
Abstract
Improved derivatives of mycophenolic acid (MPA) are necessary to reduce the frequency of adverse effects, this drug exerts in treated patients. In this study, MPA was coupled with N-(ω-hydroxyalkyl)-9-acridone-4-carboxamides or N-(ω-hydroxyalkyl)acridine-4-carboxamides to give respective ester conjugates upon Yamaguchi protocol. This esterification required protection of phenol group in MPA. Designed conjugates revealed higher potency in vitro than parent MPA. Acridine derivatives were more active than acridone analogs and length of the alkyl linker between MPA and heterocyclic units influenced the observed cytotoxicity. Derivatives 2b, 2d, 3a, 3b displayed the most promising immunosuppressive activity.
Introduction
Mycophenolic acid 1 (MPA) () is an uncompetitive inhibitor of inosine-5′-monophosphate dehydrogenase (IMPDH), the crucial enzyme in de novo purine nucleotide biosynthesis. This mechanism allows MPA 1 to decrease proliferation of lymphocytes. Hence, sodium mycophenolate [MPS, Myfortic (Novartis, Basel, Switzerland)] and MPA prodrug: mycophenolate mofetil [2-morpholinoethyl, MMF, CellCept (Roche, Basel, Switzerland)] are widely used in the clinic as immunosuppresantsCitation1–10 in the prevention of allograft rejection and treatment of autoimmune diseases. However, adverse effects related to the treatment with MPA-based drugs, such as diarrhea, leukopenia, sepsis and vomiting, are the barrier to the administration of higher doses and more effective treatment. In order to solve this issue, many structural MPA modifications followed by the assessment of the antiproliferative activity were reportedCitation11–27.
Figure 1. Structures of MPA 1 and its conjugates with acridones 2a–e and acridines 3a–e.
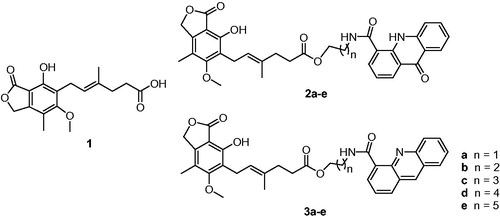
Various types of compounds were considered as potent IMPDH inhibitorsCitation28,Citation29. Among them acridines possess not only anticancer, antiviral and antibacterialCitation30–36, but also antiproliferative and immunosuppressive featuresCitation28,Citation37. Additionally, activity can be improved by conjugate formingCitation38–40. Recently, we reported potent IMPDH inhibitors bearing MPA covalently bonded to nitroacridine/nitroacridone derivatives via amide bond formationCitation41. On the other hand, there are also described promising conjugates possessing ester linkages, i.e. derivatives of muramyldipeptide with acridine/acridone moietiesCitation35 or analogs of MPA with quinic acidCitation38. In current work, we elaborated synthesis of the ester conjugates of MPA and acridones/acridines 2a–e, 3a–e, in which lack of nitro group may diminish toxicity and can help to optimize the biological activity when applied as a drug.
Methods
Chemistry
1H-NMR and 13C-NMR spectra were measured in CDCl3, solutions with Varian Gemini 500 spectrometer (Varian Inc., Palo Alto, CA), with TMS as an internal reference. IR measurements were performed with Bruker IFS66 (Billerica, MA) and UV-VIS with PerkinElmer (Waltham, MA) UV-VIS LAMBDA 18. Mass spectra were recorded with MALDI-TOF spectrometer BRUKER BIFLEX III (DHB or CCA matrix) and HRMS ESI on MaldiSYNAPT G2-S HDMS (monoisotopic masses given). Column chromatography was performed using silica gel 60 (230–400 mesh, Merck, Darmstadt, Germany) and for thin-layer chromatography (TLC) silica gel 60 F254 was used. Solid-phase extraction (SPE) analyses were performed with CHROMABOND Macherey-Nagel columns (Dűren, Germany).
N-(ω-hydroxyalkyl)-9-acridone-4-carboxamides 8a–e, N-(ω-hydroxyalkyl)acridine-4-carboxamides 9a–e were prepared according to the procedure reported in literatureCitation35.
Synthesis of tert-butyldimethylsilyl ether of MPA 6
MPA 1 (2 g, 6.2 mmol), tert-butyldimethylsilyl chloride (5.643 g, 37 mmol), imidazole (3.398 g, 50 mmol) were dissolved in dry DMF (10 mL), and the reaction mixture was stirred at room temperature. After 1 h no starting MPA 1 was observed (TLC). Subsequently, water (30 mL), diethyl ether (60 mL) were added and organic phase was separated, washed with five portions of water (20 mL each), and dried over anhydrous MgSO4. Solids were filtered off and solvent evaporated under reduced pressure.
The crude 4 was dissolved in THF (10 mL), water (10 mL), acetic acid (10 mL) and stirred at room temperature. The reaction mixture was monitored with TLC technique and after 1 h no substrate 4 was observed, and water (30 mL) and diethyl ether (60 mL) were added. Then, organic phase was separated, washed with five portions of water (20 mL each) and dried over anhydrous MgSO4. Subsequently, solid were filtered off, solvent evaporated under reduced pressure. The residue was purified with column chromatography (CH2Cl2:MeOH 50:1 to 30:1 v/v) to give product 6 in yield 77% (2.075 g, 4.8 mmol).
MP 132–133 °C, white solid
1H-NMR (CDCl3): δ[parts per million (ppm)] = 0.24 (s, CH3, 6H), 1.03 (s, CH3, 9H), 1.76 (s, CH3, 3H), 2.15 (s, CH3, 3H), 2.28–2.31 (m, CH2, 2H), 2.40–2.43 (m, CH2, 2H), 3.39 (d, J = 6.3 Hz, CH2, 2H), 3.74 (s, OCH3, 3H), 5.07 (s, CH2, 2H), 5.21 (t, J = 6.3 Hz, CHvinyl, 1H).
13C-NMR (CDCl3): δ(ppm) = −3.3, 11.6, 16.5, 19.0, 23.9, 26.3, 32.9, 34.3, 60.9, 67.9, 111.9, 118.2, 124.1, 127.8, 133.6, 146.3, 152.0, 163.4, 169.5, 179.4.
(Mycophenoyl-N-2-ethyl)-9-acridone-4-carboxamide ester 2a
N-(2-hydroxyethyl)amide of 9-acrydone-4-carboxylic acid 8a (56 mg, 0.2 mmol) was dissolved in dry chloroform (3 mL), then were added tert-butyldimethylsilyl ether of MPA 6 (87 mg, 0.2 mmol), 2,4,6-trichlorobenzoyl chloride (49 mg, 0.2 mmol), triethylamine (41 mg, 0.4 mmol), DMAP (5 mg, 0.04 mmol). The reaction mixture was stirred at room temperature for 24 h and water (5 mL), chloroform (5 mL) were added. Organic phase was separated, dried over anhydrous MgSO4. Next, solids were filtered off and solvent evaporated under reduced pressure. The crude product was purified with column chromatography or SPE (CH2Cl2:MeOH 100:1 to 80:1, 0.1 Et3N v/v) to give tert-butyldimethylsilyl ether of (mycophenoyl-N-2-ethyl)-9-acridone-4-carboxamide ester 10a in yield 58% (84 mg, 0.12 mmol).
