
Abstract
New secondary benzenesulphonamide-substituted coumarylthiazole derivatives were synthesized and their inhibitory effects on purified carbonic anhydrase I and II were evaluated using CO2 as a substrate. The result showed that all the synthesized compounds exhibited inhibitory activity on both hCA I and hCA II with N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)naphthalene-2-sulphonamide (5f, IC50 value of 5.63 and 8.48 µM, against hCA I and hCA II, respectively) as the strongest inhibitor revealed from this study. Structure–activity relationship revealed that the inhibitory activity of the synthesized compounds is related to the type of the halogen and bulky substituent on the phenyl ring. In addition, the cupric reducing antioxidant capacities (CUPRAC) and ABTS cation radical scavenging abilities of the synthesized compounds were assayed. 4-methoxy-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)benzenesulphonamide (5e) exhibited the strongest ABTS and CUPRAC activity with IC50 value of 48.83 µM and A0.50 value of 23.29 µM, respectively.
Introduction
The carbonic anhydrases (CAs; EC 4.2.1.1) are a superfamily of metalloenzymes, which catalyse the interconversion between CO2 and HCO3− by using a metal hydroxide nucleophilic mechanismCitation1–4. These metalloenzymes are known in nature as five different, genetically distinct families, the α-, β-, γ-, δ- and ζ-CAsCitation5–7. Additionally, a new genetic family of CAs, which was called the η-CA class, was discovered by Supuran's group in the last yearCitation8. The α-, β-, δ-CAs use Zn(II) ions at the active site, the γ-CAs are Fe(II) enzymes [but they are active also with bound Zn(II) or Co(II) ions], while the ζ-class uses Cd(II) or Zn(II) to perform the physiologic reaction catalysisCitation5–7. Sixteen different α-CA isoforms were isolated in mammals, where they play crucial physiological roles. Some of them are cytosolic (CA I, CA II, CA III, CA VII, CA XIII), others are membrane-bound (CA IV, CA IX, CA XII, CA XIV and CA XV), CA VA and CA VB are mitochondrial, and CA VI is secreted in saliva and milkCitation9,Citation10. The isozyme CA I is found in many tissues, and a presented study from Gao et al.Citation11 demonstrated that this enzyme is involved in retinal and cerebral edema, and its inhibition may be a valuable tool for fighting these conditions. The CA II is involved in several diseases, such as glaucoma, edema, epilepsy, and probably altitude sicknessCitation12. Although there are many studies on this enzyme, the CA enzyme family continues to capture the attention of drug discovery scientists and clinicians as the knowledge regarding the therapeutic implications associated with this enzyme class continues to growCitation4,Citation13,Citation14.
The CA inhibitor (CAI) targeting enzymes from mammals, which belong to the α-CA has been classified as (i) metal ion binders [inorganic anions; sulphonamides and their isosteres (such as the sulfamates, sulfamides, N-hydroxy-sulphonamides)]; (ii) compounds which anchor to the zinc-coordinated water molecule/hydroxide ion (phenols, polyamines, sulfocoumarins, etc.); (iii) compounds occluding the entrance of the active site (coumarins and their isosteres); and (iv) compounds which bind in an unknown manner (secondary/tertiary sulphonamides, imatinib, nilotinib, etc.)Citation15–22.
Sulphonamides are the best known CAIs and are used for the treatment of glaucoma in medicinal chemistry. Members of this class include aromatic, heterocyclic or aliphatic primary sulphonamides, but most drugs belong to the heterocyclic classCitation23,Citation24. The coumarin is a common moiety found in many biologically active natural and therapeutic products and thus represents a very important pharmacophoreCitation25–27. Besides the numerous activities of coumarin compounds, they were recently shown to constitute a novel class of inhibitors of the metalloenzyme CACitation28–30. In addition, Supuran et al.Citation31 reported that the hydrolysis of the sulfocoumarin to the vinyl sulfonic acid is mediated by the zinc hydroxide nucleophile from the CA active site, as for the coumarins which are transformed to 2-hydroxycinnamic acids. Interestingly, this sulfonic acid moiety is not coordinated to the Zn(II) ion but it is anchored to the zinc-bound water molecule/hydroxide ionCitation31.
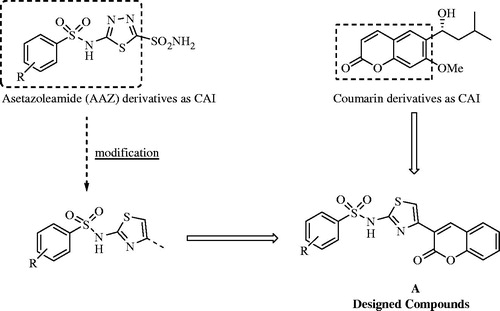
In this work, novel sulphonamides substituted coumarylthiazole were synthesized and their antioxidant activities and inhibitory effects on the activity of purified human carbonic anhydrase (hCA) I and II were evaluated. In the present study, our main aim is to check whether the new secondary sulphonamide derivatives substituted coumarin modified by the addition of thiazole ring (A) () may show higher inhibitory effect on CA enzymes with respect to the compounds previously investigated.
Figure 1. Design strategy of the reported compounds.
Methods
Chemistry
Melting points were taken on a Barnstead Electrothermal 9200 (Staffordshire, UK). IR spectra were registered on a Shimadzu Prestige-21 (200 VCE) spectrometer (Columbia, MD). 1H and 13C-NMR spectra were registered on a Varian Infinity Plus spectrometer at 300 and at 75 Hz, respectively. 1H and 13C chemical shifts are referenced to the internal deuterated solvent. The elemental analyses were carried out with a Leco CHNS-932 (St. Joseph, MI) instrument. Spectrophotometric analyses were performed by a BioTek Power Wave XS (BioTek, Winooski, VT). Sepharose 4B, l-tyrosine, sulphonamide, synthetic starting material, reagents and solvents were purchased from Merck, Alfa Easer, Sigma-Aldrich and Fluka.
