JPC = 138.1, PCHP), 109.65 (q, J = 3.7, Ar), 114.62 (q, J = 3.8, Ar), 116.98 (s, Ar), 124.24 (q, J = 271.4, CF3), 129.94 (s, Ar), 130.87 (q, J = 31.8, Ar),147.20 (s, Ar), ESI-MS m/z: calc. 333.9857 [M–H+]−, found 333.9845 [M–H+]−.
Aminoethylidenebisphosphonic acids (6). Tetraethyl ethylidenebisphosphonate (1.0 g, 3.3 mmol) was dissolved in dry THF and amine (3.3 mmol) was added. The reaction mixture was stirred for 48 h at room temperature. Volatiles were evaporated under reduced pressure to get crude tetraethyl aminoethylidenebisphosphonate. The crude intermediate was dissolved in acetonitrile and TMSBr (39 mmol) was added. The mixture was left overnight at room temperature. Subsequently, methanol was added (10 mL) and reaction was continued for 1 h. Mixture was evaporated under reduced pressure, the residue crystallized from water/ethanol.
N-phenylaminoethalidenebisphosphonic acid (6a). Yield 58%. 1H NMR D2O δ 2.34 (tt, 1H, 2JPH = 21.1, 3JHH = 7.6, PCHP), 3.59–3.71 (m, 2H, CH2), 7.35–7.45 (m, 5H, Ph), 31P NMR D2O δ 15.92, 13C NMR D2O δ 36.23 (t, 1JPC = 117.3, PCHP), 50.05 (s, CH2), 121.98, 129.08, 130.30, 135.49 (4 × s, Ph), ESI-MS m/z: calc. 280.0140 [M–H+]−, found 280.0141 [M–H+]−.
N-(4-methylphenyl)aminoethalidenebisphosphonic acid (6b). Yield 15%. 1H NMR D2O δ 2.26 (3H, s, CH3), 2.32 (tt, 1H, 2JHP = 20.9, 3JHH = 7.7, CH), 3.64 (td, 2H 3JHP = 13.9, 3JHH = 7.6, CH2), 7.26–7.30 (m, 4H Ar); 31P NMR D2O δ 19.29 13C NMR D2O δ 20.15 (s, CH3), 35.86 (t, 1JPC = 118.6, PCHP), 50.12 (s, CH2), 122.37, 130.80, 131.75, 140.65 (4 × s, Ar); ESI-MS m/z: calc. 294.0296 [M–H+]−, found 294.0294 [M–H+]−.
3,5-Difluorophenylaminoethylidenebisphosphonic acid (6o). Yield 65%. 1H NMR D2O δ 2.19 (tt, 1H, 1JHP = 21.4, 3JHH = 6.3, PCHP), 3.45 (td, 2H, 3JHP = 14.1, 3JHH = 6.8, CH2), 6.20 (1H, tt, J = 9.6, J = 2.2, Ar); 6.29 (2H, m, Ar); 31P NMR D2O δ 19.00 13C NMR D2O δ 38.74 (t, 1JPC = 114.6, PCHP), 41.09 (s, CH2), 92.75 (t, J = 24.6, Ar), 96.67 (dd, J = 22.5, J = 6.2, Ar), 150.33 (t, J = 13.1, Ar), 163.82 (dd, J = 241.8, J = 16.1, Ar); ESI-MS m/z: calc. 315.9951 [M–H+]−, found 315.9947 [M–H+]−.
Hydroxybisphosphonic acids (7 and 8). Tris(trimethylsillyl)phosphite (6 mmol) was added drop wise to acid chloride (3 mmol). The reaction mixture was stirred for 1 h at room temperature and volatiles were evaporated under reduced pressure. Product was purified using flash chromatography with C18 column (water/acetonitrile).
Phenylmethylidene-1-hydroxy-1,1-bisphosphonic acid (7a). Yield 75%. 1H NMR D2O δ 7.25, 7.59 (m, 5H, Ph), 31P NMR D2O δ 16.8, 13C NMR D2O δ 75.63 (t, 1JPC = 144.8, PCP), 125.79, 127.75, 128.11, 135.41 (4 × s, Ph), ESI-MS m/z: calc. 266.9824 [M–H+]−, found 266.9831 [M–H+]−.
