
Abstract
Four series of new bipyrazoles comprising the N-phenylpyrazole scaffold linked to polysubstituted pyrazoles or to antipyrine moiety through different amide linkages were synthesized. The synthesized compounds were evaluated for their anti-inflammatory and analgesic activities. In vitro COX-1/COX-2 inhibition study revealed that compound 16b possessed the lowest IC50 value against both COX-1 and COX-2. Moreover, the effect of the most promising compounds on inducible nitric oxide synthase (iNOS) and cyclooxygenase (COX-2) protein expression in lipopolysaccharide (LPS)-activated rat monocytes was also investigated. The results revealed that some of the synthesized compounds showed anti-inflammatory and/or analgesic activity with less ulcerogenic potential than the reference drug diclofenac sodium and are well tolerated by experimental animals. Moreover, they significantly inhibited iNOS and COX-2 protein expression induced by LPS stimulation. Compounds 16b and 18 were proved to display anti-inflammatory activity superior to diclofenac sodium and analgesic activity equivalent to it with minimal ulcerogenic potential.
Introduction
Inflammation is a complex biological response of body tissues that arises in response to traumatic, infectious, post-ischaemic, toxic, or autoimmune injury. The process normally leads to recovery from infection and to healing. However, if targeted destruction and assisted repair are not properly phased, inflammation can lead to persistent tissue damage by infiltrating leukocytes and lymphocytes or damaged collagenCitation1. Anti-inflammatory pharmacotherapy targeting cyclooxygenase (COX) enzymes is a well-established strategy to treat inflammation. Modalities of inhibiting COX enzyme-associated prostanoid synthesis fall within two categories, the use of non-steroidal anti-inflammatory drugs (NSAIDs)Citation2 or blocking COX protein synthesis using stronger anti-inflammatory drugs such as glucocorticoids.
NSAIDs are the most widely prescribed therapeutics worldwide, primarily for the treatment of pain and inflammation. The major mechanism by which NSAIDs exert their anti-inflammatory activity is the suppression of prostaglandin biosynthesis from arachidonic acid by inhibiting the COX enzyme which exists in three distinct isoforms: COX-1, COX-2, and COX-3Citation3–5. The constitutive COX-1 isoform provides cytoprotection in the gastrointestinal (GI) tract and controls renal function in the kidneys, whereas the inducible COX-2 is upregulated by pro-inflammatory mediators such as endotoxins, mitogens, or cytokines including tumor necrosis factor α (TNF-α) and interleukins (ILs) such as IL-6 and IL-1Citation6,Citation7. Furthermore, COX-3 which is a variant of COX-1 presents in central nervous system and has been proposed to be another target for anti-inflammatory agentsCitation8. GI injury is one of the major hallmarks of NSAIDs and is contraindicated in patients with history of GI bleeding or distressCitation9. Moreover, administering additional gastroprotective treatment in conjunction with NSAIDs raises the issues of polypharmacy and drug-induced organ injury in high-risk patients notably elderly and infantsCitation10. Consequently, a new generation of COX-2 selective inhibitors has been clinically used with the hope that they would exhibit a reduced risk in GI events. However, the increased cardiovascular risk associated with COX-2-selective inhibitors led to reconsideration of their appropriate useCitation11. Thus, the development of safe and effective therapy for treating inflammatory conditions still presents a major challenge to the medical community.
Pyrazoles represent a key structural motif in heterocyclic chemistry and occupy a significant position in medicinal chemistry because of their capability to exhibit a wide range of bioactivities. They were found to display anti-inflammatoryCitation12–14 and analgesic activitiesCitation15,Citation16 in addition to COX-2 inhibitory activityCitation17,Citation18. The pyrazole unit is one of the core structures in a number of selective COX-2 inhibitors such as celecoxib and SC-558 ()19–21. Moreover, extensive studies have been devoted to N-aryl pyrazole derivatives as dual anti-inflammatory and antimicrobial agentsCitation22–24. On the other hand, great attention has been focused on pyrazoline derivatives as a potent class of anti-inflammatory, analgesic, and antipyretic agentsCitation13,Citation25,Citation26 since the development of antipyrine, the first pyrazoline derivative used in the management of pain, inflammation, and fever.
Figure 1. Chemical structure of previously synthesized anti-inflammatory pyrazoles and bipyrazoles (A–D) and the newly synthesized compounds (E–H).
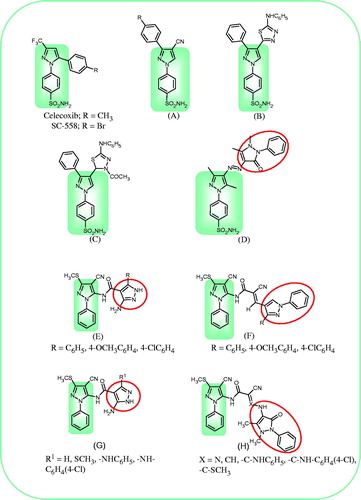
In the course of a research program aiming at finding out new lead structures that would act as potent anti-inflammatory agents, we have reported the synthesis and anti-inflammatory activities of some lead compounds comprising mainly the pyrazole counterpart substituted with various functionalities and attached to different heterocyclic ring systems through various linkagesCitation27–31. In particular, potential anti-inflammatory activity was displayed by the pyrazolyl derivatives (A–C; )Citation28,Citation30 and the bipyrazolyl lead structure (D; )Citation27.
In view of the above-mentioned facts, we report herein the synthesis, anti-inflammatory, and analgesic activities of some new amide-linked bipyrazoles. The designed compounds comprise the N-phenylpyrazole scaffold linked either to polysubstituted pyrazoles or to antipyrine moiety through different amide linkages and have the general formulae E-H (). The substitution pattern of the synthesized compounds was carefully selected so as to confer different electronic and lipophilic properties to the molecules. In addition to the targeted in vivo anti-inflammatory and analgesic activities, the in vitro COX1/COX2 inhibitory activity of the synthesized compounds and their effect on inducible nitric oxide synthase (iNOS) and COX-2 protein expression in lipopolysaccharide (LPS)-activated rat monocytes were also investigated.
Methods and materials
Chemistry
All reagents and solvents were purchased from commercial suppliers and, when necessary, were purified and dried by standard protocols. Melting points were determined in open glass capillaries using Stuart capillary melting point apparatus (Stuart Scientific Stone, Staffordshire, UK) and are uncorrected. Infrared (IR) spectra were recorded on Perkin-Elmer 1430 infrared spectrophotometer (PerkinElmer, Beaconsfield, UK) and measured by scale using KBr cell. NMR spectra were scanned on Varian Mercury VX-300 or Bruker-400 MHz spectrometer using tetramethylsilane (TMS) as internal standard and dimethyl sulfoxide d6 (DMSO-d6) as solvent (chemical shifts are given in δ ppm). Splitting patterns were designated as follows: s: singlet; d: doublet; m: multiplet. Mass spectra were run on a gas chromatograph/mass spectrometer Shimadzu GCMS-QP 2010 Plus (70 eV). Microanalyses were performed at the Regional Center for Mycology and Biotechnology, Al-Azhar University, Cairo, Egypt and the found values were within ±0.4% of the theoretical values. Follow-up of the reactions and checking the purity of the compounds was made by thin layer chromatography (TLC) on silica gel-precoated aluminum sheets (Type 60 GF254, Merck, Darmstadt, Germany) and the spots were detected by exposure to UV lamp at λ 254 nm for few seconds.
5-Amino-3-methylsulfanyl-1H-pyrazole-4-carbonitrile 2
A solution of 2-bis(methylthio)methylene)malononitrile (17 g, 100 mmol) and phenyl hydrazine (10.8 g, 100 mmol) in methanol (200 mL) was heated under reflux for 3 h. The reaction mixture was concentrated under reduced pressure and the separated solid product was filtered and recrystallized from methanol to give colorless needles (22 g, 96%), mp; 141–142 °C; reported mp: 136 °CCitation32.
2-Cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)acetamide 3
The aminopyrazole 2 (4.6 g, 20 mmol) was added to a mixture of cyanoacetic acid (2.55 g, 30 mmol) and acetic anhydride (30 mL) and heated at 50 °C for 5 h. The mixture was allowed to cool then poured into ice-cold water. The formed precipitate was filtered, washed with water, dried, and crystallized from methanol. Colorless needles (4.4 g, 74%), mp: 186–187 °C. IR (KBr, cm−1): 3253 (NH), 3048, 2950, 2907 (CH), 2224 (CN), 1704 (C=O), 1594 (C=N), 1563, 1524, 1459 (C=C), 1319, 1075 (C-S-C). 1H-NMR (300 MHz, DMSO-d6, δ ppm): 2.59 (s, 3H, SCH3), 3.99 (s, 2H, CH2), 7.46-7.60 (m, 5H, phenyl-H), 11.17 (s, 1H, NH, D2O exchangeable). 13C-NMR (75 MHz, δ ppm): 14.09 (CH3), 26.46 (CH2), 89.10 (pyrazole C4), 112.24, 115.58 (CN), 124.54 (phenyl C2,6), 129.35 (phenyl C4), 130.03 (phenyl C3,5), 137.41 (phenyl C1), 141.21 (pyrazole C3), 150.77 (pyrazole C5), 162.65 (C=O). Anal. Calcd for C14H11N5OS (297.34): C, 56.55; H, 3.73; N, 23.55. Found: C, 56.68; H, 3.72; N, 23.74.
General procedure for the synthesis of 2-Cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3-substituted acrylamides 4
To a solution of the cyanoacetamide 3 (2.97 g, 10 mmol) in absolute ethanol (25 mL) containing few drops of piperidine, the appropriate aldehyde (10 mmol) was added. The reaction mixture, was heated under reflux for 5 h. The solid products formed upon cooling was collected by filtration and crystallized from dioxane/water (3:1).
2-Cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3-phenylacrylamide 4a
Pale yellow crystals (2.77 g, 72%), mp: 205–207 °C. IR (KBr, cm−1): 3300 (NH), 3056, 2925, 2853 (CH), 2239 (CN), 1698 (C=O), 1622 (C=N), 1580, 1496, 1443 (C=C), 1306, 1075 (C-S-C). 1H-NMR (300 MHz, DMSO-d6, δ ppm): 2.57, 2.60 (2s, each 3H, 2SCH3, E and Z isomers), 7.01–8.11 (m, 22H, aromatic-H and = CH, E and Z isomers), 14.16, 14.35 (2s, each 1H, 2NH, D2O exchangeable, E and Z isomers). Anal. Calcd for C21H15N5OS (385.44): C, 65.44; H, 3.92; N, 18.17. Found: C, 65.18; H, 3.63; N, 18.53.