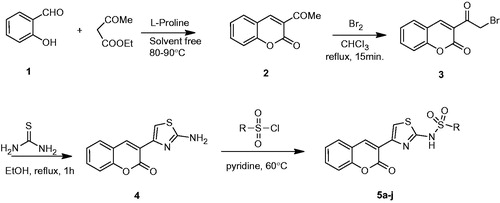
General procedure for the synthesis of 3-acetylcoumarin (2)
A mixture of benzaldehyde (3 mmol), reactive methylene compound (3 mmol) and l-proline (10 mol%) was heated under neat conditions for 0.5 h. The reaction was monitored by thin-layer chromatography (TLC). After completion of reaction, the reaction mixture was cooled and recrystallized from ethanol to get pure crystalline 3-acetylcoumarin (2) in 92% yield (0.52 g). Spectral data of this compound matched with the literatureCitation32.
General procedure for the synthesis of 3-(bromoacetyl)coumarin (3)
To a solution of 2 (0.01 mol) in 20 mL chloroform was added 0.01 mol of bromine in 5 mL chloroform, with intermittent shaking and warming to decompose an addition product. The mixture was heated for fifteen minutes on a water-bath to expel most of the hydrogen bromide, then cooled and filtered. The solid was washed with ether and recrystallizated with acetic acid. About 2.60 g product was obtained in 98% yield. Spectral data of this compound matched with the literatureCitation33.
General procedure for the synthesis of 3-(2-amino-1,3-thiazol-4-yl)coumarin (4)
Thiourea (5 mmol) was added to the solution of 3 (5 mmol) in boiling ethanol (20 mL). The mixture was refluxed for 1 h, then cooled and neutralized with aqueous ammonia. The precipitate was filtered off, washed with ethanol and used directly without crystallization or other purification. About 1.098 g product was obtained in 90% yield. Spectral data of this compound matched with the literatureCitation34.
General procedure for the synthesis of sulphonamides substituted coumarylthiazole (5a–j)
To a solution of 4 (1 mmol) in dry pyridine was added sulfonylchloride derivatives (1 mmol). The mixture was refluxed for 12 h with stirring, than cooled and added to ice water and 10% HCl. The product was filtered off, washed with water and dried under vacuum. The products were recrystallizated from ethanol over 98% purity. Compounds 5a–j were obtained with 68–82% yields.
4-Methyl-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)benzenesulphonamide (5a)
Yellow powder, 78% yield (310 mg), m.p.: 235–237 °C; IR: 3366 3287, 3042, 1712, 1634, 1599, 1522, 1365, 1271, 1134, 1081, 765 cm−1; 1H-NMR (DMSO-d6, 300 MHz) δ/ppm: 2.51 (3H, s), 7.37–7.48 (4H, m), 7.57 (1H, s), 7.67 (2H, t, J = 8.8 Hz), 7.77 (2H, t, J = 7.9 Hz), 8.63 (1H, s); 13C NMR (DMSO-d6, 75 MHz) δ/ppm: 21.6, 109.5, 116.7, 119.1, 119.4, 125.7, 126.7, 129.5, 130.2, 133.0, 133.3, 139.7, 139.8, 143.3, 153.0, 158.9, 169.5. Anal. Calcd. for C19H14N2O4S2: C, 57.27; H, 3.54; N, 7.03; found: C, 57.24; H, 3.50; N, 7.10.
2,5-Dimethyl-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)benzenesulphonamide (5b)
Orange powder, 82% yield (338 mg), m.p.: 160–162 °C; IR: 3328, 3124, 2965, 1710, 1607, 1528, 1361, 1292, 1131, 1060, 794, 593 cm−1; 1H-NMR (DMSO-d6, 300 MHz) δ/ppm: 2.35 (3H, s), 2.58 (3H, s), 7.24–7.38 (2H, m), 7.43 (2H, t, J = 7.9 Hz), 7.64 (1H, s), 7.69 (2H, t, J = 7.9 Hz), 7.77 (1H, s), 8.48 (1H, s); 13C-NMR (DMSO-d6, 75 MHz) δ/ppm: 20.2, 21.1, 111.0, 116.7, 119.0, 125.7, 126.2, 128.8, 129.5, 132.9, 133.3, 133.4, 134.0, 135.8, 139.7, 140.1, 142.9, 153.0, 158.3, 168.1. Anal. Calcd. for C20H16N2O4S2: C, 58.24; H, 3.91; N, 6.79; found: C, 58.28; H, 3.95; N, 6.75.
4-Isopropyl-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)benzenesulphonamide (5c)
Orange powder, 80% yield (340 mg), m.p.: 182–184 °C; IR: 3276, 3142, 2960, 1712, 1606, 1524, 1454, 1360, 1306, 1148, 1090, 938, 755, 569 cm−1; 1H-NMR (DMSO-d6, 300 MHz) δ/ppm: 1.21 (6H, d, J = 5.8 Hz), 2.93–2.99 (1H, m), 7.37–7.51 (4H, m), 7.63 (1H, s), 7.69 (2H, d, J = 7.6 Hz), 7.79 (2H, d, J = 8.5 Hz), 8.46 (1H, s); 13C-NMR (DMSO-d6, 75 MHz) δ/ppm: 24.1, 34.0, 111.2, 116.7, 119.9, 125.7, 126.8, 127.6, 129.5, 132.1, 133.3, 136.8, 139.6, 140.0, 143.8, 153.0, 158.4, 168.1. Anal. Calcd. for C21H18N2O4S2: C, 59.14; H, 4.25; N, 6.57; found: C, 59.10; H, 4.28; N, 6.55.