MP 82–84 °C, yellow solid
1H-NMR (CDCl3): δ(ppm) = 0.22 (s, CH3, 6H), 1.01 (s, CH3, 9H), 1.75 (s, CH3, 3H), 2.10 (s, CH3, 3H), 2.30–2.33 (m, CH2, 2H), 2.44–2.47 (m, CH2, 2H), 3.36 (d, J = 6.3 Hz, CH2, 2H), 3.68–3.74 (m, NHCH2, OCH3, 5H), 4.31 (t, J = 5.4 Hz, OCH2, 2H), 5.03 (s, OCH2, 2H), 5.18–5.20 (m, CHvinyl, 1H), 7.17–7.22 (m, NHamide, CHacr, 2H), 7.29 (dd, J = 8.3 Hz, J = 7.8 Hz, CHacr, 1H), 7.40 (d, J = 8.3 Hz, CHacr, 1H), 7.66–7.70 (m, CHacr, 1H), 7.90 (d, J = 7.8 Hz, CHacr, 1H), 8.45 (d, J = 8.3 Hz, CHacr, 1H), 8.63 (d, J = 8.3 Hz, CHacr, 1H), 12.3 (s, NHacr, 1H).
Subsequently, compound 10a was dissolved in dry THF (2 mL), treated with tetrabutylammonium fluoride (1 M solution in THF, 100 μL) and stirred at room temperature for 15 min. Then, the reaction mixture was partitioned between water (10 mL) and ethyl acetate (10 mL), and organic layer separated, dried over anhydrous MgSO4. Next, solids were filtered off and solvent evaporated under reduced pressure. The crude product was purified with column chromatography or SPE (CH2Cl2:MeOH 100:1 to 80:1, 0.1 Et3N v/v) to give 61 mg (0.104 mmol) of (mycophenoyl-N-2-ethyl)-9-acridone-4-carboxamide ester 2a (52% according to 6).
MP 84–86 °C, yellow solid
1H-NMR (CDCl3): δ(ppm) = 1.77 (s, CH3, 3H), 2.07 (s, CH3, 3H), 2.32 (t, J = 7.3 Hz, CH2, 2H), 2.46–2.49 (m, CH2, 2H), 3.32 (d, J = 6.8 Hz, CH2, 2H), 3.69 (s, OCH3, 3H), 3.72–3.74 (m, NHCH2, 2H), 4.33–4.35 (m, OCH2, 2H), 5.13 (s, OCH2, 2H), 5.20–5.23 (m, CHvinyl, 1H), 7.15 (dd, J = 7.3 Hz, J = 7.8 Hz, CHacr, 1H), 7.26–7.31 (m, NHamide, CHacr, 2H), 7.41 (d, J = 8.3 Hz, CHacr, 1H), 7.66–7.69 (m, CHacr, 1H), 7.89 (d, J = 7.3 Hz, CHacr, 1H), 8.41 (d, J = 7.8 Hz, CHacr, 1H), 8.57 (d, J = 7.8 Hz, CHacr, 1H), 12.26 (s, NHacr, 1H).
13C-NMR (CDCl3): δ(ppm) = 11.7, 16.4, 22.8, 33.1, 34.7, 40.0, 61.2, 63.1, 70.3, 106.6, 117.0, 117.5, 118.0, 120.0, 121.3, 122.1, 122.4, 122.6, 123.0, 127.0, 132.0, 132.1, 134.2, 140.4, 141.2, 144.4, 153.7, 163.8, 168.8, 173.1, 174.3, 178.1.
IR KBr ν(cm−1): 3323, 2926, 1738, 1649, 1626, 1525, 1072, 1028
λmax (CH2Cl2)/nm 256, 303, 409 (ε/dm3 mol−1 cm1 29 580, 10 597, 7744)
MS (m/z): for C33H32N2O8 calculated: 584.6 found 585.2
HRMS ESI (m/z): [M+Na] for C33H32N2O8Na calculated: 607.2056 found 607.2048
Conjugates 2b–e were obtained in the same way as 2a. Intermediates 10b–e were not isolated.
(Mycophenoyl-N-3-propyl)-9-acridone-4-carboxamide ester 2b, 69 mg, 0.116 mmol, 58% according to 6
MP 79–81.5 °C, yellow solid
1H-NMR (CDCl3): δ(ppm) = 1.81 (s, CH3, 3H), 1.96–1.98 (m, CH2, 2H), 2.08 (s, CH3, 3H), 2.34 (t, J = 7.3 Hz, CH2, 2H), 2.47 (t, J = 7.3 Hz, CH2, 2H), 3.37 (d, J = 6.4 Hz, CH2, 2H), 3.48–3.50 (m, NHCH2, 2H), 3.73 (s, OCH3, 3H), 4.24 (t, J = 5.9 Hz, OCH2, 2H), 5.15 (s, OCH2, 2H), 5.26 (t, J = 6.4 Hz, CHvinyl, 1H), 7.16 (dd, J = 7.3 Hz, J = 7.8 Hz, CHacr, 1H), 7.26–7.31 (m, NHamide, CHacr, 2H), 7.42 (d, J = 8.3 Hz, CHacr, 1H), 7.66–7.69 (m, CHacr, 1H), 7.94 (d, J = 7.3 Hz, CHacr, 1H), 8.42 (d, J = 7.8 Hz, CHacr, 1H), 8.58 (d, J = 7.8 Hz, CHacr, 1H), 12.37 (s, NHacr, 1H).
13C-NMR (CDCl3): δ(ppm) = 11.8, 16.4, 22.9, 28.9, 33.2, 34.8, 36.6, 61.2, 61.9, 70.3, 106.6, 117.0, 117.7, 118.1, 120.0, 121.4, 122.2, 122.4, 122.6, 123.0, 127.1, 131.8, 132.0, 134.2, 140.5, 141.2, 144.4, 153.8, 163.8, 168.6, 173.2, 174.3, 178.1.
IR KBr ν(cm−1): 3304, 2924, 1735, 1649, 1619, 1524, 1073, 1030
λmax (CH2Cl2)/nm 256, 304, 409 (ε/dm3 mol−1 cm1 47 748, 17 266, 12 592)
MS (m/z): for C34H34N2O8 calculated: 598.6 found 599.2
HRMS ESI (m/z): [M+Na] for C34H34N2O8Na calculated: 621.2213 found 621.2209.
(Mycophenoyl-N-4-butyl)-9-acridone-4-carboxamide ester 2c, 74 mg, 0.12 mmol, 60% according to 6
MP 67–68 °C, yellow solid
1H-NMR (CDCl3): δ(ppm) = 1.74 (m, CH2, 4H), 1.78 (s, CH3, 3H), 2.09 (s, CH3, 3H), 2.28–2.31 (m, CH2, 2H), 2.39–2.42 (m, CH2, 2H), 3.34 (d, J = 6.8 Hz, CH2, 2H), 3.54–3.55 (m, NHCH2, 2H), 3.71 (s, OCH3, 3H), 4.08 (m, OCH2, 2H), 5.16 (s, OCH2, 2H), 5.22 (t, J = 6.8 Hz, CHvinyl, 1H), 7.12 (dd, J = 7.3 Hz, J = 7.3 Hz, CHacr, 1H), 7.26–7.29 (m, NHamide, CHacr, 2H), 7.41 (d, J = 8.3 Hz, CHacr, 1H), 7.66 (dd, J = 7.3 Hz, J = 7.8 Hz, CHacr, 1H), 7.99 (d, J = 7.3 Hz, CHacr, 1H), 8.40 (d, J = 7.8 Hz, CHacr, 1H), 8.53 (d, J = 7.8 Hz, CHacr, 1H), 12.38 (s, NHacr, 1H).