3–(4-Chlorophenyl)-2-cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)acrylamide 4b
White crystals (3.1 g, 74%), mp: 225–227 °C. IR (KBr, cm−1): 3289 (NH), 3057, 2925 (CH), 2236 (CN), 1605 (C=N), 1581, 1497, 1445 (C=C), 1307, 1107 (C-S-C). 1H-NMR (300 MHz, DMSO-d6, δ ppm): 2.59, 2.62 (2s, each 3H, 2SCH3, E and Z isomers), 7.03–8.12 (m, 20H, aromatic-H and = CH, E and Z isomers), 14.24, 14.43 (2s, each 1H, 2NH, D2O exchangeable, E and Z isomers). Anal. Calcd for C21H14ClN5OS (419.89): C, 60.07; H, 3.36; N, 16.68. Found: C, 60.22; H, 3.31; N, 16.76.
2-Cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3–(4-methoxyphenyl)acrylamide 4c
White crystals (3.0 g, 72.2%), mp: 218–219 °C. IR (KBr, cm−1): 3290 (NH), 3056, 2924, 2856 (CH), 2230 (CN), 1700 (C=O), 1620 (C=N), 1580, 1488, 1456 (C=C), 1302, 1078 (C-S-C), 1266, 1024 (C-O-C). 1H-NMR (300 MHz, DMSO-d6, δ ppm): 2.57, 2.60 (2s, each 3H, 2SCH3, E and Z isomers), 3.82, 3.84 (2s, each 3H, 2OCH3, E and Z isomers), 7.01–8.14 (m, 20H, aromatic-H and = CH, E and Z isomers), 14.19, 14.33 (2s, each 1H, 2NH, D2O exchangeable, E and Z isomers). Anal. Calcd for C22H17N5O2S (415.46): C, 63.60; H, 4.12; N, 16.86. Found: C, 63.35; H, 3.86; N, 16.55.
General procedure for the synthesis of 3-Amino-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-5-substituted-1H-pyrazole-4-carboxamides 5
A mixture of compounds 4a–c (10 mmol) and hydrazine hydrate 98% (0.50 g, 10 mmol), in ethanol (25 mL) was heated under reflux for 6 h. The solid products formed upon cooling were filtered, washed with ethanol, and crystallized from dioxane/water (4:1).
3-Amino-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-5-phenyl-1H-pyrazole-4-carboxamide 5a
White crystals (2.9 g, 70%), mp: 233–234 °C . IR (KBr, cm−1): 3451, 3327, 3187 (NH), 3060, 2923 (CH), 2227 (CN), 1673 (C=O), 1625 (C=N), 1580, 1500, 1463 (C=C), 1311, 1108 (C-S-C). 1H-NMR (300 MHz, DMSO-d6, δ ppm): 2.62 (s, 3H, SCH3), 5.42 (s, 2H, NH2, D2O exchangeable), 6.86-7.54 (m, 8H, aromatic-H), 8.01 (d, J = 7.8 Hz, 2H, phenyl C2,6-H), 10.60, 11.90 (2s, each 1H, 2NH, D2O exchangeable). 13C-NMR (75 MHz, δ ppm): 14.99 (CH3), 99.28, 99.39 (pyrazole C4), 116.14 (CN), 120.73, 120.79 (phenyl C2,6), 126.41 (aminopyrazole C5), 128.27, 128.55 (phenyl C4), 129.56, 129.62 (phenyl C3,5), 139.01, 139.17 (phenyl C1), 142.21 (cyanopyrazole C3), 155.22 (cyanopyrazole C5), 158.27 (aminopyrazole C3), 165.47 (C=O). MS (m/z, %): 359 (M+.– C3H6N), 77 (100). Anal. Calcd for C21H17N7OS (415.47): C, 60.71; H, 4.12; N, 23.60. Found: C, 60.87; H, 4.19; N, 23.72.
3-Amino-5-(4-chlorophenyl)-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-1H-pyrazole-4-carboxamide 5b
White crystals (3.15 g, 70%), mp: 263–264 °C. IR (KBr, cm−1): 3432, 3298, 3150 (NH), 3071, 2908 (CH), 2228 (CN), 1670 (C=O), 1643 (C=N), 1582, 1500 (C=C), 1315, 1097 (C-S-C). 1H-NMR (300 MHz, DMSO-d6, δ ppm): 2.51 (s, 3H, SCH3), 5.41 (s, 2H, NH2, D2O exchangeable), 7.0–7.55 (m, 7H, aromatic-H), 7.95 (d, J = 7.8 Hz, 2H, chlorophenyl C2,6-H), 10.65, 11.92 (2s, each 1H, 2NH, D2O exchangeable). 13C-NMR (75 MHz, δ ppm): 14.49 (CH3), 98.62 (two pyrazole C4), 116.15 (CN), 120.14 (phenyl C2,6), 125.77 (aminopyrazole C5), 127.67 (phenyl C4), 128.87 (chlorophenyl C2,3,5,6), 128.89 (phenyl C3,5), 129.65 (chlorophenyl C1), 137.69 (chlorophenyl C4), 138.25 (phenyl C1), 141.51 (cyanopyrazole C3), 154.43 (cyanopyrazole C5), 157.53 (aminopyrazole C3), 164.45 (C=O). MS (m/z, %): 393 (M+.– C3H6N, 22.1), 64 (100). Anal. Calcd for C21H16ClN7OS (449.92): C, 56.06; H, 3.58; N, 21.79. Found: C, 56.18; H, 3.62; N, 21.95.
3-Amino-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-5-(4-methoxyphenyl)-1H-pyrazole-4-carboxamide 5c
White crystals (3.0 g, 68%) mp: 254–256 °C. IR (KBr, cm−1): 3444, 3309, 3168 (NH), 3059, 2924, 2856 (CH), 2227 (CN), 1675 (C=O), 1628 (C=N), 1586, 1525, 1468 (C=C), 1310, 1112 (C-S-C), 1262, 1229 (C-O-C). 1H-NMR (300 MHz, DMSO-d6, δ ppm): 2.55 (s, 3H, SCH3), 3.89 (s, 3H, OCH3), 5.46 (s, 2H, NH2, D2O exchangeable), 7.01–7.58 (m, 7H, aromatic-H), 8.0 (d, J = 8.0 Hz, 2H, methoxyphenyl C2,6-H), 10.58, 11.90 (2s, each 1H, 2NH, D2O exchangeable). 13C-NMR (75 MHz, δ ppm): 14.76 (CH3), 98.46, 99.24 (pyrazole C4), 113.99 (methoxyphenyl C3,5), 116.18 (CN), 120.58 (phenyl C2,6), 121.68 (methoxyphenyl C1), 126.43 (aminopyrazole C5), 128.34 (phenyl C4), 129.40 (phenyl C3,5), 131.03 (methoxyphenyl C2,6), 139.04 (phenyl C1), 142.26 (cyanopyrazole C3), 155.40 (cyanopyrazole C5), 157.88 (aminopyrazole C3), 160.12 (methoxyphenyl C4), 165.48 (C=O). Anal. Calcd for C22H19N7O2S (445.49): C, 59.31; H, 4.30; N, 22.01. Found: C, 59.67; H, 4.19; N, 22.32.
2-Cyano-N-[4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl]-2-[(2,3-dihydro-1,5-dimethyl-3-oxo-2-phenyl--1H-pyrazol-4-yl)hydrazono]acetamide 6
A well-stirred solution of 4-aminoantipyrine (1.02 g, 5 mmol) in 2 N HCl (1.5 mL) was cooled in ice salt bath and diazotized with 1 N NaNO2 solution (0.35 g, 5 mmol) in 2 mL water. The resulting cold solution of the diazonium salt was then added to a cold solution of the cyanoacetamide 3 (1.48 g, 5 mmol) in ethanol (20 mL) in the presence of sodium acetate (0.43 g, 5.2 mmol). The reaction mixture was then stirred at room temperature for 1 h and the separated product was filtered, washed with water then ethanol, dried, and crystallized from dimethylformamide (DMF)/ethanol (2:1). Red crystals (1.86 g, 73%), mp: 139–140 °C. IR (KBr, cm−1): 3308 (NH), 3062, 2924 (CH), 2215 (CN), 1652 (C=O), 1592 (C=N), 1496, 1456 (C=C), 1294, 1045 (C-S-C). 1H-NMR (300 MHz, DMSO-d6, δ ppm): 2.49 (s, 3H, SCH3), 2.56 (s, 3H, C-CH3), 3.23 (s, 3H, N-CH3), 7.30–7.57 (m, 10H, aromatic-H), 10.61, 13.90 (2s, each 1H, 2NH, D2O exchangeable). 13C-NMR (75 MHz, δ ppm): 10.60 (C-CH3), 14.49 (SCH3), 35.29 (NCH3) 96.52 (pyrazole C4), 99.68 (pyrazolinone C4), 106.21 (acrylamide C2), 113.40, 117.99 (CN), 125.39, 125.72 (phenyl C2,6), 127.56, 127.77 (phenyl C4), 129.07 (pyrazolinone C5), 129.22, 129.55 (phenyl C3,5), 133.60, 139.62 (phenyl C1), 147.91 (pyrazole C3), 150.24 (pyrazole C5), 156.63, 157.38 (C=O). MS (m/z, %): 374 (M+ – C6H7N3O, 3.87), 56 (100). Anal. Calcd for C25H21N9O2S (511.56): C, 58.70; H, 4.14; N, 24.64. Found: C, 58.87; H, 4.17; N, 24.82.
General procedure for the synthesis of 2-cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3-substituted acrylamides 7
To a solution of compound 3 (2.97 g, 10 mmol) in absolute ethanol (15 mL) containing three drops of sodium hydroxide (10%), the appropriate 3-aryl-1-phenyl-1H-pyrazole-4-carboxaldehydes (10 mmol) was added. The reaction mixture was heated under reflux for 2 h and then allowed to cool. The obtained precipitate was collected by filtration, washed with ethanol, dried, and recrystallized from a mixture of DMF/ethanol (1:1).