4-(Tert-butyl)-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)benzenesulphonamide (5d)
Orange powder, 72% yield (316 mg), m.p.: 250–252 °C; IR: 3368, 2961, 1716, 1518, 1442, 1361, 1287, 1145, 1106, 1084, 755, 578 cm−1; 1H-NMR (DMSO-d6, 300 MHz) δ/ppm: 1.28 (9H, s), 7.34–7.51 (3H, m), 7.58–7.69 (4H, m), 7.79 (2H, d, J = 8.7 Hz ), 8.46 (1H, s); 13C-NMR (DMSO-d6, 75 MHz) δ/ppm: 31.4, 35.4, 109.4, 111.2, 116.4, 116.7, 119.0, 125.3, 125.7, 126.5, 126.8, 127.7, 129.5, 133.3, 139.6, 153.0, 158.4, 168.1. Anal. Calcd. for C22H20N2O4S2: C, 59.98; H, 4.58; N, 6.36; found: C, 59.95; H, 4.56; N, 6.38.
4-Methoxy-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)benzenesulphonamide (5e)
Yellow powder, 75% yield (310 mg), m.p.: 259–261 °C; IR: 3333, 3117, 2943, 1712, 1521, 1438, 1365, 1304, 1261, 1140, 1088, 927, 749, 556 cm−1; 1H-NMR (DMSO-d6, 300 MHz) δ/ppm: 3.82 (3H, s), 7.10 (2H, d, J = 7.9 Hz), 7.37–7.46 (3H, m), 7.64–7.69 (2H, m), 7.81 (2H, d, J = 8.2 Hz), 8.46 (1H, s); 13C-NMR (DMSO-d6, 75 MHz) δ/ppm: 56.3, 109.4, 111.3, 114.9, 116.7, 119.1, 125.4, 125.7, 128.8, 129.5, 133.2, 134.1, 139.6, 153.0, 158.4, 162.8, 168.1. Anal. Calcd. for C19H14N2O5S2: C, 55.06; H, 3.40; N, 6.76; found: C, 55.09; H, 3.45; N, 6.75.
N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)naphthalene-2-sulphonamide (5f)
Cream powder, 70% yield (304 mg), m.p.: 251–253 °C; IR: 3292, 3122, 3049, 1709, 1599, 1441, 1367, 1301, 1115, 1078, 922, 752, 669 cm−1; 1H-NMR (DMSO-d6, 300 MHz) δ/ppm: 7.39 (2H, t, J = 8.4 Hz), 7.62–7.64 (5H, m), 7.84 (1H, d, J = 8.7 Hz), 8.00 (1H, d, J = 7.9 Hz), 8.09 (1H, d, J = 8.7 Hz), 8.17 (1H, d, J = 6.7 Hz), 8.42 (1H, s), 8.52 (1H, s); 13C-NMR (DMSO-d6, 75 MHz) δ/ppm: 111.3, 116.7, 119.0, 122.8, 125.7, 127.1, 128.2, 128.4, 129.3, 129.5, 129.9, 130.0, 132.3, 133.3, 134.8, 139.3, 139.7, 153.0, 158.4, 164.8. Anal. Calcd. for C22H14N2O4S2: C, 60.82; H, 3.25; N, 6.45; found: C, 60.84; H, 3.28; N, 6.40.
4-fluoro-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)benzenesulphonamide (5g)
Reddish powder, 74% yield (297 mg), m.p.: 248–250 °C; IR: 3299, 3123, 3073, 1702, 1608, 1524, 1432, 1366, 1292, 1069, 921, 753, 582 cm−1; 1H-NMR (DMSO-d6, 300 MHz) δ/ppm: 7.38–7.55 (3H, m), 7.60–7.74 (6H, m), 8.49 (1H, s); 13C-NMR (DMSO-d6, 75 MHz) δ/ppm: 111.3, 113.4, 113.7, 116.2, 116.7, 119.0, 120.0, 120.3, 122.8, 125.7, 129.5, 132.2, 132.3, 133.3, 139.7, 144.7, 144.8, 153.0, 158.3, 160.7, 163.9, 168.9. Anal. Calcd. for C18H11FN2O4S2: C, 53.72; H, 2.76; N, 6.96; found: C, 53.74; H, 2.75; N, 6.94.
4-Chloro-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)benzenesulphonamide (5h)
Yellow powder, 82% yield (342 mg), m.p.: 264–266 °C; IR: 3145, 3043, 1720, 1567, 1526, 1440, 1362, 1306, 1267, 1139, 1086, 928, 752 cm−1; 1H-NMR (DMSO-d6, 300 MHz) δ/ppm: 7.39–7.51 (3H, m), 7.64–7.70 (4H, m), 7.85 (2H, d, J = 8.2 Hz), 8.48 (1H, s); 13C-NMR (DMSO-d6, 75 MHz) δ/ppm: 111.3, 116.8, 119.0, 125.8, 128.5, 129.9, 133.4, 137.8, 139.8, 140.2, 141.7, 144.7, 153.1, 158.4, 162.7, 168.4. Anal. Calcd. for C18H11ClN2O4S2: C, 51.61; H, 2.65; N, 6.69; found: C, 51.63; H, 2.62; N, 6.67.