Phenylethylidene-1-hydroxy-1,1-bisphosphonic acid (8a). Yield 54%. 1H NMR D2O δ 3.19 (t, 2H, 3JPH = 13.4, CH2), 7.11–7.28 (m, 5H, Ph); 31P NMR D2O δ 19.36; 13C NMR D2O δ 38.19 (s, CH2), 73.62 (t, 1JCP = 146, PCP), 126.87, 127.87, 131.08 (3 × s, Ph), 135.10 (3 × s, t, 3JCP = 8.7, Ph), ESI-MS m/z: calc. 280.9980 [M–H+]−, found 280.9986 [M–H+]−.
4-Methylphenylethylidene-1-hydroxy-1,1-bisphosphonic acid (8b). Yield 80%. 1H NMR D2O δ 7.17 (d, 2H, 3JHH = 7.8, ArH), 7.03 (d, 2H, 3JHH = 7.8, ArH), 3.16 (t, 2H, 3JHP = 13.6, CHP2), 2.16 (s, 3H, CH3); 31P NMR (δ) 19.47. ESI-MS m/z: calc. 295.0136 [M–H+]−, found 295.0139 [M–H+]−.
3,4-Dichlorophenylethylidene-1-hydroxy-1,1-bisphosphonic acid (8l). Yield 72%. 1H NMR D2O δ 3.18 (t, 1H, 3JHP = 13.1, CH2), 7.18 (d, 1H, J = 8.2, Ar), 7.31 (d, 1H, J = 8.2, Ar), 7.43 (s, 1H Ar); 31P NMR D2O δ 18.24; 13C NMR D2O δ 37.57 (s, CH2), 73.58 (t, J = 145.4, PCP), 129.49, 129.99, 130.66, 130.87, 132.68 (5 × s, Ar), 136.12 (t, 3JCP = 9.2, Ar); ESI-MS m/z: calc. 348.9201 [M–H+]−, found 348.9198 [M–H+]−.
Benzylethylidene-1-hydroxy-1,1-bisphosphonic acid (8x). Yield 77%. 1H NMR D2O δ 2.04–2.20 (m, 2H, CH2), 2.77–2.82 (m, 2H, CH2Ph), 7.10–7.26 (m, 5H, Ph) 31P NMR D2O δ 19.86, 13C NMR D2O δ 29.48 (t, 3JPC = 6.7, CH2Ph), 35.74 (s, CH2CP), 73.07 (t, 1JPC = 146.8, PCP), 126.09, 128.30, 128.64, 142.04 (4 × s, Ph), ESI-MS m/z: calc. 295.0136 [M–H+]−, found 295.0136 [M–H+]−.
Protein expression
In order to obtain unadenylated MtGS, pTrc99c plasmidCitation5 with glnA1 gene being under the control of trp/lac promoter, was introduced to the competent cells prepared from genetically modified E. coli GJ4547 strain that lacks GS adenyl transferase activity. Transformed cells were grown in LB medium with addition of 100 µg/mL ampicillin at 37 °C to OD600 of 1.0, when expression was induced with 1 mM IPTG. Cells were incubated overnight (18 h) at 25 °C, then harvested by centrifugation, washed with 50 mM Hepes buffer, pH 7.5 and stored at −20 °C.
Recombinant HsGS was obtained as a product of GLUL gene (encoding HsGS from human muscle) that was cloned into pNIC-BSA4 vector and expressed in E. coli BL21 Codon Plus-RIL cells (Stratagene). Cells were grown in a phosphate-buffered medium containing per liter 2.3 g KH2PO4, 3.78 g K2HPO4, 12 g tryptone, 24 yeast extract, 4 mL glycerol, with addition of 50 µg/mL kanamycin. When optical density of the culture (at 600 nm) reached 1.0, expression was induced with 1 mM IPTG and continued for 18 h. After that, cells were harvested by centrifugation, washed with 50 mM phosphate buffer, pH 7.5 and stored at −20 °C until further use.
Protein purification
Thawed bacterial cells were suspended in binding buffer supplemented with 1.0 mM EDTA and 2.0 mM β-mercaptoethanol and were lysed by sonication. Crude extracts containing recombinant protein were separated from cell debris by centrifugation (1 h, 11 000 rpm) Supernatants were then loaded be means of ÅKTA Prime Plus Liquid Chromatography system (GE Healthcare, Warsaw, Poland) onto previously equilibrated pre-packed HisTrap™ FF column (3 × 1 mL, GE Healthcare). Unbound proteins were removed by extensive column washing with binding/wash buffer, the target protein was achieved with a linear gradient of imidazole in elution buffer. Usually, single peak of His-tagged protein with confirmed GS-activity appeared at 250–350 mM of imidazole.