2-Cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3-(1,3-diphenyl-1H-pyrazol-4-yl)acrylamide 7a
Yellowish white crystals (3.9 g, 74%), mp: 278–279 °C. IR (KBr, cm−1): 3373 (NH), 3061, 2926 (CH), 2228 (CN), 1697 (C=O), 1630 (C=N), 1582, 1523, 1498, 1458 (C=C), 1297, 1067 (C-S-C). 1H-NMR (300 MHz, DMSO-d6, δ ppm): 2.49 (s, 3H, SCH3), 7.46-7.64 (m, 13H, aromatic-H), 7.93 (d, J = 8.1 Hz, 2H, phenyl C2,6-H), 8.1 (s, 1H, methine-H), 9.21(s, 1H, pyrazole C5-H), 11.12 (s, 1H, NH, D2O exchangeable). 13C-NMR (75 MHz, δ ppm): 14.09 (SCH3), 90.32, 102.45 (pyrazole C4), 112.30 (acrylamide C2), 114.73, 116.55 (CN), 120.20, 124.17 (phenyl C2,6), 128.63, 129.38 (phenyl C4), 129.43 (phenyl C2,6), 129.52 (phenyl C4), 129.87, 130.0, 130.30 (phenyl C3,5), 130.36 (diphenylpyrazole C5), 131.10, 137.59, 138.98 (phenyl C1), 141.82, 144.79 (pyrazole C3), 150.77 (cyanopyrazole C5), 155.55 (acrylamide C3), 161.39 (C=O). MS (m/z, %): 527 (M+.,8.4), 298 (100). Anal. Calcd for C30H21N7OS (527.6): C, 68.29; H, 4.01; N, 18.58. Found: C, 68.52; H, 4.02; N, 18.65.
3-(3–(4-Chlorophenyl)-1-phenyl-1H-pyrazol-4-yl)-2-cyano-N-(4-cyano-3-methylsulfanyl-1-phenyl-1H-pyrazol-5-yl)acrylamide 7b
Yellowish white crystals (4.38 g, 78%), mp: 248–249 °C . IR (KBr, cm−1): 3376 (NH), 3058, 2926 (CH), 2232 (CN), 1704 (C=O), 1631 (C=N), 1587, 1501, 1458 (C=C), 1297, 1095 (C-S-C). 1H-NMR (300 MHz, DMSO-d6, δ ppm): 2.52 (s, 3H, SCH3), 7.48-7.65 (m, 12H, aromatic-H), 7.95 (d, J = 8.1 Hz, 2H, phenyl C2,6-H), 8.22 (s, 1H, methine-H), 9.21(s, 1H, pyrazole C5-H), 11.18 (s, 1H, NH, D2O exchangeable). 13C-NMR (75 MHz, δ ppm): 14.16 (SCH3), 90.42, 102.63 (pyrazole C4), 112.36 (CN), 114.84 (acrylamide C2), 116.56 (CN), 120.18, 124.24 (phenyl C2,6), 128.69, 129.41 (phenyl C4), 129.45 (chlorophenyl C2,6), 129.76, 129.90 (phenyl C3,5), 130.08 (chlorophenyl C3,5), 130.49 (diphenylpyrazole C5), 131.53 (chlorophenyl C1), 134.65 (chlorophenyl C4), 138.68, 139.23 (phenyl C1), 141.94, 145.02 (pyrazole C3), 150.79 (cyanopyrazole C5), 155.64 (acrylamide C3), 161.43 (C=O). MS (m/z, %): 563 (M+. + 2, 3.8), 561 (M+., 9.2), 332 (100). Anal. Calcd for C30H20ClN7OS (562.04): C, 64.11; H, 3.59; N, 17.44. Found: C, 64.18; H, 3.54; N, 17.71.
2-Cyano-N-(4-cyano-3-methylsulfanyl-1-phenyl-1H-pyrazol-5-yl)-3-(3–(4-methoxyphenyl)-1-phenyl-1H-pyrazol-4-yl)acrylamide 7c
Yellowish white needles (4.18 g, 75%), mp: 270–272 °C. IR (KBr, cm−1): 3368 (NH), 3064, 2928 (CH), 2230 (CN), 1700 (C=O), 1630 (C=N), 1584, 1511, 1488, 1453 (C=C), 1295, 1075 (C-S-C), 1260, 1025 (C-O-C). 1H-NMR (300 MHz, DMSO-d6, δ ppm): 2.49 (s, 3H, SCH3), 3.80 (s, 3H, OCH3), 7.45-7.67 (m, 12H, aromatic-H), 7.93 (d, J = 8.1 Hz, 2H, phenyl C2,6-H), 8.15 (s, 1H, methine-H), 9.20 (s, 1H, pyrazole C5-H), 11.15 (s, 1H, NH, D2O exchangeable). 13C-NMR (75 MHz, δ ppm): 14.11 (SCH3), 55.63 (OCH3), 90.38, 102.51 (pyrazole C4), 112.32 (acrylamide C2), 114.13 (methoxyphenyl C3,5), 114.68, 116.46 (CN), 120.19 (phenyl C2,6), 121.89 (methoxyphenyl C1), 123.18 (phenyl C2,6), 126.68, 126.84 (phenyl C4), 129.96, 130.18 (phenyl C3,5), 130.33 (diphenylpyrazole C5), 130.96 (methoxyphenyl C2,6), 139.73, 139.78 (phenyl C1), 141.88, 145.02 (pyrazole C3), 150.79 (cyanopyrazole C5), 155.61 (acrylamide C3), 160.12 (methoxyphenyl C4), 161.33 (C=O). Anal. Calcd for C31H23N7O2S (557.62): C, 66.77; H, 4.16; N, 17.58. Found: C, 66.43; H, 4.06; N, 17.78.
2-Cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3-(dimethylamino)acrylamide 9
A mixture of 3 (1.5 g, 5 mmol), DMF dimethyl acetal (6.55 g, 7.36 mL, 5.5 mmol) in dry dioxane (25 mL) was heated under reflux for 4 h. The separated solid product obtained on standing at room temperature was collected by filtration, washed with ethanol, and recrystallized from DMF/ethanol (3:1). White needles (1.51 g, 85%), mp: 216–218 °C. IR (KBr, cm−1): 3236 (NH), 3011, 2927 (CH), 2222, 2196 (CN), 1682 (C=O), 1612 (C=N), 1569, 1532, 1495, 1458 (C=C), 1274, 1072 (C-S-C). 1H-NMR (300 MHz, DMSO-d6, δ ppm): 2.49 (s, 3H, SCH3), 3.14, 3.21 (2s, each 3H, N(CH3)2), 7.38-7.52 (m, 5H, phenyl-H), 7.68 (s, 1H, =CH), 9.72 (s, 1H, NH, D2O exchangeable). Anal. Calcd for C17H16N6OS (352.41): C, 57.94; H, 4.58; N, 23.85. Found: C, 57.61; H, 4.63; N, 23.53.
5-Amino-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-1H-pyrazole-4-carboxamide 10
Method I: A mixture of the enaminonitrile 9 (3.52 g, 10 mmol) and hydrazine hydrate 98% (0.50 g, 10 mmol) in dry dioxane (20 mL) was heated under reflux for 5 h. The reaction mixture was concentrated and poured into ice-cold water. The separated solid product was collected by filtration, washed with ethanol, and crystallized from dioxane/ethanol (2:1). Beige crystals (2.3 g, 68%), mp: 195–196 °C.
Method II: A mixture of the ethoxyacrylamide 12 (1.0 g, 2.8 mmol) and hydrazine hydrate 98% (0.14 g, 2.8 mmol) in absolute ethanol (20 mL) was heated under reflux for 5 h. The reaction mixture was cooled to room temperature and the separated solid product was collected by filtration, washed with ethanol, and crystallized from dioxane/ethanol (2:1). Beige crystals (0.69 g, 72%). IR (KBr, cm−1): 3328, 3201 (NH), 3022, 2924 (CH), 2223 (CN), 1650 (C=O), 1612 (C=N), 1548, 1504, 1460 (C=C), 1301, 1113 (C-S-C). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 2.49 (s, 3H, SCH3), 6.09 (s, 2H, NH2, D2O exchangeable), 7.38-7.58 (m, 5H, phenyl-H), 7.78 (s, 1H, pyrazole C3-H), 10.10, 11.92 (2s, each 1H, 2NH, D2O exchangeable). 13C-NMR (100 MHz, δ ppm): 14.11 (SCH3), 90.46 (cyanopyrazole C4), 112.73 (amino pyrazole C4), 114.49 (CN), 124.0 (phenyl C2,6), 129.95 (phenyl C4), 130.29 (phenyl C3,5), 137.77 (phenyl C1), 143.01 (cyanopyrazole C3), 149.37 (aminopyrazole C3), 150.39 (cyanopyrazole C5), 152.80 (aminopyrazole C5), 163.06 (C=O). MS (m/z, %): 339 (M+., 1.6), 64 (100). Anal. Calcd for C15H13N7OS (339.38): C, 53.09; H, 3.86; N, 28.89. Found: C, 53.21; H, 3.63; N, 28.53.
2-Cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3-(2,3-dihydro-1,5-dimethyl-3-oxo-2-phenyl-1H-pyrazol-4-ylamino)acrylamide 11
Method I: A mixture of 9 (1.76 g; 5 mmol) and 4-aminoantipyrine (1.0 g; 5 mmol) was fused at 170 °C in an oil bath for 1 h. The reaction mixture was left to cool and the obtained solid residue was triturated with hot ethanol (20 mL) and filtered, then crystallized from DMF/ethanol (4:1). Grayish crystals (1.91 g, 75%), mp: 261–262 °C.
Method II: A mixture of the ethoxyacrylamide 12 (1.06 g; 3 mmol) and 4-amino antipyrine (0.61 g; 3 mmol) was fused at 170 °C in an oil bath for 1 h. The reaction mixture was left to cool and the obtained solid residue was triturated with hot ethanol (20 mL) and filtered, then crystallized from DMF/ethanol (4:1). Grayish crystals (1.12 g, 73.2%). IR (KBr, cm−1): 3339, 3287 (NH), 3045, 2922 (CH), 2227 (CN), 1666, 1631 (C=O), 1589 (C=N), 1560, 1497, 1465 (C=C), 1309, 1066 (C-S-C). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 2.43 (s, 3H, C-CH3), 2.49 (s, 3H, SCH3), 3.17 (s, 3H, N-CH3), 7.39-7.57 (m, 11H, aromatic-H and = CH), 11.23, 12.74 (2s, each 1H, 2NH, D2O exchangeable). 13C-NMR (100 MHz, δ ppm): 10.36 (C-CH3), 12.97 (SCH3), 35.83 (NCH3), 94.67 (pyrazole C4), 99.17 (acrylamide C2), 109.95 (pyrazolinone C4), 116.85 (two CN), 124.20, 125.75 (phenyl C2,6), 127.98, 128.26 (phenyl C4), 129.65, 129.75 (phenyl C3,5), 134.65 (pyrazolinone C5), 137.50, 138.49 (phenyl C1), 143.06 (pyrazole C3), 145.98 (pyrazole C5), 150.85 (acrylamide C3), 151.22, 160.55 (C=O). MS (m/z, %): 510 (M+. – C4H2, 3.8), 56 (100). Anal. Calcd for C26H22N8O2S (510.57): C, 61.16; H, 4.34; N, 21.95. Found: C, 61.34; H, 4.31; N, 22.17.