4-Bromo-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)benzenesulphonamide (5i)
Reddish powder, 76% yield (351 mg), m.p.: 266–268 °C; IR: 3275, 3043, 1726, 1605, 1529, 1441, 1366, 1307, 1269, 1139, 1083, 936, 759, 553 cm−1; 1H-NMR (DMSO-d6, 300 MHz) δ/ppm: 7.35–7.45 (4H, m), 7.50–7.76 (5H, m), 8.45 (1H, s); 13C-NMR (DMSO-d6, 75 MHz) δ/ppm: 111.2, 116.2, 116.8, 119.0, 122.7, 125.7, 128.6, 129.6, 132.8, 133.4, 139.8, 141.0, 153.1, 158.3, 162.4, 168.6. Anal. Calcd. for C18H11BrN2O4S2: C, 46.66; H, 2.39; N, 6.05; found: C, 46.64; H, 2.35; N, 6.08.
4-Iodo-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)benzenesulphonamide (5j)
Dark yellow powder, 68% yield (347 mg), m.p.: 244–246 °C; IR: 3382, 3154, 3042, 1701, 1605, 1532, 1438, 1361, 1265, 1140, 1086, 941, 759, 554 cm−1; 1H-NMR (DMSO-d6, 300 MHz) δ/ppm: 7.36–7.45 (2H, m), 7.58–7.67 (5H, m), 7.94 (2H, d, J = 7.6 Hz), 8.46 (1H, s); 13C-NMR (DMSO-d6, 75 MHz) δ/ppm: 111.2, 116.8, 119.0, 125.7, 128.3, 129.6, 133.4, 137.5, 138.6, 139.8, 142.2, 147.4, 153.1, 158.4, 161.1, 168.6. Anal. Calcd. for C18H11IN2O4S2: C, 42.36; H, 2.17; N, 5.49; found: C, 42.39; H, 2.15; N, 5.47.
Preparation and purification of haemolysate from blood red cells
Preparation and purification of haemolysate from blood red cells made by the literatureCitation35. Blood samples (25 mL) were taken from healthy human volunteers. They were anticoagulated with acid–citrate–dextrose, centrifuged at 2000 g for 20 min at 4 °C and the supernatant was removed. The packed erythrocytes were washed three times with 0.9% NaCl and then haemolysed in cold water. The ghosts and any intact cells were removed by centrifugation at 2000 g for 25 min at 4 °C, and the pH of the haemolysate was adjusted to pH 8.5 with solid Tris-base. The 25 mL haemolysate was applied to an affinity column containing l-tyrosine-sulphonamide-Sepharose-4B equilibrated with 25 mM Tris–HCl/0.1 M Na2SO4 (pH 8.5). The affinity gel was washed with 50 mL of 25 mM Tris–HCl/22 mM Na2SO4 (pH 8.5). The human CA (hCA) isozymes were eluted with 0.1 M NaCl/25 mM Na2HPO4 (pH 6.3) and 0.1 M CH3COONa/0.5 M NaClO4 (pH 5.6), which recovered hCA-I and hCA-II, respectively. Fractions of 3 mL were collected and their absorbance measured at 280 nm.
CA enzyme assay
CA activity was measured by the Maren method which is based on determination of the time required for the pH to decrease from 10.0 to 7.4 due to CO2 hydrationCitation36. The assay solution was 0.5 M Na2CO3/0.1 M NaHCO3 (pH 10.0) and Phenol Red was added as the pH indicator. CO2-hydratase activity [enzyme units (EU)] was calculated by using the equation t0−tc/tc where t0 and tc are the times for pH change of the non-enzymatic and the enzymatic reactions, respectively.
In vitro inhibition studies
For the inhibition studies of sulphonamides, five different concentrations of these compounds were added to the medium in 4.2 mL of total reaction volume including the enzyme solution. Duration (in seconds) of the colour change from red to yellow in solution was measured in a 10-ml glass tube with 1 cm diameter. CA enzyme activity without a compound solution was accepted as 100% activity. All compounds were tested in triplicate at each concentration used. Activity % values of CA for different concentrations of each compound were determined by regression analysis using Microsoft Office 2000 Excel. For the compounds having an inhibition affect, the inhibitor concentration causing up to 50% inhibition (IC50 values) was determined from the graphs.
Antioxidant activity assays
In CUPRAC assay, the absorbance values were used to calculate for A0.50, but in ABTS assay, inhibition (%) values were used to calculate for IC50.
ABTS cation radical decolourization assay
ABTS•+ scavenging activities of the synthesized compounds were determined as previously reportedCitation37. The solution of ABTS•+ radical was generated by dissolving 19.2 mg of 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (7 mM ABTS) and 3.3 mg K2S2O3 in distilled water (5 mL). This solution was kept in dark for 24 h at room temperature, and the absorbance of the solution was fixed to ∼0.70 at 734 nm by dilution. The solutions of the samples were prepared in n-propanol at a concentration of 1000 µg/mL. The absorbance was measured at room temperature at 734 nm, after 6 min from ABTS•+ addition. The decrease in the absorption was used to calculate the activities. The results were expressed as IC50. Propyl alcohol was used as a control solvent.
Cupric reducing antioxidant capacity assay (CUPRAC)
Cupric reducing antioxidant capacities of the synthesized compounds were determined in accordance with the literature methodCitation38. The solutions of compounds and standards were prepared in n-propanol at a concentration of 1000 µg/mL. Different volumes (1000 mg/L and 54.5 mL) of the sample were added to a solution prepared by adding 61.0 µL of 10 mM CuCl2, 61.0 µL 7.5 mM neocuproine and 61.0 µL of 1.0 mM NH4CH3COO buffer (pH 7), respectively. The absorbance was measured at room temperature at 450 nm, after an hour. The results were calculated as A0.50. Propyl alcohol was used as a solvent to controls.