13C-NMR (CDCl3): δ(ppm) = 11.8, 16.4, 22.8, 26.2, 26.5, 33.2, 34.8, 39.9, 61.2, 64.0, 70.3, 106.5, 117.0, 117.9, 118.1, 120.0, 121.2, 122.3, 122.4, 122.9, 127.00, 131.7, 132.1, 134.2, 134.3, 140.4, 141.2, 144.3, 153.8, 163.8, 168.8, 173.2, 173.7, 178.0.
IR KBr ν(cm−1): 3323, 2925, 1733, 1649, 1619, 1524, 1074, 1029
λmax (CH2Cl2)/nm 256, 304, 408 (ε/dm3 mol−1 cm1 29 821, 10 892, 8019)
MS (m/z): for C35H36N2O8 calculated: 612.7 found 613.3
HRMS ESI (m/z): [M+Na] for C35H36N2O8Na calculated: 635.2369 found 635.2366.
(Mycophenoyl-N-5-pentyl)-9-acridone-4-carboxamide ester 2d, 60 mg, 0.96 mmol, 48% according to 6
MP 175–177 °C, yellow solid
H-NMR (CDCl3): δ(ppm) = 1.44–1.50 (m, CH2, 2H), 1.68 (t, J = 7.3 Hz, CH2, 2H), 1.71–1.74 (m, CH2, 2H), 1.78 (s, CH3, 3H), 2.19 (s, CH3, 3H), 2.29 (t, J = 7.3 Hz, CH2, 2H), 2.38–2.41 (m, CH2, 2H), 3.36 (d, J = 6.8 Hz, CH2, 2H), 3.51–3.55 (m, NHCH2, 2H), 3.73 (s, OCH3, 3H), 4.05 (d, J = 6.3 Hz, OCH2, 2H), 5.18–5.21 (m, OCH2, CHvinyl, 3H), 6.89 (s, OH, 1H), 7.21 (dd, J = 7.3 Hz, J = 7.3 Hz, CHacr, 1H), 7.30 (dd, J = 7.3 Hz, J = 7.8 Hz, CHacr, 1H), 7.43 (d, J = 8.3 Hz, CHacr, 1H), 7.68–7.71 (m, NHamide, CHacr, 2H), 7.99 (d, J = 7.3 Hz, CHacr, 1H), 8.47 (d, J = 7.8 Hz, CHacr, 1H), 8.63 (d, J = 7.8 Hz, CHacr, 1H), 12.44 (s, NHacr, 1H).
13C-NMR (CDCl3): δ(ppm) = 11.8, 16.3, 22.8, 23.6, 28.6, 29.2, 33.2, 34.8, 40.2, 61.2, 64.1, 70.3, 106.5, 117.0, 117.9, 118.00, 119.9, 121.4, 122.3, 122.4, 122.9, 127.1, 131.7, 132.0, 134.1, 134.3, 140.5, 141.3, 144.3, 153.9, 163.9, 168.8, 173.2, 173.7, 178.1.
IR KBr ν(cm−1): 3330, 2925, 1733, 1650, 1523, 1070, 1027
λmax (CH2Cl2)/nm 256, 303, 409 (ε/dm3 mol−1 cm1 35 225, 12 515, 9215)
MS (m/z): for C36H38N2O8 calculated: 626.7, found 627.3
HRMS ESI (m/z): [M+Na] for C36H38N2O8Na calculated: 649.2526 found 649.2520.
(Mycophenoyl-N-6-hexyl)-9-acridone-4-carboxamide ester 2e, 69 mg, 0.108 mmol, 54%, according to 6
MP 94–96 °C, yellow solid
1H-NMR (CDCl3): δ(ppm) = 1.36–1.49 (m, CH2, 4H), 1.59–1.65 (m, CH2, 2H), 1.68–1.74 (m, CH2, 2H), 1.79 (s, CH3, 3H), 2.11 (s, CH3, 3H), 2.30 (t, J = 7.3 Hz, CH2, 2H), 2.39–2.42 (m, CH2, 2H), 3.36 (d, J = 6.8 Hz, CH2, 2H), 3.51–3.55 (m, NHCH2, 2H), 3.73 (s, OCH3, 3H), 4.03 (d, J = 6.8 Hz, OCH2, 2H), 5.18 (s, OCH2, 2H), 5.21–5.24 (m, CHvinyl, 1H), 6.91 (s, OH, 1H), 7.22 (dd, J = 7.8 Hz, J = 7.8 Hz, CHacr, 1H), 7.31 (dd, J = 7.3 Hz, J = 7.8 Hz, CHacr, 1H), 7.43 (d, J = 8.3 Hz, CHacr, 1H), 7.68–7.72 (m, NHamide, CHacr, 2H), 8.00 (d, J = 6.8 Hz, CHacr, 1H), 8.48 (d, J = 7.8 Hz, CHacr, 1H), 8.66 (d, J = 7.8 Hz, CHacr, 1H), 12.48 (s, NHacr, 1H).
13C-NMR (CDCl3): δ(ppm) = 11.8, 16.4, 22.8, 25.8, 26.8, 28.8, 29.6, 33.2, 34.8, 40.2, 61.2, 64.4, 70.4, 106.5, 117.0, 117.9, 118.1, 119.4, 120.0, 121.2, 122.4, 122.4, 122.9, 127.1, 131.9, 132.0, 134.3, 140.5, 141.2, 144.2, 153.8, 163.9, 168.7, 173.4, 173.8, 178.2.
IR KBr ν(cm−1): 3336, 2936, 1734, 1645, 1619, 1596, 1524, 1077, 1030
λmax (CH2Cl2)/nm 256, 303, 408 (ε/dm3 mol−1 cm−1 38 230, 13 623, 9976)
MS (m/z): for C37H40N2O8 calculated: 640.7 found 641.3
HRMS ESI (m/z): [M+Na] for C37H40N2O8Na calculated: 663.2682 found 663.2676.
Synthesis of tert-butyldiphenylsilyl ether of MPA 7
MPA 1 (2 g, 6.2 mmol), tert-butyldiphenylsilyl chloride (10.23 g, 37 mmol), imidazole (3.398 g, 50 mmol), DMAP (10 mg, 0.08 mmol) were dissolved in dry DMF (10 mL), and the reaction mixture was stirred at room temperature. After 24 h no starting MPA 1 was observed (TLC). Subsequently, water (30 mL) and diethyl ether (60 mL) were added and organic phase was separated, washed with 2 N HCl (20 ml) and dried over anhydrous MgSO4. Solids were filtered off and solvent evaporated under reduced pressure.
The crude 5 was dissolved in THF (10 mL), acetic acid (10 mL), 1 N HCl (10 mL) and stirred at room temperature. The reaction mixture was monitored with TLC technique and after 12 h no substrate 5 was observed, and water (30 mL) and diethyl ether (60 mL) were added. Then, organic phase was separated, washed with five portions of water (20 mL each), and dried over anhydrous MgSO4. Subsequently, solid were filtered off, solvent evaporated under reduced pressure. The residue was purified with column chromatography (CH2Cl2:MeOH 50:1 to 30:1 v/v, white foam during evaporation) to give product 7 in yield 89% (3.083 g, 5.52 mmol).