2-Cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3-ethoxyacrylamide 12
A mixture of the cyanoacetamide 3 (2.97 g, 10 mmol), triethyl orthoformate (1.48 g, 1.66 mL, 10 mmol) in dry dioxane (5 mL) containing few drops of piperidine was heated under reflux for 5 h. The reaction mixture was left to attain room temperature then poured into ice-cold water and stirred for 30 min. The separated solid product was filtered, washed with water, dried, and crystallized from ethanol. White needles (2.3 g, 65%), mp: 237–238 °C. IR (KBr, cm−1): 3197 (NH), 3079, 3036, 2931 (CH), 2226 (CN), 1685 (C=O), 1593 (C=N), 1527, 1463, 1430 (C=C), 1316, 1072 (C-S-C), 1264, 1024 (C-O-C). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 1.23 (t, J = 6.9 Hz, 3H, CH3), 2.59 (s, 3H, SCH3), 3.99 (q, J = 6.9 Hz, 2H, CH2), 7.49-7.56 (m, 5H, phenyl-H), 7.94 (s, 1H, =CH), 10.58 (s, 1H, NH, D2O exchangeable). Anal. Calcd for C17H15N5O2S (353.4): C, 57.78; H, 4.28; N, 19.82. Found: C, 57.41; H, 4.53; N, 19.49.
N1-(4-Cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)malonamide 13
This compound was obtained as a major product in the first trial to prepare compounds 14a, b upon treatment of the cyanoacetamide 3 (1.19 g; 4 mmol) with the selected isothiocyanate (4 mmol) in DMF (8 mL) containing finely divided KOH (0.22 g; 4 mmol) at room temperature followed by the addition of dimethyl sulfate (0.5 g, 0.38 mL, 4 mmol) and stirring for an overnight at room temperature. It was crystallized from DMF/ethanol (3:1). White crystals (1.07 g, 85%), mp: 259–260 °C. IR (KBr, cm−1): 3433, 3328 (NH), 3066, 2984, 2924 (CH), 2213 (CN), 1638 (C=O), 1592 (C=N), 1502, 1463, 1434 (C=C), 1316, 1115 (C-S-C). 1H-NMR (400 MHz, DMSO-d6, δ ppm): 2.68 (s, 3H, SCH3), 3.98 (s, 2H, CH2), 7.15 (s, 2H, NH2, D2O exchangeable), 7.33 (t, J = 8.2 Hz, 1H, phenyl C4-H), 7.53 (t, J = 7.6 Hz, 2H, phenyl C3,5-H), 8.13 (dd, J = 8.2 Hz, 1. 0 Hz, 2H, phenyl C2,6-H), 11.4 (s, 1H, NH, D2O-exchangeable). 13C-NMR (100 MHz, δ ppm): 15.73 (CH3), 54.89 (CH2), 100.64 (pyrazole C4), 115.50 (CN), 120.91 (phenyl C2,6), 126.59 (phenyl C4), 129.59 (phenyl C3,5), 138.96 (phenyl C1), 142.07 (pyrazole C3), 150.74 (pyrazole C5), 154.07, 166.55 (C=O). Anal. Calcd for C14H13N5O2S (315.35): C, 53.32; H, 4.16; N, 22.21. Found: C, 53.41; H, 4.26; N, 22.49.
General procedure for the synthesis of 2-Cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3-(methylsulfanyl)-3-(substituted amino) acrylamides 14
To a well-stirred ice-cold suspension of finely divided KOH (0.22 g; 4 mmol) in dry DMF (8 mL) was added the cyanoacetamide derivative 3 (1.19 g; 4 mmol) followed by phenyl; or 4-chlorophenyl isothiocyanate (4 mmol). The mixture was stirred at 0–5 °C for 2 h, left to attain room temperature then stirring is continued for an overnight. Dimethyl sulfate (0.5 g, 0.38 mL, 4 mmol) was then added and stirring was maintained for 24 h. The reaction mixture was poured into ice-cold water and the separated product was filtered, washed with ethanol, dried, and crystallized from DMF/ethanol (2:1).
2-Cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3-(methylsulfanyl)-3-(phenylamino)acrylamide 14a
Yellowish crystals (1.0 g, 56%), mp: 197–198 °C. IR (KBr, cm−1): 3215, 3195 (NH), 3062, 2926 (CH), 2226, 2195 (CN), 1630 (C=O), 1597 (C=N), 1542, 1502, 1448 (C=C), 1275, 1245, 1086 (C-S-C). 1H-NMR (300 MHz, DMSO-d6, δ ppm): 2.34, 2.59 (2s, each 3H, 2SCH3), 6.78-7.55 (m, 10H, aromatic-H), 8.79, 11.43 (2s, each1H, 2NH, D2O exchangeable). Anal. Calcd for C22H18N6OS2 (446.55): C, 59.17; H, 4.06; N, 18.82. Found: C, 59.28; H, 4.12; N, 18.95.
3–(4-Chlorophenylamino)-2-cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3-(methylsulfanyl)acrylamide 14b
Beige crystals (1.25 g, 65%), mp: 204–206 °C . IR (KBr, cm−1): 3224, 3197 (NH), 3059, 2925 (CH), 2226, 2196 (CN), 1630 (C=O), 1591 (C=N), 1540, 1501, 1448 (C=C), 1280, 1245, 1088 (C-S-C). 1H-NMR (300 MHz, DMSO-d6, δ ppm): 2.24, 2.60 (2s, each 3H, 2SCH3), 7.33-7.51 (m, 5H, phenyl-H), 7.55, 7.56 (2d, J = 7.8 Hz, each 2H, chlorophenyl-H), 9.25, 11.48 (2s, each1H, 2NH, D2O exchangeable). Anal. Calcd for C22H17ClN6OS2 (480.99): C, 54.94; H, 3.56; N, 17.47. Found: C, 55.02; H, 3.59; N, 17.56.
General procedure for the synthesis of 5-Amino-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3-(substituted amino)-1H-pyrazole-4-carboxamides 15
A mixture of the acrylamide 14 (5 mmol) and hydrazine hydrate 98% (0.25 g; 5 mmol) was heated on a water bath for 2 h. Then, ethanol (20 mL) was added and the reaction mixture was refluxed for further 2 h. The reaction mixture was left to stand overnight and the separated solid product was filtered, washed with ethanol, dried, and crystallized from ethanol.
5-Amino-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3-(phenylamino)-1H-pyrazole-4-carboxamide 15a
White needles (1.46 g, 68%), mp: 199–200 °C. IR (KBr, cm−1): 3373, 3225, 3163 (NH), 3068, 2926 (CH), 2222 (CN), 1681 (C=O), 1626 (C=N), 1570, 1515, 1488 (C=C), 1275, 1078 (C-S-C). 1H-NMR (300 MHz, DMSO-d6) δ 2.59 (s, 3H, SCH3), 6.12 (s, 2H, NH2, D2O exchangeable), 6.76 (t, J = 7.8 Hz, 1H, phenylamino C4-H), 7.05-7.59 (m, 9H, aromatic-H), 8.05, 9.31, 11.30 (3s, each 1H, 3NH, D2O exchangeable). 13C-NMR (75 MHz, DMSO-d6) δ 14.16 (CH3), 87.10, 88.87 (pyrazole C4), 112.94 (CN), 116.60 (phenylamino C2,6), 119.86 (phenylamino C4), 124.34 (phenyl C2,6), 129.19 (phenyl C4), 129.21 (phenyl C3,5), 129.92 (phenylamino C3,5), 137.51 (phenyl C1), 142.65 (phenylamino C1), 143.53, 149.92 (pyrazole C3), 150.51, 150.69 (pyrazole C5), 162.62 (C=O). MS (m/z, %): 256 (M+. – C9H10N4, 2.8), 80 (100). Anal. Calcd for C21H18N8OS (430.49): C, 58.59; H, 4.21; N, 26.03. Found: C, 58.77; H, 4.18; N, 26.28.
3–(4-Chlorophenylamino)-5-amino-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-1H-pyrazole-4-carboxamide 15b
White needles (1.63 g, 70.1%), mp: 215–216 °C . IR (KBr, cm−1): 3347, 3220, 3166 (NH), 3062, 2956 (CH), 2224 (CN), 1686 (C=O), 1621 (C=N), 1567, 1517, 1490 (C=C), 1313, 1077 (C-S-C). 1H-NMR (300 MHz, DMSO-d6) δ 2.60 (s, 3H, SCH3), 6.20 (s, 2H, NH2, D2O exchangeable), 7.19-7.55 (m, 9H, aromatic-H), 8.38, 9.40, 11.44 (3s, each 1H, 3NH, D2O exchangeable). 13C-NMR (75 MHz, DMSO-d6) δ 13.69 (CH3), 86.55, 88.40 (pyrazole C4), 112.29 (CN), 117.43 (chlorophenyl C2,6), 122.62 (chlorophenyl C4), 123.65 (phenyl C2,6), 123.90 (phenyl C4), 128.21 (chlorophenyl C3,5), 129.17 (phenyl C3,5), 136.84 (phenyl C1), 141.74 (chlorophenyl C1), 141.96, 149.34 (pyrazole C3), 149.60, 149.80 (pyrazole C5), 161.89 (C=O). MS (m/z, %): 464 (M+ 0.13), 208 (100). Anal. Calcd for C21H17ClN8OS (464.93): C, 54.25; H, 3.69; N, 24.10. Found: C, 54.41; H, 3.63; N, 24.49.
General procedure for the synthesis of 2-Cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3-(2,3-dihydro-1,5-dimethyl-3-oxo-2-phenyl-1H-pyrazol-4-ylamino)-3-(substituted amino)acrylamides 16
A mixture of the acrylamide 14 (5 mmol) and 4-aminoantipyrine (1 g; 5 mmol) was fused at 170–180 °C in oil bath for 2 h. Ethanol (20 mL) was added and the reaction mixture was refluxed for further 2 h then concentrated and left to stand for an overnight. The separated solid product was filtered, washed with ethanol, dried, and crystallized from ethanol–DMF (4:1).