In case of MtGS expressed in E. coli GJ4647, the wash/binding buffer was composed of 50 mM HEPES, pH 7.5, 300 mM NaCl, 10% glycerol, 25 mM imidazole and elution buffer was 50 mM HEPES, pH 7.5 with 300 mM NaCl, 10% glycerol and 450 mM imidazole.
During purification of human GS expressed in E. coli BL21 Codon PLUS-RIL strain, the following buffers were used: the wash/binding buffer consisted of 50 mM phosphate, 7.5, 500 mM NaCl, 10% glycerol, 25 mM imidazole, while elution was done with the same buffer containing 450 mM imidazole.
In the next step, obtained active fractions of the target proteins (MtGS or HsGS) were pooled together, concentrated on Amicon Ultra-15 Centrifugal Filter Units (NMWL 10 kDa, Merck, Warsaw, Poland) and then loaded onto Sephacryl S-300 HR 26/60 column. During gel-filtration step 20 mM HEPES, pH 7.5 with the addition of 150 mM NaCl and 10% glycerol was used as the working buffer (Figure S1 and S2). Identification of GS-containing protein fraction was performed by means of biosynthetic assay.
Biosynthetic assay
Biosynthetic activity of GS was measured by means of malachite green-acid molybdate assay enabling detection of inorganic phosphate that is released during glutamine synthesis. Colorimetric reagent was composed of 6.3 mM malachite green dye and 34 mM ammonium molybdate dissolved in 4M HCl. Both of these components were mixed with each other in the 1:1 ratio and finally diluted with two volumes of distilled water. Colorimetric solution was stirred for 20 min, stabilized by addition of CHAPS (2 mg/mL) and centrifuged for 3 min at 2500 g in order to remove undissolved particles.
Reaction mixture optimized for MtGS contained enzyme solution, 100 mM HEPES buffer pH 7.5, 50 mM l-glutamate, 2.5 mM ATP, 10 mM MgCl2, 10 mM NH4Cl, yielding a final volume of 100 µL. In case of HsGS, the highest enzymatic activity was observed when 100 mM HEPES, pH 8.0, 20 mM MgCl2, 100 mM l-glutamate, 2.5 mM ATP and 10 mM NH4Cl were used.
In both cases, reaction was started by the addition of the enzyme. At the end of incubation, 1 mL of colorimetric reagent was added to the reaction mixture and after 1-min assay was accomplished by the addition of 100 µL of 34% citrate. Optical density of each probe was determined spectrophotometrically at 630 nm. Quantitative analysis of GS activity was done on the basis of a standard curve obtained with proper dilutions of a 2 mM solution of KH2PO4.
Inhibitory studies
IC50 determinations were conducted under the optimized conditions of biosynthetic assay. GS inhibition was evaluated by introduction of an appropriate dilution of freshly prepared 25 mM stock solution of the analyzed compound (pH of dissolved inhibitor was adjusted to 7.0–7.5) to the reaction mixture. Reaction was started by the addition of the enzyme and had been run for 30 min at 37 °C in water bath. At least three measurements were performed for each dose of tested inhibitor. IC50 parameter was calculated using linear regression method by means of Prism 5 software (GraphPad, La Jolla, CA). The activity of enzyme was expressed as percentage of untreated control plotted against logarithm of used inhibitor concentration.
For kinetic evaluations, MtGS was assayed in the presence of increasing concentrations of tested compounds and substrate concentrations ranging from 5 to 100 mM in case of glutamate and from 0.25 to 5 mM for ATP. At least five inhibitor concentrations ranging from 0.5 to 2-fold of IC50 value were evaluated in triplicate for one concentration of substrate and fixed concentration of the enzyme. The model of inhibition towards ATP and glutamate was determined on the basis of Lineweaver–Burk plots and curve-fitting method based on non-linear regression analysis using GraphPad Prism 5 software.
Reversibility of inhibition was determined by measuring the recovery of enzymatic activity after a rapid and large dilution of the enzyme–inhibitor complex. Target enzyme at a concentration of 100-fold over the concentration used in activity assay was pre-incubated with inhibitor used at concentration equivalent to 10-fold the IC50. After 15-min equilibration time period, this mixture was diluted 100-fold into reaction buffer containing all necessary substrates to initiate reaction. The progress curve for tested sample was measured and compared to that of control sample that was pre-incubated in the absence of inhibitor and diluted the same way.