Results and discussion
Sulphonamide derivatives are known as the strongest CAIsCitation23. The asetazolamide (AAZ) derivatives have also been reported as potent CAICitation4. Our design strategy is that the 1,3,4-thidiazole ring of AAZ was modified to thiazole ring and was added to sulphonamides in order to increase inhibitory activity by forming hydrogen bond. On the other hand, we think that the presence of coumarin moiety contributes to inhibitor activity by hydrolysing to 2-hydroxycinnamic acids or interacting with the enzyme active sites ().
The synthetic procedures employed to obtain the target compounds 5a–j are depicted in . Compound 4 was synthesized from salicylaldehydes in three steps according to the literatureCitation39, and then it was reacted with various benzenesulfonyl choloride derivatives in pyridine to get product sulphonamides substituted coumarylthiazole (5a–j).
Scheme 1. Synthesis of new sulphonamides substituted coumarylthiazole (5a–j) derivatives.
All the new compounds were characterized by 1H-NMR, 13C-NMR, IR and elemental analysis. In the infrared spectra of the synthesized compounds, it was possible to observe the absorptions ∼3300 cm−1 relating to NH stretch of sulphonamide groups, ∼1520 cm−1 relating to C=N stretch for thiazole, absorptions in ∼1710 cm−1 from coumarin carbonyl moiety stretch and absorptions ∼1360 cm−1 relating to SO2 antisym stretch in sulphonamides. From the 1H-NMR spectra, the resonance due to the hydrogen attached to the sulphonamide nitrogen was not detected. The signals for aromatic hydrogens were observed between 7.10 and 8.17 ppm, the signal of proton at thiazole ring was detected at ∼8.48 ppm. From the 13C-NMR spectra, the signals can be seen ∼158 and 168 ppm relating to coumarin carbonyl and thiazole ring, respectively.
For evaluation, the physiologically relevant human CA isozyme hCA I and II inhibitory activity, all the synthesized compounds were subjected to CA inhibition assay with CO2 as a substrate.
The result showed that all synthesized compounds (5a–j) inhibited the hCA I and II enzyme activity. The IC50 values for hCA I and II inhibitions are summarized in . The IC50 values were between 5.63 and 22.63 µM for hCA I and between 8.48 and 23.87 µM for hCA II inhibitory activity. Among synthesized compounds, 5f (IC50 = 5.63 and 8.48 µM, for hCA I and hCA II, respectively) showed the highest inhibitory activity against hCA I and II. Most of the primary sulphonamide compounds are mentioned very potent inhibitor of the cytosolic isoform hCAsCitation6. All the synthesized sulphonamides in this study are moderate inhibitory activity for the hCAs, because they are secondary sulphonamides and contain big bulky groups. Up to now, many sulphonamide and coumarin derivatives have been synthesized as strong CAIs by Supuran's groupCitation28–30,Citation40–45. The inhibition values of them ranged from low micromolar to nanomolar. These compounds have stronger inhibitory effect than the sulphonamides substituted coumarylthiazole in this study. But, 5f (IC50 = 5.63 and 8.48 µM, for hCA I and hCA II, respectively) showed higher inhibitory properties against hCA I and II compared to some sulphonamide and coumarin derivatives, reported in our previously studyCitation46–51, with an IC50 value of between 6.79 and 620 µM, for hCA I; between 6.54 and 51.45 µM, for hCA II.
Table 1. In vitro inhibition IC50 and A0.50 values (µM) of sulphonamides substituted coumarylthiazole (5a–j) for hCA I and hCA II and antioxidant activities.
The following structure–activity relationship (SAR) observations can be drawn from data of : (i) the best inhibitor among the newly synthesized and investigated compounds was naphthalene substituted derivative (5f) for hCA I and II; (ii) the alkyl series at the phenyl ring showed a qualitative relationship between increasing inhibitory activity and bulky group for hCA I and II [5d (R = 4-tert-butyl-benzene, IC50 = 6.21 and 10.91 µM for hCA I and II, respectively) > 5c (R = 4-isopropyl-benzene, IC50 = 6.46 and 11.12 µM for hCA I and II, respectively) > 5b (R = 2,5-dimethyl-benzene, IC50 = 8.64 and 11.68 µM for hCA I and II, respectively) > 5a (R = 4-methyl-benzene, IC50 = 22.63 and 23.87 µM for hCA I and II, respectively)]; and (iii) the inhibitory activity on both hCA I and II seems to be strongly dependent on the size and polarizability of the halogen substituent at the para-position of the phenyl ring [for size and polarizability, I > Br > Cl > F, for inhibitory activity, 5j (R = 4-iodobenzene, IC50 = 5.88 and 8.69 µM for hCA I and II, respectively) > 5i (R = 4-bromobenzene, IC50 = 7.67 and 11.35 µM for hCA I and II, respectively) > 5h (R = 4-cholorobenzene, IC50 = 9.55 and 15.72 µM for hCA I and II, respectively) > 5g (R = 4-fluorobenzene, IC50 = 16.21 and 18.30 µM for hCA I and II, respectively)].
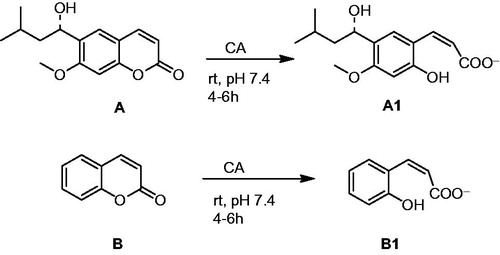
According to SAR study, it is clear that the bulky substituents (such as tert-butyl, naphthalene and iodine) increase inhibitory activity of the compound due to steric effect. The sulphonamide drug binds in deprotonated form to the catalytically critical Zn(II) ion, also participating in extensive hydrogen bond and van der Waals interactions with amino acid residues both in the hydrophobic and hydrophilic halves of the enzyme active site, as shown by X-ray crystallographic studies of enzyme–inhibitor complexesCitation6. The inhibition mechanism of coumarins is different compared to that of the classical CAIs of the sulphonamide typeCitation40. Several kinetic and X-ray crystallographic studies have revealed that coumarins are mechanism-based inhibitors, which undergo hydrolysis under the influence of the zinc hydroxide, nucleophilically active species of the enzyme, with generation of substituted-2-hydroxycinnamic acids ()Citation28,Citation41,Citation42. Supuran's group reported that inhibitor (as for coumarin/sulfocoumarin) and enzyme solutions were pre-incubated together for ∼6 h prior to assay in order to allow for the formation of the E−I complex or for the eventual active site mediated hydrolysis of the inhibitorCitation31.