2-Cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3-(2,3-dihydro-1,5-dimethyl-3-oxo-2-phenyl-1H-pyrazol-4-ylamino)-3-(phenylamino)acrylamide 16a
Light brown crystals (2.1 g, 70%), mp: 279–280 °C. IR (KBr, cm−1): 3376, 3312, 3187 (NH), 3038, 2921 (CH), 2225 (CN), 1662, 1631 (C=O), 1590 (C=N), 1560, 1498 (C=C), 1310, 1067 (C-S-C). 1H-NMR (300 MHz, DMSO-d6, δ ppm): 2.39 (s, 3H, C-CH3), 2.51 (s, 3H, SCH3), 3.15 (s, 3H, N-CH3), 7.33-7.57 (m, 15 H, aromatic-H), 7.69, 11.20, 12.40 (3s, each 1H, 3NH, D2O exchangeable). 13C-NMR (75 MHz, δ ppm): 13.65 (C-CH3), 14.95 (SCH3), 35.84 (N-CH3), 97.51 (pyrazole C4), 99.15 (acrylamide C2), 99.55 (pyrazolinone C4), 112.80, 116.84 (CN), 120.36 (pyrazolinone phenyl C2,6), 123.73 (phenylamino C2,6), 124.23 (phenylamino C4), 125.73 (pyrazolinone phenyl C4), 127.21 (phenyl C4), 128.01 (pyrazolinone C5), 129.13 (phenyl C2,6), 129.40 (pyrazolinone phenyl C3,5), 129.65 (phenyl C3,5), 129.80 (phenylamino C3,5), 139.07 (pyrazolinone phenyl C1), 139.43 (phenyl C1), 142.26 (phenylamino C1), 148.69 (pyrazole C3), 150.21 (pyrazole C5), 155.39, 157.48 (C=O), 161.50 (acrylamide C3). MS (m/z, %): 460 (M+. -C10H7N, 3.7), 56 (100). Anal. Calcd for C32H27N9O2S (601.68): C, 63.88; H, 4.52; N, 20.95. Found: C, 63.97; H, 4.61; N, 21.17.
3-(Chlorophenylamino)-2-cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3-(2,3-dihydro-1,5-dimethyl-3-oxo-2-phenyl-1H-pyrazol-4-ylamino)acrylamide 16b
Light brown crystals (2.35 g, 74%), mp: 294–295 °C. IR (KBr, cm−1): 3376, 3312, 3187 (NH), 3041, 2923 (CH), 2224 (CN), 1662, 1631 (C=O), 1590 (C=N), 1561, 1496 (C=C), 1308, 1093 (C-S-C). 1H-NMR (300 MHz, DMSO-d6, δ ppm): 2.38 (s, 3H, C-CH3), 2.52 (s, 3H, S-CH3), 3.17 (s, 3H, N-CH3), 7.35-7.59 (m, 14 H, aromatic-H), 7.82, 11.30, 12.43 (3s, each 1H, 3NH, D2O exchangeable). 13C-NMR (75 MHz, δ ppm): 13.58 (C-CH3), 14.94 (SCH3), 35.88 (N-CH3), 97.56 (pyrazole C4), 99.25 (acrylamide C2), 99.73 (pyrazolinone C4), 113.04, 116.92 (CN), 120.56 (pyrazolinone phenyl C2,6), 123.94 (chlorophenylamino C2,6), 124.38 (pyrazolinone phenyl C4), 125.73 (phenyl C4), 127.71 (phenyl C2,6), 128.13 (Chlorophenylamino C4), 129.24 (pyrazolinone C5), 129.54 (pyrazolinone phenyl C3,5), 129.83 (phenyl C3,5), 129.89 (phenylamino C3,5), 139.14 (pyrazolinonephenyl C1), 139.69 (phenyl C1), 142.45 (phenylamino C1), 149.02 (pyrazole C3), 150.28 (pyrazole C5), 155.44, 157.57 (C=O), 161.53 (acrylamide C3). MS (m/z, %): 460 (M+. – C10H6ClN, 11.3), 56 (100). Anal. Calcd for C32H26ClN9O2S (636.13): C, 60.42; H, 4.12; N, 19.82. Found: C, 60.19; H, 4.33; N, 19.59.
2-Cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3,3-bis(methylsulfanyl)acrylamide 17
To a well-stirred ice-cold suspension of 3 (2.97 g, 10 mmol) and finely powdered potassium hydroxide (0.56 g, 10 mmol) in dry dimethyformamide (10 mL), carbon disulfide (0.91 g, 0.72 mL, 12 mmol) was gradually added over a period of 1 h. The reaction mixture was stirred at room temperature for 3 h, then cooled again to 0 °C and treated with dimethyl sulfate (1.5 g, 1.14 mL, 12 mmol) dropwise then stirred at room temperature for an additional 6 h. The reaction mixture was poured into ice/water and the resulting precipitate was filtered off, dried, and crystallized from dioxane. Yellowish crystals (3.0 g, 75%), mp: 230–232 °C . IR (KBr, cm−1): 3338 (NH), 3038, 2995, 2925, 2855 (CH), 2206 (CN), 1660, (C=O), 1599 (C=N), 1567, 1515, 1455 (C=C), 1303, 1118 (C-S-C). 1H-NMR (300 MHz, DMSO-d6, δ ppm): 2.60, 2.62, 2.71 (3s, each 3H, 3SCH3), 7.45-7.59 (m, 3H, phenyl C3,4,5-H), 7.65 (d, J = 7.2 Hz, 2H, phenyl C2,6-H), 11.81(s, 1H, NH, D2O exchangeable). Anal. Calcd for C17H15N5OS3 (401.53): C, 50.85; H, 3.77; N, 17.44. Found: C, 51.04; H, 3.72; N, 17.61.
5-Amino-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3-(methylsulfanyl)-1H-pyrazole-4-carboxamide 18
A mixture of the acrylamide 17 (2 g, 5 mmol) and hydrazine hydrate 80% (0.25 g, 5 mmol) in ethanol (20 mL) was heated under reflux for 2 h. The reaction mixture was left to cool and the precipitated solid product was filtered, dried, and crystallized from dioxane. Beige crystals (1.34 g, 70%), mp: 235–236 °C. IR (KBr, cm−1): 3353, 3299, 3208 (NH), 3058, 2922 (CH), 2225 (CN), 1646, (C=O), 1613 (C=N), 1548, 1520, 1434 (C=C), 1289, 1213, 1106, 1031 (C-S-C). 1H-NMR (300 MHz, DMSO-d6, δ ppm): 2.59, 2.65 (2s, each 3H, 2SCH3), 5.84 (s, 2H, NH2, D2O exchangeable), 7.30–7.78 (m, 5H, phenyl-H), 9.55, 11.25 (2s, each 1H, 2NH, D2O exchangeable). 13C-NMR (75 MHz, δ ppm): 14.12 (CH3), 92.10, 94.87 (pyrazole C4), 123.24 (phenyl C2,6), 128.89 (phenyl C4), 129.23 (phenyl C3,5), 138.45(phenyl C1), 141.15(aminopyrazole C5), 141.86 (pyrazole C3), 150.56, 152.69 (pyrazole C5), 162.62 (C=O). MS (m/z, %): 385 (M+., 3.0), 129 (100). Anal. Calcd for C16H15N7OS2 (385.47): C, 49.85; H, 3.92; N, 25.44. Found: C, 49.48; H, 4.13; N, 25.29.
2-Cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3-(2,3-dihydro-1,5-dimethyl-3-oxo-2-phenyl-1H-pyrazol-4-ylamino)- 3-(methylsulfanyl)acrylamide 19
A mixture of 17 (2 g; 5 mmol) and 4-aminoantipyrine (1 g; 5 mmol) was fused at 140 °C in an oil bath for 1 h. The reaction mixture was left to cool and the obtained solid residue was triturated with ethanol (20 mL) and filtered, then crystallized from dioxane. Brown fine crystals (1.9 g, 68.6%), mp: 221–222 °C. IR (KBr, cm−1): 3334, 3286 (NH), 3055, 2926 (CH), 2227 (CN), 1665, 1630 (C=O), 1590 (C=N), 1550, 1498, 1450 (C=C), 1310, 1023 (C-S-C). 1H-NMR (300 MHz, DMSO-d6, δ ppm): 2.59, 2.65, 2.69 (3s, each 3H, 2SCH3, and C-CH3), 3.15 (s, 3H, N-CH3), 7.30–7.56 (m, 10H, aromatic-H), 11.20, 12.68 (2s, each 1H, 2NH, D2O exchangeable). 13C-NMR (75 MHz, δ ppm): 11.29 (C-CH3), 13.14 (SCH3), 35.78 (NCH3), 92.01 (pyrazole C4), 99.14 (acrylamide C2), 109.58 (pyrazolinone C4), 113.16, 116.89 (CN), 123.99 (phenyl C2,6), 125.46 (phenyl C4), 127.88 (phenyl C2,6), 128.29 (phenyl C4), 129.54, 129.83 (phenyl C3,5), 129.95 (pyrazolinone C5), 137.58, 138.64 (phenyl C1), 142.98 (pyrazole C3), 146.02 (pyrazole C5), 155.20, 157.63 (C=O), 163.06 (acrylamide C3). MS (m/z, %): 459 (M+. – C5H5S, 11.3), 56 (100). Anal. Calcd for C27H24N8O2S2 (556.66): C, 58.26; H, 4.35; N, 20.13. Found: C, 58.41; H, 4.63; N, 20.49.
Biological screening
Carrageenan-induced mouse paw edema
Swiss albino mice of both sexes, weighing 20–35 g, were used throughout the work. The mice were kept in an animal house under standard conditions of light and temperature with free access to food and water. The animals were randomly divided into groups of six mice each. In the control group, only the vehicle (polyethylene glycol (PEG), acetone, and DMSO) was administered. Positive control groups were treated with diclofenac Na (50 mg/kg body weight) as the reference drug. The tested compounds were given in equimolar doses to that of diclofenac. All drugs and the vehicle were administered intraperitoneally. After 1 h, the mice were challenged with subcutaneous injection of 0.025 mL of 2% w/v solution of carrageenan into the plantar side of the right hind paw. The volume of the injected paw was determined plethysmographically using a pressure transducer transforming the volume being increased by immersion of the paw into a proportional voltage recorded on a computer. The paw volume was measured immediately after carrageenan injection (zero time) and 4 h laterCitation33. The difference between the two readings gave the actual edema volumes to calculate the percentage protection. The anti-inflammatory activity of the test compounds relative to that of diclofenac was also calculated.