MP 61–64 °C, white foam
1H-NMR (CDCl3): δ(ppm) = 1.10 (s, CH3, 9H), 1.50 (s, CH3, 3H), 2.12 (s, CH3, 3H), 2.18–2.21 (m, CH2, 2H), 2.32–2.35 (m, CH2, 2H), 3.18 (d, J = 5.9 Hz, CH2, 2H), 3.62 (s, OCH3, 3H), 4.96–4.99 (m, CH2, CHvinyl, 3H), 7.32 (dd, J = 6.8 Hz, J = 7.8 Hz, CHarom, 4H), 7.38 (t, J = 7.3 Hz, CHarom, 2H), 7.70 (J = 6.8 Hz, CHarom, 4H).
13C-NMR (CDCl3): δ(ppm) = 11.6, 16.4, 20.6, 24.5, 26.8, 32.8, 34.2, 60.8, 67.7, 111.4, 118.1, 124.2, 127.4, 127.6, 129.7, 133.5, 133.8, 135.2, 146.2, 151.8, 163.4, 168.6, 179.4.
(Mycophenoyl-N-2-ethyl)-acridine-4-carboxamide ester 3a
N-(2-hydroxyethyl)acridine-4-carboxamide 9a (53 mg, 0.2 mmol) was dissolved in dry chloroform (3 mL), then were added tert-butyldiphenylsilyl ether of MPA 7 (103 mg, 0.2 mmol), 2,4,6-trichlorobenzoyl chloride (49 mg, 0.2 mmol), triethylamine (41 mg, 0.4 mmol), DMAP (5 mg, 0.04 mmol). The reaction mixture was stirred at room temperature for 24 h and water (5 mL), chloroform (5 mL) were added. Organic phase was separated, dried over anhydrous MgSO4. Next, solids were filtered off and solvent evaporated under reduced pressure. The crude product was purified with column chromatography or SPE (CH2Cl2:MeOH 100:1 to 80:1, 0.1 Et3N v/v) to give tert-butyldiphenylsilyl ether of (mycophenoyl-N-2-ethyl)-acridine-4-carboxamide ester 11a in yield 61% (93 mg, 0.12 mmol).
MP 73–75 °C, yellow solid
1H-NMR (CDCl3): δ(ppm) = 1.06 (s, CH3, 9H), 1.42 (s, CH3, 3H), 2.08 (s, CH3, 3H), 2.21–2.24 (m, CH2, 2H), 2.40–2.43 (m, CH2, 2H), 3.09 (d, J = 5.9 Hz, CH2, 2H), 3.57 (s, OCH3, 3H), 3.916–3.925 (m, NHCH2, 2H), 4.41 (t, J = 5.4 Hz, OCH2, 2H), 4.91–4.94 (m, OCH2, CHvinyl, 3H), 7.25–7.28 (m, CHarom, 4H), 7.33 (t, J = 7.3 Hz, CHarom, 2H), 7.58 (dd, J = 7.3 Hz, J = 7.3 Hz, CHacr, 1H), 7.65 (d, J = 6.8 Hz, CHarom, 4H), 7.70–7.73 (m, CHacr, 1H), 7.76 (dd, J = 7.3 Hz, J = 7.8 Hz, CHacr, 1H), 8.05 (d, J = 8.3 Hz, CHacr, 1H), 8.14 (d, J = 8.8 Hz, CHacr, 1H), 8.17 (d, J = 8.3 Hz, CHacr, 1H), 8.93 (m, CHacr, 1H), 9.04 (s, NHamide, 1H), 12.06 (s, CHacr, 1H).
Subsequently, compound 11a was dissolved in dry THF (2 mL), treated with tetrabutylammonium fluoride (1 M solution in THF, 100 μL), and stirred at room temperature for 15 min. Then the reaction mixture was partitioned between water (10 mL) and ethyl acetate (10 mL), and organic layer separated, dried over anhydrous MgSO4. Next, solids were filtered off and solvent evaporated under reduced pressure. The crude product was purified with column chromatography or SPE (CH2Cl2:MeOH 100:1 to 80:1, 0.1 Et3N v/v) to give 58 mg (0.102 mmol) of (mycophenoyl-N-2-ethyl)-acridine-4-carboxamide ester 3a (51% according to 7).
MP 136–138 °C, yellow solid
1H-NMR (CDCl3): δ(ppm) = 1.74 (s, CH3, 3H), 2.08 (s, CH3, 3H), 2.33–2.36 (m, CH2, 2H), 2.50–2.54 (m, CH2, 2H), 3.33 (d, J = 6.8 Hz, CH2, 2H), 3.70 (s, OCH3, 3H), 3.93–3.94 (m, NHCH2, 2H), 4.41 (t, J = 5.4 Hz, OCH2, 2H), 5.13 (s, OCH2, 2H), 5.20–5.22 (m, CHvinyl, 1H), 7.58–7.61 (m, OH, CHacr, 2H), 7.66 (dd, J = 7.3 Hz, J = 7.3 Hz, CHacr, 1H), 7.80 (dd, J = 7.3 Hz, J = 7.3 Hz, CHacr, 1H), 8.04 (d, J = 8.3 Hz, CHacr, 1H), 8.14–8.17 (m, CHacr, 2H), 8.90 (m, CHacr, 1H), 8.98 (s, NHamide, 1H), 12.07 (s, CHacr, 1H).
13C-NMR (CDCl3): δ(ppm) = 11.8, 16.4, 22.8, 33.3, 34.8, 39.0, 61.2, 63.8, 70.2, 106.5, 116.9, 122.3, 122.9, 125.6, 125.9, 126.2, 126.7, 128.5, 128.7, 128.8, 131.8, 132.8, 134.3, 135.9, 138.3, 144.2, 146.0, 147.2, 153.8, 163.9, 166.4, 173.1, 173.5.
λmax (CH2Cl2)/nm 250, 305, 342, 359, 377 (ε/dm3 mol−1 cm1 92 249, 5376, 6653, 11 172, 5669).
IR KBr ν(cm−1): 3405, 2922, 1736, 1655, 1625, 1565, 1552, 1524, 1164, 1138, 738.
MS (m/z): [M]+ for C33H32N2O7 calculated: 568.6, found: 569.0
HRMS ESI (m/z): [M+Na]+ for C33H32N2O7Na calculated: 591.2107 found 591.2111.
Conjugates 3b–e were obtained in the same way as 3a. Intermediates 11b–e were not isolated.
(Mycophenoyl-N-3-propyl)-acridine-4-carboxamide ester 3b, 48 mg, 0.082 mmol, 41% according to 7
MP 48–50 °C, yellow solid
1H-NMR (CDCl3): δ(ppm) = 1.66 (s, CH3, 3H), 1.99 (s, CH3, 3H), 1.93–2.18 (m, CH2, 4H), 2.29–2.36 (m, CH2, 2H), 3.24 (d, J = 6.4 Hz, CH2, 2H), 3.61 (s, OCH3, 3H), 3.70–3.94 (m, NHCH2, 2H), 4.17–4.23 (m, OCH2, 2H), 5.02–5.05 (m, CHvinyl, 1H), 5.19 (s, OCH2, 2H), 7.66–7.79 (m, OH, CHacr, 3H), 7.95 (dd, J = 6.8 Hz, J = 7.9 Hz, CHacr, 1H), 8.20–8.39 (m, CHacr, 3H), 8.72 (d, J = 6.8 Hz, CHacr, 1H), 9.32–9.36 (m, NHamide, 1H), 11.35 (s, CHacr, 1H).