Molecular modeling
Molecular modeling calculations were performed using Discover Studio 3.1 program package (Accelrys, San Diego, CA). The crystal structure of M. tuberculosis GS (PDB id 2BVC) was used as starting point for calculationsCitation7. Initially, protons were added to the protein molecule assuming pH 7. The model of bisphosphonate inhibitor was constructed and placed in ATP binding site. Various positioning of bisphosphonate molecule was tested. Partial charges were computed using Momany-Rone algorithm. Minimizations were performed using CHARMm forcefield and Smart Minimizer algorithm up to Energy change 0.0 or RMS gradient 0.1. All residues that are not forming the active site cleft were frozen.
Results and discussion
We have previously shown aminomethylenebisphosphonic acids to be effective inhibitors of plant GSCitation15. The bisphosphonic group is postulated to be an analogue of the terminal pyrophosphate fragment of ATP, and is indispensable for potent inhibitory activity. This mechanism was proven by the inactivity of a structurally similar aminophosphonic acid bearing one phosphonic acid moiety. In the context of these studies, a broad screening of various structures bearing the bisphosphonic moiety against MtGS has been envisaged ().
Figure 3. Scaffolds and range of substituents tested against Mycobacterium tuberculosis GS.
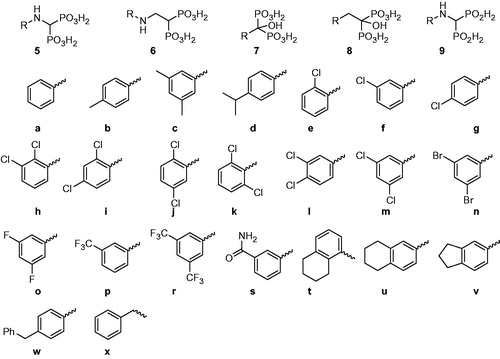
In addition to the aminomethylenebisphosphonic acids (5)Citation26, related structures containing bisphosphonic fragments (6–8) were examined. The position of hydrogen bond donor/acceptor (NH or OH groups) and the linker length (zero to two atoms between the methylenebisphosphonic group and the aromatic fragments) were varied. Preliminary experiments (data not shown) showed bisphosphonates with aliphatic substituents to be much less effective than those bearing aromatic groups. As a consequence, all of the proposed substitution patterns were based on modified phenyl rings (fragments a–y). The aim of the screening was to establish the influence of both the bisphosphonic scaffold and the presence and positioning of substituents on the aromatic rings.
All bisphosphonic compounds were synthesized using standard literature methods (). Briefly, aminomethylenebisphosphonic acids were obtained using Kafarski’s method (tri-component condensation of aromatic amine, diethyl phosphite and triethyl orthoformate and subsequent transesterification followed by hydrolysis)Citation26. The analogous reaction done with ethyl diethoxymethyl-H-phosphinate followed by HBr/AcOH hydrolysis yielded aminomethylenebis-H-phosphinic acids 9Citation24. The application of Hutchinson's method (addition of aromatic amine to tetraethyl ethylidenebisphosphonate and removal of ethyl esters using TMSBr and MeOH) gave compounds 6Citation27. Hydroxybisphosphonic acids 7 and 8 were synthesized by Lecouvey's method (the reaction of acid chlorides with tris(trimethylsilyl)phosphite and subsequent hydrolysis)Citation28.
Figure 4. Synthesis of bisphosphonic acids 5–9.