The percentage protection against inflammation was calculated as follows:
where, Vc is the increase in paw volume in the absence of test compound (control) and Vd is the increase in paw volume after injection of the test compound. Data were expressed as the mean ± SD. Significant difference between the control and treated groups was determined using one-way analysis of variance (ANOVA) followed by Tukey simultaneous comparison t-values. The difference in results was considered significant when p < 0.01.
Animal care and all experimental procedures used in this study were performed in accordance with the regulations and guidelines stipulated by the Institutional Animal Care and Use Guidelines at Beirut Arab University, Lebanon.
Gastric-ulcerogenic effect
After the employment of anti-inflammatory activity experiment, mice were killed under deep ether anesthesia and the stomach of each mouse was removed. An opening at the greater curvature was made and the stomach was cleaned by washing with cold saline and inspected for any evidence of hyperemia, hemorrhage, erosion, or ulcer. An arbitrary scale was used to calculate the ulcer index (UI) which indicates the severity of stomach lesionsCitation34. The UI of each group was calculated as the sum of the following three figures:
UI = UN + US + [UP/10]
UN = average of number of ulcers per animal
US = average of severity score
UP = percentage of animals with ulcers
Severity was recorded with the following scores:
• 0 = no ulcer
• 1 = superficial ulcers
• 2 = deep ulcers
• 3 = perforation.
Acute toxicity
Animals employed in the carrageenan-induced paw edema experiment were observed for morbidity or mortality every 24 h for 14 d after receiving the tested compoundsCitation35.
In vitro COX study
The inhibitory COX activity of the tested compounds and the reference was assayed using Cayman colorimetric COX (ovine) inhibitor screening assay kit (Catalog No. 760111, Cayman Chemicals, Ann Arbor, MI) according to the manufacturer’s instructionsCitation36. Aliquots (170 μL) of the assay buffer (0.1 N Tris-HCl, pH 8.0), heme, and enzyme ovine (COX-1 or COX-2) were placed in a 96-well plate. The test compounds were added to the aliquots. The plate was carefully shaken for a few seconds and then incubated for 5 min at 25 °C. The colorimetric substrate N,N,N′,N′-tetramethyl-p-phenylenediamine (20 μL, TMPD) and arachidonic acid (20 μL) were added to the aliquots. The plate was carefully shaken for a few seconds and then incubated for 5 min at 25 °C. The absorbance was measured at 590 nm using a 96-well Tecan Safire plate reader. The mixture of 160 μL of assay buffer and 10 μL of heme served as a background control. The mixture of 150 μL of assay buffer, 10 μL of heme, and 10 μL of ovine COX-1 or COX-2 showed 100% initial activity as a control experiment in the absence of inhibitor. Diclofenac and celecoxib were used as reference standards in the study. The assays were performed in triplicate and the IC50 values were calculated from the concentration curves by means of GraphPad software PRISM.
Cell isolation and culture
Fresh peripheral blood mononuclear cells (PBMCs), consisting of lymphocytes and monocytes, were isolated from EDTA (Sigma Chemical Co., St. Louis, MO) treated blood samples of healthy young male Sprague-Dawley rats of 6 weeks age weighing around 250 g. All procedures on rats were done according to the IRB guidelines of the Lebanese American University. Blood samples were mixed with EDTA, to a final concentration of 4 mM to prevent coagulation, diluted 1/2 with sterile and warm phosphate-buffered saline (PBS) (1X) (Sigma-Aldrich, St. Louis, MO), and centrifuged over Ficoll (Histopaque-1077, Sigma-Aldrich). PBMCs were washed twice in sterile warm PBS (1X) before seeding. The mononuclear cells were seeded in 6-well culture plates (Corning Costar) at a density of approximately 4 × 105 cells/cm2, in a final volume of 2 mL. The cells were allowed to adhere to plastic dishes overnight at 37 °C, 5% CO2, in complete RPMI medium (Sigma-Aldrich) supplemented with fetal bovine serum (10%) (Sigma-Aldrich), penicillin and streptomycin (Sigma-Aldrich), 100 U/mL and 100 μg/mL respectively. The next day, by vigorous washes with PBS (three times), the non-adherent cells (mainly lymphocytes) were removed. Macrophages obtained in this fashion are greater than 95% pure. Cell viability was determined by trypan blue exclusion. PMBCs were then cultured in RPMI serum-free media and stimulated with factors for 6 h prior to cell lysis and western blot analysis.
Cell stimulation protocol
Monocytes were cultured in a total volume of 2 mL for 6 h in 6-well plates in the presence of LPS (Sigma-Aldrich) and increasing concentrations of either of the following compounds 7a, 11, 15a, 16a, 16b, 18, and 19. Macrophages are cultured in RPMI-free media and incubated with either vehicle alone, or solvent alone (consist of methanol 1:2 and DMSO 1:2, then PEG 400 is added 1:1), or LPS (100 ng/mL) alone (Sigma-Aldrich), or 25 μM, 50 μM, or 100 μM respectively of each compound for 30 min prior to stimulation with LPS (100 ng/mL) for 6 h.
Preparation of cell extracts and western blotting
Cells in each well were lysed in RIPA Buffer containing 50 mM Tris-HCL (pH 7.5) (Sigma-Aldrich), 150 mM NaCl (Fisher Scientific, Pittsburgh, PA), 1% Tween 20 (Himedia), 0.5% sodium dodecyl sulfate (SDS) (Himedia, Paris, France), and 2% beta-mercaptoethanol (Sigma-Aldrich). Then 10 μL of the protein lysate was used to measure the total protein concentration using NanoDrop 2000c Spectrophotometer (Thermo Scientific, Pittsburgh, PA). Equal amount of 50 μg of total protein from each sample was analyzed on 10% SDS–polyacrylamide gel electrophoresis (PAGE). Protein samples were diluted 1:1 in 2× Laemmli sample buffer before loading onto 10% SDS-PAGE. Protein bands in the gel were transferred onto polyvinylidene fluoride membrane (Sigma-Aldrich). After blocking of the membrane in 5% skimmed milk in Tris-buffered saline containing 0.05% Tween-20 for 1 h at room temperature, the membranes were incubated overnight at 4 °C with rabbit polyclonal anti-rat COX-2 (Abcam, Cambridge, UK; 1:1000 dilution) or rabbit anti-rat iNOS (Abcam; 1:1000 dilution) in TTBS (1X) containing 5% skimmed milk. The second goat polyclonal anti-rabbit antibody-horseradish peroxidase conjugate (Abcam, 1:2000 dilution) was subsequently incubated with membranes for 1 h at room temperature. Membranes are then washed extensively for 1–2 h with TTBS (1X). Following incubation with ECL chemiluminescence reagent (Clarity Western ECL Substrate, Bio-Rad, Hercules, CA), protein bands on the membrane were visualized by ChemiDocTM MP imaging system (Bio-Rad). Data for the in vitro analysis were statistically analyzed using one-way ANOVA followed by Tukey simultaneous comparison t-values using SPSS. Values denote mean (n = 3) and statistical differences were considered significant at *p < 0.05 (Figures S3–S10 in Supplementary Material).
Analgesy-meter induced force in rats
Analgesic activity was tested in rats using an analgesy-meter according to a previously described procedureCitation37. Male albino rats weighing 120–150 g were used throughout this assay. They were kept in the animal house under standard conditions of light and temperature with free access to food and water. The animals were randomly divided into groups each of six rats. One group of six rats was kept as a control and another group received the standard drug diclofenac sodium (at a dose of 50 mg/kg body weight i.p.). The tested compounds were administrated intraperitoneally at a dose equimolar to that of diclofenac sodium. The rats were gently placed between the plinth and plunger. Mechanical pain was induced onto the paw of the rats by the use of analgesy-meter (Ugo Basile, Varese, Italy) which exerted force at a constant rate. The rats were restrained while their tails were placed between the plinth and the plunger. The force was continuously monitored by a pointer moving along a linear scale. Pain response was taken to be the point at which the rat struggles to set itself free or its paw slips off the plinth of the instrument. The weight causing pain before treatment (0) and then 4 h after treatment to each rat in all the groups was recorded. Percentage pain inhibition (increase in pain threshold) produced relative to the control group was calculated for each group over the period of study using the formula:
where Ft is the weight of the induced force at which the animal tries to free itself after administration of the test compound, Fo is the weight of force at which the animal tries to free itself before administration of the test compound.
Statistical analysis
Data were statistically analyzed using one-way ANOVA followed by Tukey simultaneous comparison t-values. Statistical differences were considered to be significant at p < 0.01.
Results and discussion
Chemistry
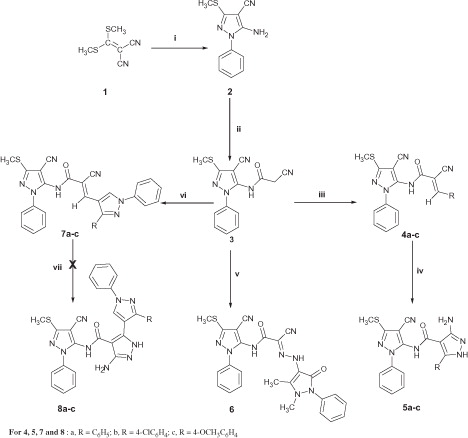
The synthetic strategies adopted for the synthesis of the intermediate and target compounds are depicted in Schemes 1–3. In Scheme 1, the starting 5-amino-3-methylsulfanyl-1H-pyrazole-4-carbonitrile 2 was prepared as previously describedCitation32. Synthesis of the cyanoacetamide derivative 3 was achieved by pouring the pyrazolecarbonitrile 2 into a solution of the cyanoacetylating reagent (cyanoacetic acid and acetic anhydride) followed by heating the mixture at 50 °C for 5 h modifying the reaction conditions previously reported for synthesis of allied compoundsCitation38. It should be noted here that heating the reaction mixture at 95 °C for 30 min. as previously reportedCitation38 resulted in a black tar which was very difficult to work up or purify, whereas, heating at 50 °C for a longer time gave a pure product in a good yield. Condensation of 3 with aromatic aldehydes in boiling ethanol containing a catalytic amount of piperidine afforded the corresponding acrylamides 4a–c. 1H-NMR of the latter compounds revealed presence of double the number of the expected protons indicating presence of these compounds as a mixture of E and Z isomers. Reaction of compounds 4a–c with hydrazine hydrate following reaction conditionsCitation39 described for synthesis of analogous compounds yielded the respective 3-aminopyrazoles 5a–c in a moderate yield. The reaction mechanism involves β-attack on the –CO–C=C moiety in 4 with subsequent 1,5-intramolecular dipolar cyclization and concomitant aromatizationCitation39. Microanalysis and spectral data of 5a–c were fully consistent with the proposed structures. On the other hand, coupling of compound 3 with the diazonium salt of 4-aminoantipyrine in ethanol buffered with sodium acetate adopting a published procedureCitation40 gave the corresponding antipyrinyl hydrazone derivative 6 in a good yield. Condensation of 3 with 3-aryl-1-phenyl-1H-pyrazole-4-carboxaldehydesCitation41 in ethanolic NaOH 10% afforded the corresponding acrylamides 7a–c. Trials to cyclize compounds 7a–c into the corresponding 3-aminopyrazoles 8a–c using hydrazine hydrate were unsuccessful. In fact, follow-up of the reaction by TLC revealed complex reaction mixture and extensive degradation. However, on cooling the reaction mixture, the major decomposition product separated out as a white solid. 1H-NMR of the separated decomposition products confirmed their structures as the corresponding 3-aryl-1-phenyl-1H-pyrazole-4-carboxaldehydes.