13C NMR (CDCl3): δ(ppm) = 11.2, 16.1, 22.6, 28.7, 32.5, 34.3, 36.4, 60.8, 62.3, 68.9, 107.2, 116.2, 122.5, 123.2, 125.6, 125.9, 126.7, 126.9, 128.4, 128.7, 128.8, 132.3, 133.2, 133.4, 134.8, 139.2, 145.6, 146.0, 147.2, 153.0, 162.8, 165.1, 170.4, 172.9.
IR KBr ν(cm−1): 3421, 2928, 1731, 1654, 1623, 1562, 1522, 1162, 1132, 741.
λmax (CH2Cl2)/nm 251, 305, 342, 359, 377 (ε/dm3 mol−1 cm1 85 875, 4692, 5749, 9995, 5096)
MS (m/z): [M]+ for C34H34N2O7: calculated: 582.6, found: 583.1
HRMS ESI (m/z): [M]+ for C34H35N2O7 calculated: 583.2444 found 583.2445
(Mycophenoyl-N-4-butyl)-acridine-4-carboxamide ester 3c, 72 mg, 0.12 mmol, 62% according to 7
MP 51–53 °C, yellow solid
1H-NMR (CDCl3): δ(ppm) = 1.67 (s, CH3, 3H), 1.72–1.79 (m, CH2, 4H), 1.99 (s, CH3, 3H), 2.16 (t, J = 7.3 Hz, CH2, 2H), 2.34 (t, J = 7.3 Hz, CH2, 2H), 3.22 (d, J = 6.8 Hz, CH2, 2H), 3.52–3.55 (m, NHCH2, 2H), 3.60 (s, OCH3, 3H), 4.04–4.06 (m, OCH2, 2H), 5.07–5.10 (m, CHvinyl, 1H), 5.18 (s, OCH2, 2H), 7.66 (dd, J = 7.3 Hz, J = 7.8 Hz, CHacr, 1H), 7.72 (dd, J = 7.3 Hz, J = 7.8 Hz, CHacr, 1H), 7.89–7.93 (m, CHacr, 1H), 8.19–8.21 (m, OH, CHacr, 2H), 8.34 (d, J = 7.3 Hz, CHacr, 1H), 8.69–8.70 (m, CHacr, 1H), 9.27 (s, CHacr, 1H), 9.29 (s, NHamide, 1H), 11.36–11.38 (m, CHacr, 1H).
13C NMR (CDCl3): δ(ppm) = 11.7, 16.5, 23.0, 26.4, 26.7, 33.1, 34.8, 39.4, 61.2, 64.2, 69.3, 107.6, 116.6, 122.9, 123.7, 126.0, 126.3, 127.2, 127.2, 129.1, 129.2, 129.2, 132.6, 133.5, 133.9, 135.0, 139.2, 146.0, 146.8, 147.3, 153.8, 163.8, 165.2, 170.2, 173.9.
IR KBr ν(cm−1): 3418, 2923, 1732, 1652, 1624, 1562, 1522, 1161, 1132, 740.
λmax (CH2Cl2)/nm 251, 305, 342, 359, 377 (ε/dm3 mol−1 cm1 58 658, 3378, 3897, 6723, 3371)
MS (m/z): [M]+ for C35H36N2O7: calculated: 596.7 found: 597.0
HRMS ESI (m/z): [M+Na]+ for C35H36N2O7Na calculated: 619.2420 found 619.2418.
(Mycophenoyl-N-5-pentyl)-acridine-4-carboxamide ester 3d, 72 mg, 0.118 mmol, 59% according to 7
MP 54–56 °C, yellow solid
1H-NMR (CDCl3): δ(ppm) = 1.50–1.72 (m, CH2, 6H), 1.60 (s, CH3, 3H), 2.01 (s, CH3, 3H), 2.04–2.07 (m, CH2, 2H), 2.19–2.22 (m, CH2, 2H), 3.21 (d, J = 6.8 Hz, CH2, 2H), 3.51–3.56 (m, NHCH2, 2H), 3.60 (s, OCH3, 3H), 3.96 (t, J = 6.3 Hz, OCH2, 2H), 5.00–5.02 (m, CHvinyl, 1H), 5.20 (s, OCH2, 2H), 7.67 (dd, J = 6.8 Hz, J = 7.8 Hz, CHacr, 1H), 7.73 (dd, J = 6.8 Hz, J = 7.8 Hz, CHacr, 1H), 7.91–7.94 (m, CHacr, 1H), 8.19–8.20 (m, OH, CHacr, 2H), 8.34 (d, J = 8.3 Hz, CHacr, 1H), 8.71 (d, J = 6.8 Hz, CHacr, 1H), 9.25–9.30 (m, CHacr, 1H), 9.33 (s, NHamide, 1H), 11.43 (s, CHacr, 1H).
13C NMR (CDCl3): δ(ppm) = 11.7, 16.5, 23.0, 24.0, 28.6, 29.4, 33.0, 34.7, 61.2, 64.4, 64.6, 69.3, 107.6, 116.6, 123.0, 123.6, 126.0, 126.3, 127.2, 127.2, 129.0, 129.1, 129.2, 132.7, 133.5, 133.8, 135.3, 139.4, 146.2, 146.4, 147.7, 153.4, 163.2, 165.3, 170.8, 173.1.
IR KBr ν(cm−1): 3421, 2928, 1732, 1653, 1624, 1563, 1523, 1162, 1133, 741.
λmax (CH2Cl2)/nm 251, 305, 342, 359, 377 (ε / dm3 mol−1 cm1 68 208, 4887, 5394, 9045, 4646)
MS (m/z): [M]+ for C36H38N2O7 calculated: 610.7, found: 611.1
HRMS ESI (m/z): [M]+ for C36H39N2O7 calculated: 611.2757 found 611.2750.
(Mycophenoyl-N-6-hexyl)-acridine-4-carboxamide ester 3e, 73 mg, 0.118 mmol, 59%, according to 7
MP 57–59 °C, yellow solid
1H-NMR (CDCl3): δ(ppm) = 1.37–1.62 (m, CH2, 8H), 1.68 (s, CH3, 3H), 2.00 (s, CH3, 3H), 2.13 (t, J = 7.3 Hz, CH2, 2H), 2.29 (t, J = 7.3 Hz, CH2, 2H), 3.22 (d, J = 6.8 Hz, CH2, 2H), 3.52–3.54 (m, NHCH2, 2H), 3.61 (s, OCH3, 3H), 3.92 (t, J = 6.3 Hz, OCH2, 2H), 5.07–5.09 (m, CHvinyl, 1H), 5.19 (s, OCH2, 2H), 7.67–7.75 (m, CHacr, 2H), 7.92–7.95 (m, CHacr, 1H), 8.18–8.22 (m, OH, CHacr, 2H), 8.35 (d, J = 7.8 Hz, CHacr, 1H), 8.71 (d, J = 7.3 Hz, CHacr, 1H), 9.302–9.307 (m, CHacr, NHamide, 2H), 11.42 (s, CHacr, 1H).