Figure 2. Formation of 2-hydroxy-cinnamic acids A1 and B1 by the CA-mediated hydrolysis of coumarin A and BCitation28,Citation41,Citation42.
On the other hand, the two endocyclic nitrogen of the 1,3,4-thidiazole ring of AAZ derivatives participates in two hydrogen bonds with the OH of Thr200Citation4.
Based on the above consideration, we consider that the coumarin ring should not undergo ring opening without pre-incubation on enzyme and inhibitor. Also it is difficult that the sulphonamide moiety of the synthesized compounds could bind to zinc cation in traditional manner due to contain big bulky groups. So we suppose that the bulky substituents (such as tert-butyl, naphthalene and iodine), increasing inhibitory activity, could locate at the entrance of the enzyme active site. We also think that the nitrogen of the thiazole ring of the synthesized compounds contribute to inhibition by forming hydrogen bonds.
Antioxidant activity assay
It is known that many natural and synthetic coumarin, sulphonamide and thiazole derivatives have various pharmacological properties. Among these properties, their antioxidant effects were examinedCitation52–54. Also, Gocer et al.Citation53 have recently reported that some sulphonamides demonstrated effective antioxidant properties (for Cu2+ reducing capabilities and ABTS•+). In addition, Supuran's group investigated the the interaction of CA isozymes with some antioxidant compounds, such as resveratrol, dobutamine, curcumin, catechin, silymarin, salicyclates and quercetin. It was reported that all these antioxidant compounds showed effective hCA I and II inhibitory activityCitation55–58. In view of these findings, it was considered that the synthesized compounds might possess certain antioxidant activity due to include sulphonamide, coumarin and thiazole moiety containing sulfonyl, carbonyl, imine and methoxy groups.
The ABTS method is based on the ability of hydrogen or electron-donating antioxidants to decolourize the performed radical monocation of 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) generated due to oxidation of ABTS with potassium persulfateCitation37. The results in indicated that all synthesized compounds exhibited moderate radical scavenging ability. 5e and 5g exhibited the strongest ABTS activity with IC50 values of 48.83 and 50.45 µM, respectively. These potencies of 5e and 5g was 3.3-fold less than that of quercetin (IC50 = 15.49 µM) used as the reference compound.
The halogen substituent at the para-position of the phenyl ring showed an inverse relationship for increasing ABTS activity with growing size and polarizability [for size and polarizability, I > Br > Cl > F; for ABTS activity, 5j (R = 4-iodobenzene, IC50 = 87.65 µM) < 5i (R = 4-bromobenzene, IC50 =81.29 µM) < 5h (R = 4-cholorobenzene, IC50 = 60.40 µM) < 5g (R = 4-fluorobenzene, IC50 = 50.45 µM)].
CUPRAC assays have a distinct advantage over other electron-transfer based assays (e.g. Folin, FRAP, ABTS, DPPH). This advantage is its realistic pH close to that physiological, favourable redox potential, accessibility and stability of reagents and applicability to lipophilic antioxidants as well as hydrophilic onesCitation38. The cupric reducing antioxidant capacities of the synthesized compounds (5a–j) were determined according to the literature methodCitation38 using quercetin as the reference compound. Among the synthesized compounds, only 5e (A0.50 = 23.29 µM) showed close cupric reducing antioxidant activity to quercetin (A0.50 = 18.47 µM). The others have less the cupric reducing antioxidant capacity than quercetin.
Interestingly, in contrast ABTS activity, the CUPRAC activity seems to be strongly dependent on the increasing size and polarizability of the halogen substituent at the para-position of the phenyl ring [for size and polarizability, I > Br > Cl > F; for CUPRAC activity, 5j (R = 4-iodobenzene, A0.50 =41.96 µM) > 5i (R = 4-bromobenzene, A0.50 = 49.58 µM) > 5h (R = 4-cholorobenzene, A0.50 = 71.06 µM) > 5g (R = 4-fluorobenzene, A0.50 = 81.19 µM)].
Conclusions
A series of new secondary benzenesulphonamide substituted coumarylthiazole derivatives (5a–j) was synthesized and their activities as hCA I and hCA II inhibitors and structure–activity relationship were examined. All synthesized compounds inhibited hCA I and II. 5f exhibited the strongest inhibition against hCA I and II with IC50 value of 5.63 and 848 µM, respectively. The SARs revealed that the inhibitory activity of the synthesized compounds could also be affected by the type of the halogen substituent on the phenyl ring. Also the bulky substituents (such as tert-butyl, naphthalene and iodine) increase inhibitory activity of the compounds due to steric effect. Additionaly, the synthesized compounds showed moderate antioxidant activity.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
This work was supported by the Sakarya Research Fund of the Sakarya University (2014-28-00-001).