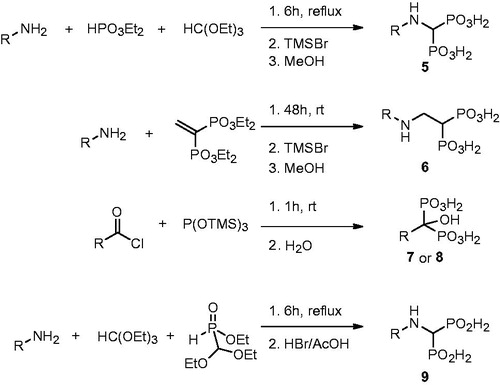
The inhibitory properties of 39 bisphosphonic (5–8) and four bis-H-phosphinic (9) acids were analysed against the unadenylated form of recombinant MtGS using a colorimetric assay based on detection of phosphate released in physiologically relevant biosynthetic reactions (). To obtain unadenylated MtGS, the vector pTrc99c bearing the glnA1 gene of M. tuberculosis (H37Rv strain) was used to transform an E. coli GJ4547 strain lacking adenyl transferase activity, and thus unable to modify Tyr-406 residues of MtGS subunitsCitation9. Enzymatic tests revealed that almost all the bisphosphonates (with the exceptions of 5n and 5r) exhibited inhibitory effects on MtGS with IC50 values in the micromolar range, similar to the reference compound MetSox (1). Comparison of the activities of the unsubstituted phenyl bisphosphonate compounds 5a, 6a, 7a and 8a indicated the aminoethylidenebisphosphonic acid scaffold (compounds 6) to be the most effective, although only slightly so. Moreover, the aminomethylenebis-H-phosphinic acids (9) were found to be completely inactive, indicating that all three oxygen atoms of the phosphonate groups on 5–8 are required for successful inhibition of the enzyme.
Table 1. Inhibitory activities of bisphosphonic and bis-H-phosphinic acids against recombinant MtGS.
In general, substitution of the phenyl ring did not significantly influence the inhibitory activity. Halogen atoms, alkyl groups or even larger substituents like benzyl groups placed in various positions on the aromatic ring yielded similar IC50 values in the majority of cases. However, it was noted that scaffolds with shorter linkers (5 and 7) are slightly more sensitive to the presence of substituents than the more flexible compounds (6 and 8). For example, compound 5r showed no inhibitory activity, while its homologue 6r (IC50 = 168 µM) was equipotent to the unsubstituted derivative 6a. Among all of the tested bisphosphonates, three compounds (6m, 6o and 8l) exhibited IC50 values lower than the reference compound 1.
To gain insight into the mechanism of inhibition, detailed kinetics studies on the most active compounds (6m, 6o and 8l) were undertaken (). Due to the complexity of the possible interactions between the enzyme, the inhibitor and the substrates of the reaction, a clear interpretation of the results is not obvious. The most consistent mechanism that can be proposed based on linear and non-linear regression analyses is that bisphosphonates act like a mixed-type inhibitor with respect to glutamate, and as uncompetitive inhibitors with respect to ATP. This characteristic denotes the ability of these inhibitors to interact with both the free enzyme and the enzyme–glutamate complex at a site other than the glutamate-binding site. Moreover, the inhibitors interact with the enzyme after ATP binding.
Figure 5. Kinetic analysis of MtGS inhibition by compound 6m at varying concentration of glutamine (panel A) and ATP (panel B).
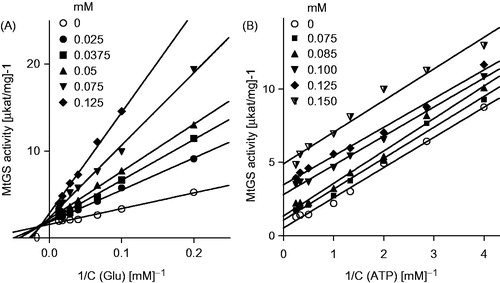
When the tested inhibitors were applied at concentrations higher than 150 μM, GS activity failed to display typical enzyme kinetics, most likely because of non-specific interactions between the inhibitor and metal ions present in the reaction mixture, which reduce the availability of the metal ions for the enzyme. These deviations from the linear models hindered further analysis of the mode of inhibition (Figure S3).
The most active bisphosphonic derivatives were found to be equipotent with the reference GS inhibitor MetSox (1), which was tested under the same conditions (). When assayed against HsGS, selected bisphosphonates were remarkably less active. Such a decrease may be explained by the structural differences between eukaryotic and prokaryotic GSs (which share less than 20% sequence identity), in particular by the differences in their ATP binding architecturesCitation7. Although the shapes of ATP-binding clefts in HsGS and MtGS are analogous, none of amino acid residues forming this site are conserved. Therefore, bisphosphonates substituted with aromatic moiety may serve as potential scaffolds for inhibitors capable of differentiating between bacterial and human GS.
Table 2. Inhibition constants (Ki) against MtGS and IC50 values against HsGS for selected compounds.