13C NMR (CDCl3): δ(ppm) = 11.7, 16.5, 23.0, 25.9, 27.1, 29.7, 33.0, 34.8, 61.2, 64.4, 64.5, 69.3, 107.6, 116.6, 119.8, 123.0, 123.7, 126.0, 126.3, 127.2, 127.2, 129.0, 129.3, 132.7, 133.5, 133.9, 135.3, 139.4, 146.2, 146.4, 147.7, 153.4, 163.2, 165.3, 170.8, 173.1.
IR KBr ν(cm−1): 3422, 2925, 1732, 1651, 1624, 1564, 1523, 1162, 1134, 741.
λmax (CH2Cl2)/nm 251, 305, 342, 359, 377 (ε/dm3 mol−1 cm1 10 3670, 6576, 7236, 12 002, 6230)
MS (m/z): [M]+ for C37H40N2O7 calculated: 624.7, found: 625.0
HRMS ESI (m/z): [M]+ for C37H41N2O7 calculated: 625.2914 found 625.2906.
Biological activity evaluation
The following cells were used: Jurkat cell line (ATCC collection) or PBMC obtained from buffy coats of healthy volunteers by standard ficoll gradient centrifugation. The cells were disposed 5 × 104 cells per well in RPMI medium containing 10% fetal calf serum in an atmosphere of 5% CO2 at 37 °C in triplicates with the following final concentration of the examined conjugates: 0 (control), 0.0001, 0.001, 0.01 and 0.1 mg/ml.
Viability tests IC50
The readout of the test was performed with colorimetric MTT assay. After 24 h of the incubation, (3–4.5 dimethylthiazol-2-yl)-2.5 diphenyl-tetrazolium bromide (MTT) was added to the wells in the final concentration of 1 mg/ml and the plates were incubated for additional 4 h. Then, the reaction was stopped by addition of 100 µl of isopropanol. Optical density was read at 570 nm on the automated plate reader (Victor 4, PerkinElmer, Waltham, MA).
Proliferation tests EC50
The readout of the test was performed with tritium assay. The proliferative ability of the cells was measured in response to CD3/CD28 Dynabeads (Dynal, Dynabeads®, Life Technologies, Carlsbad, CA) in 1:1 ratio cultured for 4 days. Proliferation was measured after 3H-thymidine (0.5 μCi/well) incubation for the last 16 h [Liquid Scintillation Counter (LSC) reader, PerkinElmer, Waltham, MA] as counts per minute units.
The results from both tests were then adjusted with SigmaPlot Software (Systat Software Inc., San Jose, CA) in order to obtain IC50 or EC50 values for each compound.
Results
Chemistry
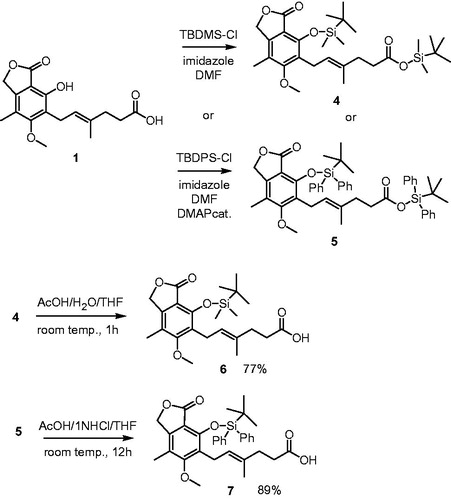
Direct condensation of MPA 1 with N-(ω-hydroxyalkyl)-9-acridone-4-carboxamides 8a–e (35) or N-(ω-hydroxyalkyl)acridine-4-carboxamides 9a–e (35) occurred to be laborious, probably due to participation of free phenolic group in undesired esterifications or macrolactonization. As a result, isolation of pure compounds 2a–e and 3a–e was difficult. Therefore, the preparation of MPA bearing blocked phenolic group was performed similarly to procedures reported in literature (14,26) (). Thus, MPA 1 was treated with excess of tert-butyldimethylsilyl chloride and both phenol and carboxylic groups underwent silylation to 4. Subsequently, tert-butyldimethylsilyl ester was hydrolyzed in the presence of tert-butyldimethylsilyl ether to MPA derivative 6 bearing free carboxylic group, but protected phenolic group.
Scheme 1. Synthesis of tert-butyldimethylsilyl ether of MPA 6 and tert-butyldiphenylsilyl ether of MPA 7.
We tested several methods for esterification including commercially available coupling agents and the best results were achieved in case of Yamaguchi protocolCitation42,Citation43 (). The reaction of blocked MPA 6 with N-(ω-hydroxyalkyl)-9-acridone-4-carboxamides 8a–e led to esters 10a–e, followed by deprotection of phenolic group by tetrabutylammonium fluoride treatment to conjugates 2a–e in moderate yields (48–60%, according to 6).
Scheme 2. Synthesis of conjugates of MPA with acridones 2a–e and conjugates of MPA with acridones 3a–e.
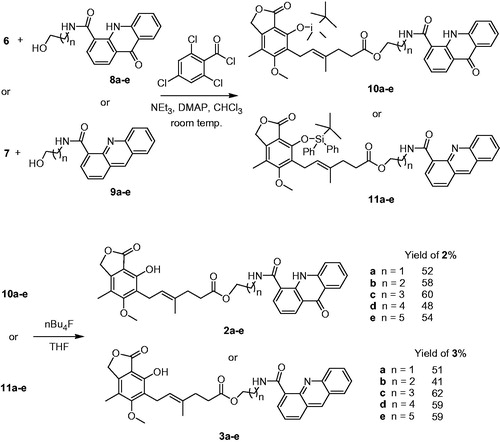
Similarly to this synthetic pathway, MPA derivative 6 was treated with N-(ω-hydroxyalkyl)acridine-4-carboxamides 9a–e towards conjugates 3a–e. However, we observed unexpected recovering of phenolic group during this esterification. The unprompted removal of tert-butyldimethylsilyl ether caused contamination of product 3, which was difficult to purify. It suggested clearly, that nitrogen atom in acridine ring participated in this undesired deprotection.
It was widely reported, that stability of silyl ethers depends strongly on substituents at silicon atomCitation44. Particularly, bulky alkyl or aryl groups increase resistance towards basic reagents and this correlation is a one possible basis in selective recovering of hydroxyl groups blocked with silyl ethers. For instance, tert-butyldimethylsilyl ether can be cleaved under basic conditions in the presence of tert-butyldiphenylsilyl ether, which is more stable due to steric reasonsCitation44.
Then we decided to obtain MPA bearing protected phenolic group as more stable tert-butyldiphenylsilyl ether 7 (). The synthetic pathway was similar to 6, but silylation of MPA 1 to 5 with tert-butyldiphenylsilyl required catalytic amount of DMAP, and hydrolysis of tert-butyldipfenylsilyl ester 5 to 7 needed more acidic conditions. Noteworthy, we observed no interrupting deprotection in the course of this esterification and conjugates 3a–e were obtained in moderate yields (41–62%, according to 7).