In Scheme 2, reaction of the cyanoacetamide 3 with DMF dimethyl acetal in dry dioxane resulted in the corresponding enaminonitrile 9 in a good yield. Refluxing the enaminonitrile 9 with hydrazine hydrate in ethanol as previously described for the synthesis of analogous compoundsCitation42 gave rise to the corresponding 5-aminopyrazole 10 (Method I). Inspection of the 1H-NMR spectrum enabled establishing structure 10 as 5-aminopyrazole derivative since the pyrazole C3-H appeared as a singlet at 7.78 ppm which is characterized from the tautomeric 3-aminopyrazole that would reveal pyrazole C5-H as a doubletCitation43. Fusion of 9 with 4-aminoantipyrine in an oil bath at 170 °C gave rise to the corresponding pyrazole derivative 11 (Method I). Synthesis of the ethoxyacrylamide derivative 12 was achieved by heating 3 with triethyl orthoformate in dioxane containing few drops of piperidine. At this stage, it seemed of interest to investigate the possibility of using compound 12 as a precursor for the alternative synthesis of compounds 10 and 11 (Method II). Accordingly, heating 12 with hydrazine hydrate in ethanol furnished the corresponding 5-aminopyrazole 10 whereas, fusion of 12 with 4-aminoantipyrine at 170 °C yielded the corresponding pyrazole derivative 11. Compounds 10 and 11 from both methods gave identical Rf value in TLC, melting point and IR spectra.
In Scheme 3, trials to prepare the ketene N, S-acetals 14a, b through treatment of the cyanoacetamide 3 with aryl isothiocyanates in DMF containing potassium hydroxide under cooling till complete addition of the isothiocyanate as previously describedCitation44 were unsuccessful and the corresponding malonamide 13 was separated instead. The chemical structure of 13 was confirmed on the basis of 1H-NMR which portrayed a D2O exchangeable singlet at 7.15 ppm attributed to the CONH2 protons. Additionally, its 13C-NMR showed two signals at 154.07 and 166.55 ppm characteristic for two C=O groups. To prevent hydrolysis of the CN group to the corresponding CONH2, the reaction mixture was cooled at 0–5 °C during addition of the isothiocyanate and cooling was maintained for further 2 h then the reaction mixture was left at room temperature over night till complete reaction with aryl isothiocyanates followed by addition of dimethyl sulfate to achieve methylation. In this case, the ketene N,S acetals 14a, b were separated in a good yield and purity. Microanalysis and spectral data of 14a, b were in agreement with their proposed structures. Reaction of 14a, b with hydrazine hydrate in ethanol yielded the requisite 5-aminopyrazoles 15a, b. It should be noted here that a literature survey revealed that some authors reported the formation of the isomeric 3-aminopyrazoles under the same reaction conditionsCitation44, while others claimed the separated products to be the isomeric 5-aminopyrazolesCitation45. In our study, the preferential formation of the isomeric 5-amino pyrazoles (15a, b) was supported from molecular orbital calculations utilizing semi-empirical PM6 method implemented in Gaussian 09 programCitation46, which revealed that the heat of formation of the 5-aminopyrazole isomer is lower than the isomeric 3-aminopyrazole by 1.6 kcal/mol. On the other hand, fusion of 14a, b with 4-aminoantipyrine at 170 °C resulted in the corresponding pyrazoles 16a, b. Treatment of 3 with carbon disulfide in DMF containing potassium hydroxide at 0–5 °C, followed by methylation using dimethyl sulfate at room temperature gave rise to the bis(methylsulfanyl) acrylamide derivative 17 which upon condensation with hydrazine hydrate furnished the 5-aminopyrazole derivative 18 as described for the synthesis of analogous compoundsCitation47. Finally, fusion of 17 with 4-aminoantipyrine at 140 °C gave rise to the corresponding derivative 19 in a good yield. Spectral data of compounds 18 and 19 were in agreement with their assigned structures.
Biological evaluation
Anti-inflammatory activity
The anti-inflammatory activity of the synthesized compounds was evaluated utilizing the carrageenan-induced paw edema bioassay in mouse using diclofenac sodium (50 mg/kg) as a reference standard anti-inflammatory agentCitation33. The data obtained are presented in and expressed as means ± SD. Statistical differences of control and test groups were carried out using one-way ANOVA followed by Tukey simultaneous comparison t-values. The difference in results was considered significant when p < 0.01.
Table 1. Anti-inflammatory activity of the synthesized compounds in carrageenan-induced mouse paw edema bioassay.
Carrageenan-induced paw edema bioassay
In this inflammatory model, each test compound was administered intraperitoneally (at a dose equimolar to that of diclofenac sodium) 1 h prior to the induction of inflammation by carrageenan injection. Diclofenac sodium was utilized as a reference drug at a dose of 50 mg/kg, i.p. Preliminary study of the time response curve for the synthesized compounds revealed that the maximum activity was observed after 4 h, therefore the anti-inflammatory activity was calculated after 4 h and presented in as the mean paw volume (mL) in addition to the percentage of anti-inflammatory activity (AI%). The anti-inflammatory activity of the test compounds relative to that of diclofenac was also calculated.
A comparative study of the anti-inflammatory activity of the test compounds relative to the reference drug after 4 h () revealed that compounds 16b and 18 showed anti-inflammatory activity (% protection = 78% and 75% respectively) superior to that of diclofenac sodium (% protection = 53%), whereas compounds 7a, 7c, 11, 15a, and 16a were nearly equivalent to the reference drug in inhibiting paw edema with percentage activity of (58–50%). The other compounds showed moderate to weak activity.
A deep insight in the structures of the synthesized compounds revealed that they represent four different series of compounds (E-H; ); series E (compounds 5a–c) comprising the 3-amino-5-substituted pyrazole moiety linked to the N-phenyl pyrazole through amide link, series F (compounds 7a–c) comprising the 1-phenyl-3-substituted pyrazole linked to the N-phenylpyrazole scaffold through 2-cyano acrylamide moiety, series G (compounds 10, 15a, 15b, and 18) comprising the 5-amino-3-substituted pyrazole attached to the N-phenylpyrazole through amide bridge and series H (compounds 6, 11, 16a, 16b, and 19) comprising the 4-aminoantipyrinyl moiety attached to the N-phenylpyrazole through -X = C(CN)-CONH- spacer. The preliminary anti-inflammatory results revealed that the 3-aminopyrazolyl derivatives (5a–c, series E) did not show significant anti-inflammatory activity. Among compounds 7a–c (series F), the phenyl derivative 7a displayed nearly equipotent activity to diclofenac sodium. Substitution on the phenyl ring with a methoxy group (compound 7c) did not significantly alter the activity, whereas, substitution with a chloro group (compound 7b) markedly decreased the activity. Regarding the 5-amino-3-substituted pyrazole derivatives (series G), the 5-amino-3-(un)substituted pyrazolyl derivative (compound 10) was found to be the least active. Substitution at position 3 with phenylamino, 4-chlorophenyl amino or methylsulfanyl group greatly improved the activity in the order methylsulfanyl ≫ phenylamino ≫ 4-chlorophenylamino. In this context, compound 15a comprising the phenylamino moiety displayed activity equivalent to diclofenac sodium, whereas, compound 18 with a methylsulfanyl moiety was superior to the reference drug (% protection = 75). In series H comprising the antipyrinyl moiety, the anti-inflammatory activity appears to be closely related to the nature of the (X) group. When X was a nitrogen atom (compound 6), the activity was weak. Replacement of the nitrogen atom with CH group (compound 11) increased the activity to approach that of diclofenac sodium. Substitution of the hydrogen atom of the CH group with phenylamino or methylsulfanyl moieties (compounds 16a and 19 respectively) did not result in a significant change in activity, whereas, substitution with 4-chlorophenylamino group (compound 16b) noticeably improved the activity to surpass that of diclofenac sodium. In this view, compound 16b emerged with the highest activity in this study (% protection = 78).
The most active compounds (7a, 7c, 11, 15a, 16a, 16b, and 18) were further tested at different doses in order to determine their ED50 values after 4 h interval (). In terms of ED50, compounds 7a, 7c, 16b, and 18 (ED50 = 98.28–139.17 μmol/Kg) were more active than diclofenac sodium (ED50 = 148.52 μmol/Kg), however, compounds 16b and 18 were found to be the most potent (ED50 = 98.28 and 108.64 respectively, ).
Ulcerogenic activity
All the synthesized compounds were further evaluated for their gastric ulcerogenic potential in mice using diclofenac sodium as reference drugCitation34. Results () revealed that the tested compounds were less ulcerogenic than diclofenac sodium except for compounds 6, 7b, and 7c which possessed nearly the same ulcerogenic potential.
Table 2. Ulcerogenic activity of the tested compounds and diclofenac sodium in mice.
Toxicity
No morbidity or mortality was observed in animals that received the tested compounds. All mice showed no toxicity after being observed every 24 h for 14 d after receiving the tested compounds at the doses used for anti-inflammatory activityCitation35.
In vitro COX-1/COX-2 inhibition assay
The synthesized compounds were further evaluated for their ability to inhibit COX-1 and COX-2 isoenzymes using the in vitro colorimetric COX (ovine) inhibitor assay methodCitation36, which utilizes the peroxidase component of cyclooxygenase. The peroxidase activity is assayed colorimetrically by monitoring the appearance of oxidized TMPD, which is produced during the reduction of PGG2 to PGH2, at 590 nm. The efficacies of the tested compounds were determined as the concentration causing 50% enzyme inhibition (IC50) and recorded in . The data revealed that some of the test compounds inhibit COX isoenzymes in a high concentration range compared to celecoxib. The results also showed that compounds 7b, 7c, 15a were non selective, while compounds 7a, 11, 16a, 16b, 18, and 19 showed relatively slight selectivity toward COX-2 than COX-1. Among the tested compounds, compound 16b demonstrated the highest inhibitory activity as it showed IC50 value of 20.3 μM for COX-1 and 11.6 μM for COX-2. These results indicate that further investigation is needed in order to gain insight into the mechanism of action of the examined compounds.