Molecular modelling
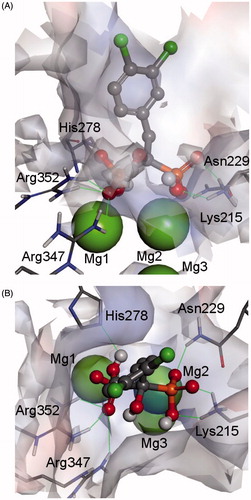
To elucidate the molecular mode of binding of the most active bisphosphonic MtGS inhibitors, the structure of 8l-MtGS was modeled (). The bisphosphonate fragment of the inhibitor is tightly bound by two magnesium ions and a net of hydrogen bonds formed by the side chains of Lys215, Asn229, His278, Arg347 and Arg352. Importantly, all of the phosphonate oxygen atoms are engaged in interactions with the enzyme, a fact that is consistent with the lack of inhibitory activity of the bis-H-phosphinates with two oxygen atoms on phosphorus atom. The aromatic fragment of the inhibitor is located in a cleft formed by Arg364, Ala362, Thr356, Ile355, Pro98, Leu70 and Ala52. The space available in this site does not significantly restrict the sizes and positions of the phenyl ring substituents. This phenomenon also correlates well with the experimental observations showing no direct dependence of inhibitory activity on the phenyl substitution pattern.
Figure 6. Modelled complex of MtGS and hydroxybisphosphonate 8l (front view – panel A and top view – panel B). The inhibitor molecule is shown in ball-and-stick representation, enzyme residues with stick representation, and metal ions in CPK representation. Hydrogen bonds between the ligand and the enzyme are marked by thin green lines. The enzyme surface is colored according to interpolated charge (blue – positive, gray – neutral, red – negative) (color figure can be viewed in the online issue).
Conclusions
In summation, extensive screening of bisphosphonic acids representing varied, albeit similar, structural scaffolds resulted in the discovery of effective MtGS inhibitors with potencies similar to the reference compound L-methionine-S-sulfoximine (1). Importantly, the binding mode of the bisphosphonic inhibitors differs significantly from that of compound 1. While L-methionine-S-sulfoximine is a typical transition state analogue of glutamate, bisphosphonates bind to the active site from the opposite side, partially overlapping the ATP binding site. Because of the low degree of conservation in this portion of enzyme among species, different effects against bacterial and human orthologs were observed. The combination of high efficiencies, selectivities and bone-targeting properties makes these bisphosphonate GS inhibitors an attractive class of compounds in the development of treatments for osseous tuberculosis.
Supplementary material available online
IENZ_1070846_supp.pdf
Download PDF (272.9 KB)Acknowledgements
The authors would like to thank Dr. Jayaraman Gowrishankar, Centre for DNA Fingerprinting and Diagnostics, Hyderabad, India, for the kind gift of GJ4547 E. coli strain and Prof. Sherry L. Mowbray from Uppsala University for recombinant plasmids encoding MtGS and HsGS. The Discovery Studio package was used under a Polish country-wide license. The use of software resources (Discovery Studio program package) of the Wrocław Centre for Networking and Supercomputing is also kindly acknowledged.
Declaration of interest
Ł. B. acknowledges the financial support from the Polish Ministry of Science and Higher Education (grant N N302 159937). The project was supported by Wroclaw Centre of Biotechnology, programme The Leading National Research Centre (KNOW) for years 2014–2018. The authors report no declarations of interest.