Biological results
Antiproliferative activity of the obtained compounds was measured with Jurkat cell line and PBMC (peripheral blood mononuclear cells from healthy donors). Data collected in present EC50 of conjugates 2a–e, 3a–e. These results were compared to starting acridones 8a–e, acridines 9a–e respectively, and MPA 1. Acridine conjugates 3a–e occurred to suppress proliferation more than acridone conjugates 2a–e, starting acridines 9a–e, and MPA 1 towards Jurkat cell line. In case of experiments with PBMC, both type of conjugates 2a–e and 3a–e revealed comparable suppressive activity, apart from 2b which gave EC50 better than 3b.
Table 1. EC50 [µM] values of conjugates 2a–e, 3, a–e, N-(ω-hydroxyalkyl)-9-acridone-4-carboxamides 8a–e, N-(ω-hydroxyalkyl)acridine-4-carboxamides 9a–e, 1 for cell line Jurkat and activated PBMC, obtained in antiproliferation test, p statistical significance.
Subsequently, toxicity measured as IC50 of compounds 2a–e, 3a–e were investigated. These results are presented in and show, that acridine conjugates 3a–e were more cytotoxic with Jurkat cell line than acridone conjugates 2a–e and starting acridines 9a–e, but less toxic if compared to MPA 1. Conjugates 2a–e, 3a–e gave slightly higher cytotoxicity than MPA 1 towards PBMC, whereas acridine derivatives 3b–e were mostly more toxic than acridone analogs 2b–e, apart from compounds 2a and 3a.
Table 2. IC50 [µM] values of conjugates 2a–e, 3, a–e, N-(ω-hydroxyalkyl)-9-acridone-4-carboxamides 8a–e, N-(ω-hydroxyalkyl)acridine-4-carboxamides 9a–e, 1 for cell line Jurkat and activated PBMC, obtained in MTT test, p statistical significance.
Discussion
The question arose on the influence of covalent bond between MPA and acridone/acridine moieties on observed antiproliferative activity.
Previously reported amide conjugates of MPA with nitroacridine/nitroacridone derivatives gave activities lower or comparable than their structural parts tested separately, but still better than parent MPACitation41. In other words, the observed effect was rather additive, not synergistic.
In contrast to that, EC50 values of the most ester conjugates (2b, d, e, 3a–e) was higher than EC50 of mixture of MPA 1 and relevant acridone 8b, d, e or acridine 9a–e (). These results strongly suggest, that covalent bond between both structural units is important for biological activity of conjugates 2a–e, 3a–e. Noteworthy, mixtures of MPA with 8a–e, 9d gave intermediate EC50 values, whereas mixtures of MPA with 9a–c, e exhibited lower activity than both separate components (), which work with another mechanism of action and observed activities were not always additive.
Table 3. Comparison of EC50 [µM] values of conjugates 2a–e, 3, a–e, with mixtures of MPA and respective N-(ω-hydroxyalkyl)-9-acridone-4-carboxamides 8a–e, N-(ω-hydroxyalkyl)acridine-4-carboxamides 9a–e, 1 for cell line Jurkat, p statistical significance.
In order to estimate efficacy of conjugates 2a–e, 3, a–e as potential drugs, we calculated their selective index (SI). These values are provided in . MPA 1 gave SI value of 1.4 against Jurkat cell line and >780 against PBMC. The acridone derivatives 2b (n = 3) and 2d (n = 5) revealed the most interesting results from among conjugates 2a–e. Compound 2b exhibited SI value of 7.1 against Jurkat cell line and >8355 against PBMC. Similarly, analog 2d displayed SI value of 12 against Jurkat cell line and 7980 against PBMC.
Table 4. Selectivity index (IC50/EC50) calculated for conjugates 2a–e, 3, a–e.
In the series of acridine conjugates 3a–e a short linker occurred to be the most promising. Derivative 3a (n = 1) provided SI value of 17 against Jurkat cell line and 3518 against PBMC. The highest value of SI against Jurkat cell line (86, ) was observed in the case of compound 3b (n = 2).
In the preliminary experiment we investigated mechanism of action of compounds 2a–e, 3a–e. It was reported in literature, that the addition of guanosine monophosphate (GMP), which is the main product of IMPDH activity, reverses suppression of cell proliferation in the presence of this enzyme inhibitorCitation1,Citation12. In the similar test done in the presence of conjugates 2a–e, 3a–e we observed, that the addition of 50 μM of GMP increased cells proliferation (results provided in Supplementary Figures 1 and 2). These results suggest that all compounds 2a–e, 3a–e worked as IMPDH inhibitors.
Conclusion
In summary, synthesis of novel MPA and acridone/acridine conjugates via Yamaguchi esterification was elaborated. For this purpose, synthesis of MPA bearing free carboxylic group and blocked phenol was optimized. The coupling of MPA with N-(ω-hydroxyalkyl)acridine-4-carboxamides 9a–e required conversion of phenol group in MPA to bulky tert-butyldiphenylsilyl ether. Obtained conjugates 2a–e, 3a–e are IMPDH inhibitors and indicated interesting antiproliferative activity in vitro in comparison to parent MPA 1. In most cases, acridine derivatives 3a–e were more active than acridone analogs 2a–e. Length of linker between MPA and heterocyclic units also influenced the observed activity. The most promising compounds 2b, 2d, 3a, 3b are considered to farther examinations as potential immunosuppressive agents.
Supplementary Material available online
Supplementary Figures 1 and 2
IENZ_A_1077821_SUPP.pdf
Download PDF (209.6 KB)Acknowledgements
Authors would like to thank Dr W.J. Watkins, Gilead Sciences Inc., Foster City, CA for MPA sample.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
This work has been supported by The National Centre for Research and Development (Poland, grant no. LIDER/07/58/L-2/10/NCBiR/2011) and Medical University of Gdansk (grant no. ST49).