References
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77
- Supuran CT. Carbonic anhydrase inhibitors and activators for novel therapeutic applications. Future Med Chem 2011;3:1165–80
- Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72
- Monti SM, Maresca A, Viparelli F, et al. Dithiocarbamates are strong inhibitors of the beta-class fungal carbonic anhydrases from Cryptococcus neoformans, Candida albicans and Candida glabrata. Bioorg Med Chem Lett 2012;22:859–62
- D'Ambrosio K, Smaine FZ, Carta F, et al. Development of potent carbonic anhydrase inhibitors incorporating both sulfonamide and sulfamide groups. J Med Chem 2012;55:6776–83
- Vullo D, Kupriyanova EV, Scozzafava A, et al. Anion inhibition study of the β-carbonic anhydrase (CahB1) from the cyanobacterium Coleofasciculus chthonoplastes (ex-Microcoleus chthonoplastes). Bioorg Med Chem 2014;22:1667–71
- Prete SD, Vullo D, Fisher GM, et al. Discovery of a new family of carbonic anhydrases in the malaria pathogen Plasmodium falciparum-the η-carbonic anhydrases. Bioorg Med Chem Lett 2014;24:4389–96
- Supuran CT, Scozzafava A. Carbonic anhydrases as targets for medicinal chemistry. Bioorg Med Chem 2007;15:4336–50
- Husain A, Madhesia D. Heterocyclic compounds as carbonic anhydrase inhibitor. J Enzyme Inhib Med Chem 2012;27:773–83
- Gao BB, Clermont A, Rook S, et al. Extracellular carbonic anhydrase mediates hemorrhagic retinal and cerebral vascular permeability through prekallikrean activation. Nat Med 2007;13:181–8
- Alterio V, Di Fiore A, D'Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms. Chem Rev 2012;112:4421–68
- Poulsen SA. Carbonic anhydrase inhibition as a cancer therapy: a review of patent literature, 2007–2009. Exp Opin Ther Pat 2010;20:795–806
- Krall N, Pretto F, Decurtins W, et al. A small-molecule drug conjugate for the treatment of carbonic anhydrase IX expressing tumors. Angew Chem Int Ed 2014;53;4231–5
- Congiu C, Onnis V, Balboni G, Supuran CT. Synthesis and carbonic anhydrase I, II, IX and XII inhibition studies of 4-N,N-disubstituted sulfanilamides incorporating 4,4,4-trifluoro-3-oxo-but-1-enyl, phenacylthiourea and imidazol-2(3H)-one/thione moieties. Bioorg Med Chem Lett 2014;24:1776–9
- Durdagi S, Senturk M, Ekinci D, et al. Kinetic and docking studies of phenol-based inhibitors of carbonic anhydrase isoforms I, II, IX and XII evidence a new binding mode within the enzyme active site. Bioorg Med Chem 2011;19:1381–9
- De Simone G, Alterio V, Supuran CT. Exploiting the hydrophobic and hydrophilic binding sites for designing carbonic anhydrase inhibitors. Expert Opin Drug Discov 2013;8:793–810
- Supuran CT. Carbonic anhydrase inhibitors: an editorial. Expert Opin Ther Pat 2013;23:677–9
- Aggarwal M, Kondeti B, McKenna R. Anticonvulsant/antiepileptic carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Pat 2013;23:717–24
- Liu F, Martin-Mingot A, Lecornué F, et al. Carbonic Anhydrases inhibitory effects of new benzenesulfonamides synthesized by using superacid chemistry. J Enzyme Inhib Med Chem 2012;27:886–91
- Metayer B, Mingot A, Vullo D, et al. New superacid synthesized (fluorinated) tertiary benzenesulfonamides acting as selective hCA IX inhibitors: toward a new mode of carbonic anhydrase inhibition by sulphonamides. Chem Commun 2013;49:6015–17
- Metayer B, Martin-Mingot A, Vullo D, et al. Superacid synthesized tertiary benzenesulfonamides and benzofuzed sultams act as selective hCA IX inhibitors: toward understanding a new mode of inhibition by tertiary sulphonamides. Org Biomol Chem 2013;11:7540–9
- Supuran CT, Winum JY. Selectivity issues in the design of CA inhibitors. Drug design of zinc-enzyme inhibitors: functional, structural, and disease applications. Vol. 13. New Jersey: John Wiley & Sons; 2009. p. 473
- Guler OO, De Simone G, Supuran CT. Drug design studies of the novel antitumor targets carbonic anhydrase IX and XII. Curr Med Chem 2010;17:1516–26
- Kontogiorgis C, Detsi A, Hadjipavlou-Litina D. Coumarin-based drugs: a patent review (2008–present). Expert Opin Ther Pat 2012;22:437–54
- Kumar A, Gupta MK, Kumar M. An efficient non-ionic surfactant catalysed multicomponent synthesis of novel Benzylamino coumarin derivative via Mannich type reaction in water. Tetrahedron Lett 2011;52:4521–5
- Parvez A, Meshram J, Tiwari V, et al. Pharmacophores modeling in terms of prediction of theoretical physico-chemical properties and verification by experimental correlations of novel coumarin derivatives produced via Betti's protocol. Eur J Med Chem 2010;45:4370–8
- Maresca A, Temperini C, Vu H, et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–62
- Maresca A, Temperini C, Pochet L, et al. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem 2010;53:335–44
- Carta F, Maresca A, Scozzafava A, Supuran CT. Novel coumarins and 2-thioxo-coumarins as inhibitors of the tumor-associated carbonic anhydrases IX and XII. Bioorg Med Chem 2012;20:2266–73