Related Research Data
References
- Eisenberg D, Gill HS, Pfluegl GM, Rotstein SH. Structure-function relationships of glutamine synthetases. Biochim Biophys Acta 2000;1477:122–45
- Stadtman ER. The story of glutamine synthetase regulation. J Biol Chem 2001;276:44357–64
- Kumada Y, Benson DR, Hillemann D, et al. Evolution of the glutamine synthetase gene, one of the oldest existing and functioning genes. Proc Natl Acad Sci USA 1993;90:3009–13
- Almassy RJ, Janson CA, Hamlin R, et al. Novel subunit-subunit interactions in the structure of glutamine synthetase. Nature 1986;323:304–9
- Yamashita MM, Almassy RJ, Janson CA, et al. Refined atomic model of glutamine synthetase at 3.5 Å resolution. J Biol Chem 1989;264:17681–90
- Llorca O, Betti M, Gonzalez JM, et al. The three-dimensional structure of an eukaryotic glutamine synthetase: functional implications of its oligomeric structure. J Struct Biol 2006;156:469–79
- Krajewski WW, Collins R, Holmberg-Schiavone L, et al. Crystal structures of mammalian glutamine synthetases illustrate substrate-induced conformational changes and provide opportunities for drug and herbicide design. J Mol Biol 2008;375:217–28
- van Rooyen JM, Abratt VR, Belrhali H, Sewell T. Crystal structure of Type III glutamine synthetase: surprising reversal of the inter-ring interface. Structure 2011;19:471–83
- Krajewski WW, Jones AT, Mowbray SL. Structure of Mycobacterium tuberculosis glutamine synthetase in complex with a transition-state mimic provides functional insights. Proc Natl Acad Sci USA 2005;102:10499–504
- Pesole G, Gissi C, Lanave C, Saccone C. Glutamine synthetase gene evolution in bacteria. Mol Biol Evol 1995;12:189–97
- Berlicki Ł. Inhibitors of glutamine synthetase and their potential application in medicine. Mini-reviews. Med Chem 2008;8:869–78
- Ronzio RA, Meister A. Phosphorylation of methionine sulfoximine by glutamine synthetase. Proc Natl Acad Sci USA 1968;59:164–70
- Berlicki Ł, Kafarski P. Computer-aided analysis of the interactions of glutamine synthetase with its inhibitors. Bioorg Med Chem 2006;14:4578–85
- Obojska A, Berlicki Ł, Kafarski P, et al. Herbicidal pyridyl derivatives of aminomethylene-bisphosphonic acid inhibit plant glutamine synthetase. J Agric Food Chem 2004;52:3337–44
- Occhipinti A, Berlicki Ł, Giberti S, et al. Effectiveness and mode of action of phosphonate inhibitors of plant glutamine synthetase. Pest Manag Sci 2010;66:51–8
- Nilsson MT, Krajewski WW, Yellagunda S, et al. Structural basis for the inhibition of Mycobacterium tuberculosis glutamine synthetase by novel ATP-competitive inhibitors. J Mol Biol 2009;293:504–13
- Gising J, Nilsson MT, Odell LR, et al. Trisubstituted imidazoles as Mycobacterium tuberculosis glutamine synthetase inhibitors. J Med Chem 2012;55:2894–8
- Harth G, Horwitz MA. An inhibitor of exported Mycobacterium tuberculosis glutamine synthetase selectively blocks the growth of pathogenic mycobacteria in axenic culture and in human monocytes: extracellular proteins as potential novel drug targets. J Exp Med 1999;189:1425–36
- Tullius MV, Harth G, Horwitz MA. Glutamine synthetase GlnA1 is essential for growth of Mycobacterium tuberculosis in human THP-1 macrophages and guinea pigs. Infect Immun 2003;71:3927–36
- Harth G, Horwitz MA. Inhibition of Mycobacterium tuberculosis glutamine synthetase as a novel antibiotic strategy against tuberculosis: demonstration of efficacy in vivo. Infect Immun 2003;71:456–64
- Suarez-Garcia I, Noguerado A. Drug treatment of multidrug-resistant osteoarticular tuberculosis: a systematic literature review. Int J Inf Dis 2002;16:e774–8
- Cremers SC, Pillai G, Papapoulos SE. Pharmacokinetics/pharmacodynamics of bisphosphonates: use for optimisation of intermittent therapy for osteoporosis. Clin Pharmacokinet 2005;44:551–70
- Forlani G, Berlicki Ł, Duò M, et al. Synthesis and evaluation of effective inhibitors of plant δ1-pyrroline-5-carboxylate reductase. J Agric Food Chem 2013;61:6792–8
- Bochno M, Berlicki Ł. A three-component synthesis of aminomethylenebis-H-phosphinates. Tetrahedron Lett 2014;55:219–23
- Gross H, Costisella B, Gnauk T, Brenneke L. Derivate der aminomethan-bis-phosphonsäure. J Prakt Chem 1976;318:116–26
- Dąbrowska E, Burzyńska A, Mucha A, et al. Insight into the mechanism of three component condensation leading to aminomethylenebisphosphonates. J Organomet Chem 2009;694:3806–13
- Hutchinson DW, Thornton DM. Michael addition reactions of ethenylidenebisphosphonates. J Organomet Chem 1988;346:341–8
- Lecouvey M, Mallard I, Bailly T, et al. A mild and efficient one-pot synthesis of 1-hydroxymethylene-1,1-bisphosphonic acids. Preparation of new tripod ligands. Tetrahedron Lett 2001;42:8475–8