Related Research Data
References
- Bentley R. Mycophenolic acid: a one hundred year odyssey from antibiotic to immunosuppressant. Chem Rev 2000;100:3801–25
- Cholewinski G, Malachowska-Ugarte M, Dzierzbicka K. The chemistry of mycophenolic acid – synthesis and modifications towards desired biological activity. Curr Med Chem 2010;17:1926–41
- Papageorgiou, C. Enterohepatic recirculation: a powerful incentive for drug discovery in the inosine monophosphate dehydrogenase field. Mini-Rev Med Chem 2001;1:71–7
- Hedstrom L. IMP dehydrogenase: structure, mechanism and inhibition. Chem Rev 2009;109:2903–28
- Sintchak MD, Nimmesgern E. The structure of inosine 5′-monophosphate dehydrogenase and the design of novel inhibitors. Immunopharmocol 2000;47:163–84
- Kaufmann DB, Shapiro R, Lucey MR, et al. Immunosuppression: practice and trends. Am J Transplant 2004;4:38–53
- Yang N, Wang J, Wang Z-W, et al. Computional insights into the inhibition of inosine 5′-monophosphate dehydrogenase by mycophenolic acid analogs: three dimensional quantitative structure-activity relationship and molecular docking studies. Chem Biol Drug Des 2012;79:1063–71
- Kamar N, Oufroukhi L, Faure P, et al. Questionnaire-based evaluation of gastrointestinal disorders in de novo renal-transplant patients receiving either mycophenolate mofetil or enteric-coated mycophenolate sodium. Nephrol Dial Transplant 2005;20:2231–6
- Aptaramanov B, Seyahi N, Alagoz S, et al. A comparison of mycophenolate mofetil with mycophenolate sodium in renal transplant recipients on tacrolimus-based treatment. Transplant Proc 2011;43:833–6
- Silva HT Jr, Yang HC, Abouljoud M, et al. One-year results with extended-release tacrolimus/MMF, tacrolimus/MMF and cyclosporine/MMF in de novo kidney transplant recipients. Am J Transplant 2007;7:595–608
- Felczak K, Chen L, Wilson D, et al. Cofactor-type inhibitors of inosine monophosphate dehydrogenase via modular approach: targeting the pyrophosphate binding sub-domain. Bioorg Med Chem 2011;19:1594–605
- Mitsuhashi S, Takenaka J, Iwamori K, et al. Structure-activity relationships for inhibition of inosine monophosphate dehydrogenase and differentiation induction of K562 cells among the mycophenolic acid derivatives. Bioorg Med Chem Lett 2010;18:8106–11
- Habib E, León F, Bauer JD, et al. Mycophenolic derivatives from Eupenicillium parvum. J Nat Prod 2008;71:1915–18
- Nelson PH, Carr SF, Devens BH, et al. Structure-activity relationships for inhibition of inosine monophosphatedehydrogenase by nuclear variants of mycophenolic acid. J Med Chem 1996;39:4181–96
- Chen Z, Zheng Z, Huang H, et al. Penicacids A–C, three new mycophenolic acid derivatives and immunosuppressive activities from the marine-derived fungus Penicillium sp. SOF07. Bioorg Med Chem Lett 2012;22:3332–5
- Iwaszkiewicz-Grzes D, Cholewinski G, Kot-Wasik A, et al. Synthesis and biological activity of mycophenolic acid-amino acid derivatives. Eur J Med Chem 2013;69:863–71
- Malachowska-Ugarte M, Cholewinski G, Chojnacki J, Dzierzbicka K. 4-[(tert-Butyldimethylsilyl)oxy]-6-methoxy-7-methyl-5-(oxiran-2-ylmethyl)-2-benzofuran-3(1H)-one. Acta Crystallogr E 2011;67:o3393
- Watkins WJ, Chen JM, Cho A, et al. Phosphonic acid-containing analogues of mycophenolic acid as inhibitors of IMPDH. Bioorg Med Chem Lett 2006;16:3479–83
- Fardis M, Mertzman M, Thomas W, et al. Use of benzofuran for concomitant protection of aldehyde and phenol groups in the preparation of mycophenolic acid analogues. J Org Chem 2006;71:4835–9
- Chen L, Wilson D, Jayaram HN, Pankiewicz KW. Dual inhibitors of IMP-dehydrogenase and histone deacetylases for cancer treatment. J Med Chem 2007;50:6685–91
- Rohloff JC, Gardner JO, Towne RW. Mycophenolate dianions. Tetrahedron Lett 1995;43:7803–6
- Smith DB, Waltos AM, Loughhead DG, et al. Asymmetric synthesis and stereochemical assignment of RS-97613, a potent immunosuppressive and antiinflammatory agent. J Org Chem 1996;61:2236–41
- Jones DF, Moore RH, Crawley GC. Microbial modifications of mycophenolic acid. J Chem Soc 1970;1725–1737
- Jones DF, Mills SD. Preparation and antitumor properties of analogs and derivatives of mycophenolic acid. J Med Chem 1971;14:305–11
- Suzuki S, Takaku S, Mori T. Antitumor activity of derivatives of mycophenolic acid. J Antibiotics 1976;3:275–85
- Sunohara K, Mitsuhashi S, Shigetomi K, Ubukata M. Discovery of N-(2,3,5-triazoyl)mycophenolic amide and mycophenolic epoxyketone as novel inhibitors of human IMPDH. Bioorg Med Chem Lett 2013;23:5140–4
- Felczak K, Vince R, Pankiewicz KW. NAD-based inhibitors with anticancer potential. Bioorg Med Chem Lett 2014;24:332–6
- Petrelli R, Vita P, Torquati I, et al. Novel inhibitors of inosine monophospate dehydrogenase in patent literature of the last decade. Recent Pat Anticancer Drug Discov 2013;8:103–25
- Cholewinski G, Iwaszkiewicz Grzes D, Prejs M, et al. Synthesis of the inosine 5′-monophosphate dehydrogenase (IMPDH) inhibitors. J Enzyme Inhib Med Chem 2015;30:550–63
- Cholewinski G, Dzierzbicka K, Kolodziejczyk AM. Natural and synthetic acridines/acridones as antitumor agents: their biological activities and methods of synthesis. Pharmacol Rep 2011;63:305–36
- Denny WA. Acridine derivatives as chemotherapeutic agents. Curr Med Chem 2002;9:1655–65
- Dzierzbicka K, Gozdowska, M, Wysocka-Skrzela B, Kołodziejczyk AM. Synthesis and anticancer activites of new MDP conjugates with amino-acridine/acridone derivatives. Drug Res 1999;56:20–4
- Dzierzbicka K, Kołodziejczyk AM, Wysocka-Skrzela B, Kołodziejczyk AS. Synthesis of muramyl dipeptide conjugated with acridine derivatives, showing anti-HIV-1 and anticancer activity. Polish J Chem 1994;68:37–45
- Dzierzbicka K, Kołodziejczyk AM, Wysocka-Skrzela B, et al. Synthesis and antitumor activity of conjugates of muramyldipeptides, normuramyldipeptide, and desmuramylpeptides with acridine/acridone derivatives. J Med Chem 2001;44:3606–15
- Dzierzbicka K, Kołodziejczyk AM. Synthesis and antitumor activity of conjugates of muramyldipeptide or normuramyldipeptide with hydroxyacridine/acridone derivatives. J Med Chem 2003:46:183–9
- Kukowska-Kaszuba M, Dzierzbicka K, Serocki M, Skladanowski A. Solid phase synthesis and biological activity of tuftsin conjugates. J Med Chem 2011;54:2447–54
- Hess FK, Steward PB. Preparation of a new immunosuppressant 4,5-bis(aminomethyl)acridine. J Med Chem 1975;18:320–1
- Wu H, Pagadala J, Yates ChR, et al. Synthesis and characterization of an anti-apoptotic immunosuppressive compound for improving the outcome of islet transplantation. Bioconjugate Chem 2013;24:2036–44
- Dzierzbicka K, Kolodziejczyk AM. Adjuvants-essential components of new generation vaccines. Post Bioch 2006;52:204–11
- Wardowska A, Dzierzbicka K, Mysliwski A. Tuftsin-new analogues and properties. Post Bioch 2007;53:60–5
- Malachowska-Ugarte M, Cholewinski G, Dzierzbicka K, Trzonkowski P. Synthesis and biological activity of novel mycophenolic acid conjugates containing nitro-acridine/acridone derivatives. Eur J Med Chem 2012;54:197–201
- Inanaga J, Hirata K, Saeki H, et al. A rapid esterification by means of mixed anhydride and its application to large-ring lactonization. Bull Chem Soc Jpn 1979;52:1989–93
- Dhimitruka I, SantaLucia J Jr. Investigation of the Yamaguchi esterification mechanism. Synthesis of the Lux-S enzyme inhibitor using an improved esterification method. Org Lett 2006;8:47–50
- Crouch RD. Selective monodeprotection of bis-silyl ethers. Tetrahedron 2004;60:5833–71