- Tars K, Vullo D, Kazaks A, et al. Sulfocoumarins (1,2-benzoxathiine-2,2-dioxides): a class of potent and isoform-selective inhibitors of tumor-associated carbonic anhydrases. J Med Chem 2013;56:293–300
- Karade N, Gampawar SN, Shinde SV, Jadhav WN. L-Proline catalyzed solvent-free Knoevenagel condensation for the synthesis of 3-substituted coumarins. Chinese J Chem 2007;25:1686–9
- Koelsch CF. Bromination of 3-acetocoumarin. J Am Chem Soc 1950;72:2993–5
- Zhuravel OI, Kovalenko SM, Vlasov SV, Chernykh VP. Solution-phase synthesis of a combinatorial library of 3-[4-(coumarin-3-yl)-1,3-thiazol-2-ylcarbamoyl]propanoic acid amides. Molecules 2005;10:444–56
- Arslan O, Nalbantoglu B, Demir N, et al. A new method for the purification of carbonic anhydrase isozymes by affinity choromatography. Turk J Med Sci 1996;26:163–6
- Maren TH. A simplified micromethod for the determination of carbonic anhydrase and its inhibitors. J Pharm Exp Ther 1960;130:2629–34
- Re R, Pellegrini N, Proteggente A, et al. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radical Bio Med 1999;26:1231–7
- Apak R, Guclu K, Ozyurek M, Karademir SE. Novel total antioxidant capacity index for dietary polyphenols and vitamins C and E, using their cupric ion reducing capability in the presence of neocuproine: CUPRAC method. J Agr Food Chem 2004;52:7970–81
- Kurt BZ, Gazioglu I, Sonmez F, Kucukislamoglu M. Synthesis, antioxidant and anticholinesterase activities of novel coumarylthiazole derivatives. Bioorg Chem 2015; 59:80–90
- Grandane A, Tanc M, Zalubovskis R, Supuran CT. 6-Triazolyl-substituted sulfocoumarins are potent, selective inhibitors of the tumor-associated carbonic anhydrases IX and XII. Bioorg Med Chem Lett 2014;24:1256–60
- Maresca A, Scozzafava A, Supuran CT. 7,8-Disubstituted- but not 6,7-disubstituted coumarins selectively inhibit the transmembrane, tumor-associated carbonic anhydrase isoforms IX and XII over the cytosolic ones I and II in the low nanomolar/subnanomolar range. Bioorg Med Chem Lett 2010;20:7255–8
- Wagner J, Avvaru BS, Robbins AH, et al. Coumarinyl-substituted sulfonamides strongly inhibit several human carbonic anhydrase isoforms: solution and crystallographic investigations. Bioorg Med Chem 2010;18:4873–8
- Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Arylsulfonylureido- and arylureido-substituted aromatic and heterocyclic sulfonamides: towards selective inhibitors of carbonic anhydrase isozyme I. J Enzyme Inhib Med Chem 1999;14:343–63
- Pacchiano F, Carta F, McDonald PC, et al. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J Med Chem 2011;54:1896–902
- Pacchiano F, Aggarwal M, Avvaru BS, et al. Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureido-benzenesulfonamides and correlate to inhibitor potency. Chem Commun 2010;46:8371–3
- Celik F, Arslan M, Yavuz E, et al. Synthesis and carbonic anhydrase inhibitory properties of novel 1,4-dihydropyrimidinone substituted diarylureas. J Enzyme Inhib Med Chem 2014;29:18–22
- Karatas MO, Alici B, Cakir U, et al. New coumarin derivatives as carbonic anhydrase inhibitors. Artif Cells Nanomed Biotechnol 2014;42:192–8
- Karatas MO, Alici B, Cakir U, et al. Synthesis and carbonic anhydrase inhibitory properties of novel coumarin derivatives. J Enzyme Inhib Med Chem 2013;28:299–304
- Celik F, Arslan M, Kaya MO, et al. Synthesis and carbonic anhydrase inhibitory properties of tetrazole- and oxadiazole substituted 1,4-dihydropyrimidinone compounds. Artif Cells Nanomed Biotechnol 2014;42:58–62
- Sonmez F, Bilen C, Sumersan S, et al. In vitro inhibition effect and structure-activity relationships of some saccharin derivatives on erythrocyte carbonic anhydrase I and II. J Enzyme Inhib Med Chem 2014;29:118–23
- Kuday H, Sonmez F, Bilen C, et al. Synthesis and in vitro inhibition effect of new pyrido[2,3-d]pyrimidine derivatives on erythrocyte carbonic anhydrase I and II. Biomed Res Int 2014;2014:594879
- Kostava I. Synthetic and natural coumarins as antioxidants. Mini Rev Med Chem 2006;6:365–74
- Göçer H, Akıncıoglu A, Öztaskın N, et al. Synthesis, antioxidant, and antiacetylcholinesterase activities of sulfonamide derivatives of dopamine-related compounds. Arch Pharm Chem Life Sci 2013;346:783–92
- Geronikaki AA, Pitta EP, Liaras KS. Thiazoles and tthiazolidinones as antioxidants. Curr Med Chem 2013;20:4460–80
- Ekinci D, Karagoz L, Ekinci D, et al. Carbonic anhydrase inhibitors: in vitro inhibition of α isoforms (hCA I, hCA II, bCA III, hCA IV) by flavonoids. J Enzyme Inhib Med Chem 2013;28:283–8
- Innocenti A, Gülçin I, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Antioxidant polyphenols effectively inhibit mammalian isoforms I–XV. Bioorg Med Chem Lett 2010;20:5050–3
- Sentürk M, Gülçin I, Dastan A, et al. Carbonic anhydrase inhibitors. Inhibition of human erythrocyte isozymes I and II with a series of antioxidant phenols. Bioorg Med Chem 2009;17:3207–11
- Sentürk M, Gülçin I, Beydemir S, et al. In vitro inhibition of human carbonic anhydrase I and II isozymes with natural phenolic compounds. Chem Biol Drug Des 2011;77:494–9