Table 3. In vitro COX-2 selectivity index of the synthesized compounds.
Amide-linked bipyrazoles reduced LPS-induced iNOS and COX-2 protein expression
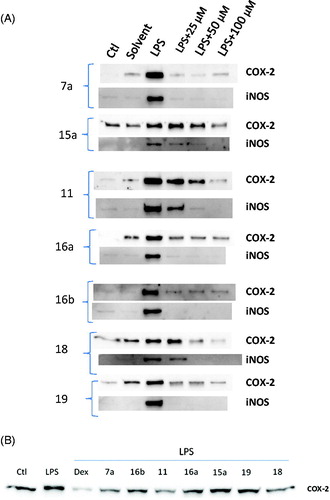
iNOS and COX-2 are key proinflammatory enzymes induced by LPS or cytokines and were found to work in concert in a number of similar pathophysiological activities and inflammatory diseaseCitation48,Citation49. Under basal condition, the products of iNOS and COX-2, including nitric oxide (NO) and prostaglandins (PGs), are involved in modulation of cellular functions and homeostasis. However, during inflammation, NO and PGs are released simultaneously in large amounts up to micromolar concentrationCitation50. Previous study has shown that NO directly increases COXs activity and leads to a remarkable sevenfold increase in PGE2 formationCitation49; further studies suggest that there is a considerable cross talk between NO and PGs biosynthetic pathwaysCitation51,Citation42. Therefore, a compound with dual inhibitory effect on iNOS and COX-2 expression would hold tremendous potential in advancing the treatment of inflammatory or chronic immune disorders. Hence, the most promising compounds were evaluated for their molecular anti-inflammatory effect on the level of protein expression of iNOS and COX-2 () in LPS-activated rat PBMCs. Additionally, the inhibitory potency of the compounds on COX-2 protein upregulation was compared to the anti-inflammatory glucocorticoid dexamethasone (). The results in revealed that iNOS and COX-2 proteins were slightly detectable in unstimulated cells. After treatment with LPS for 6 h, the protein expression of iNOS and COX-2 was markedly increased. Co-treatment with the test compounds dose-dependently (25, 50, and 100 μM) inhibited iNOS and COX-2 protein expression induced by LPS. All the tested compounds significantly suppressed COX-2 expression at a concentration of 100 μM, and five of them (11, 15a, 16b, 18, and 19) reduced COX-2 protein expression to levels comparable to control unstimulated cells (Figures S3–S10 in Supplementary Material). However, at a concentration of 50 μM, compounds 7a, 11, 15a, 16b, and 18 reduced COX-2 protein expression to levels comparable to control (Figures S3–S10 in Supplementary Material). When a concentration of 25 μM was used, all compounds showed detectable inhibitory effect on COX-2 protein expression, and three of them (7a, 15a, and 19) inhibited COX-2 expression by at least 80% (Figures S3–S10 in Supplementary Material) when compared to cells treated with LPS alone. On the other hand, all compounds at 100 μM, completely reduced iNOS protein expression to levels similar to control non-stimulated cells. Same results were obtained with these compounds when used at 50 μM except for compounds 11 and 16b. However, when the compounds were used at 25 μM, only compounds 7a, 16a, 15a, and 19 were able to strongly inhibit iNOS expression. Finally, it could be concluded that the most active compounds might have exerted their anti-inflammatory effect in part through inhibition of COX-2 and iNOS proteins expression. However, further investigation in order to rule out the detailed mechanisms by which these compounds block COX-2 and iNOS protein translation is in progress.
Figure 2. Amide-linked bipyrazoles blocked LPS-induced COX-2 and iNOS protein expression in rat monocytes. In (A), PMBCs were incubated with vehicle alone, or with solvent alone, or with LPS (100 ng/mL) alone, or with 25 µm, 50 µm or 100 µm of each of the test compounds respectively for 30 min prior to stimulation with LPS (100 ng/mL) for 6 h as indicated. Cell lysates were resolved by SDS-PAGE and immunoblotted with anti-COX-2 and anti-iNOS antibodies. In (B), PBMCs were incubated with vehicle alone, or LPS (100 ng/mL) alone, or LPS (100 ng/mL) + dexamethasone (40 μM), or LPS (100 ng/mL) + 7a (50 μM), or LPS (100 ng/mL) + 16b (50 μM), or LPS (100 ng/mL) +11(50 μM), or LPS (100 ng/mL) + 16a (50 μM), or LPS (100 ng/mL) + 15a (50 μM), or LPS (100 ng/mL) + 19 (50 μM), or LPS (100 ng/mL) + 18 (50 μM). Expression levels were normalized to total amount of protein loaded onto the gel and assessed by densitometric analysis (n = 3). *p < 0.05 versus vehicle control.
Analgesic activity
The analgesic activity of the synthesized compounds was evaluated in rats using the analgesy-meter-induced pain method and utilizing diclofenac sodium (50 mg/kg, i.p.) as reference drugCitation36. The results were recorded as the pre and post treatment (4 h) weight causing pain for each rat and the percentage protection. The results were presented in and expressed as means ± SEM. Statistical differences of control and test groups were carried out as described under section “Anti-inflammatory activity”.
Scheme 1. Synthesis of compounds 2–7. Reagents and conditions: (i) phenylhydrazine/ethanol/reflux; (ii) cyanoacetic acid/acetic anhydride/50 °C; (iii) the appropriate aldehyde/ethanol/piperidine/reflux; (iv) N2H4 98%/ethanol/reflux; (v) 4-amino antipyrine/NaNO2/ethanol/sodium acetate/r.t.; (vi) 3-aryl-1-phenyl-1H-pyrazole-4-carboxaldehyde/ethanol/NaOH/reflux; (vii) N2H4 98%/ethanol/reflux. For 4, 5, 7 and 8: a, R = C6H5; b, R = 4-ClC6H4; c, R = 4-OCH3C6H4.
Scheme 2. Synthesis of compounds 9–12. Reagents and conditions: (i) DMF-DMA/dry dioxane/reflux; (ii) N2H4 98%/dry dioxane/reflux; (iii) 4-aminoantipyrine/oil bath/170 °C; (iv) triethyl orthoformate/piperidine/dioxane/reflux; (v) N2H4 98%/ethanol/reflux; (vi) 4-aminoantipyrine/oil bath/170 °C.
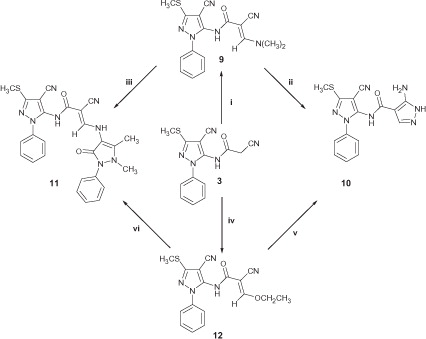
Scheme 3. Synthesis of compounds 13–19. Reagents and conditions: (i) RNCS/KOH/DMF/r.t.; (ii) RNCS/KOH/DMF/0 °C; (iii) N2H4 98%/ethanol/reflux; (iv) 4-aminoantipyrine/oil bath/170 °C; (v) CS2/KOH/DMF/0 °C then (CH3)2SO4/r.t.; (vi) N2H4 80%/ethanol/reflux; (vii) 4-aminoantipyrine/oil bath/140 °C. For 14–16: a, R = C6H5; b, R = ClC6H4.
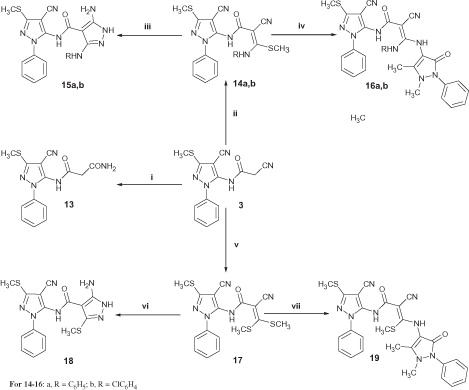
Table 4. Analgesic activity of the synthesized compounds using the analgesy-meter-induced pain in rats.
A comparative study of the analgesic activity of the test compounds relative to the reference drug revealed that most of the tested compounds exhibited significant analgesic activity; however, none of them was superior to diclofenac sodium. Seven compounds namely; 7c, 11, 15a, 16a, 16b, 18, and 19 displayed analgesic activity (% protection = 47–54) nearly equivalent to the reference drug (% protection = 52). The other compounds showed moderate to weak activity.
Further interpretation of the results revealed that the 3-aminopyrazolyl derivatives (5a–c, series E) did not display considerable analgesic activity. Among compounds 7a–c (series F), the phenyl derivative 7a displayed moderate activity (% protection = 42). Substitution on the phenyl ring with chloro group (compound 7b) decreased the activity, whereas, introduction of a methoxy group (compound 7c) slightly increased the activity to approach that of diclofenac sodium. Regarding the 5-amino-3-substituted pyrazolyl derivatives (series G), the unsubstituted 5-aminopyrazolyl derivative (compound 10) was the least active (% protection = 13). However, substitution at position 3 with phenylamino (compound 15a) or methylsulfanyl group (compound 18) greatly improved the activity to be close to that of diclofenac sodium whereas substitution with 4-chlorophenyl amino group (compound 15b) did not alter the activity. In series H comprising the antipyrinyl moiety, the anti-inflammatory activity appears to be closely related to the nature of the (X) group. When X was a nitrogen atom (compound 6), the activity was moderate (% protection = 43). Replacement of the nitrogen atom with CH group (compound 11) improved the activity (% protection = 48) to approach that of diclofenac sodium. On the other hand, substitution of the hydrogen atom of the CH moiety with phenylamino or methylsulfanyl groups (compounds 16a and 19) did not alter the activity, whereas, substitution with a 4-chlorophenylamino moiety (compound 16b) slightly improved the activity.
Conclusion
The aim of the present study was to synthesize and investigate the anti-inflammatory and analgesic activities of new amide-linked bipyrazoles with the hope of discovering new lead structures devoid of the GI side effects associated with conventional NSAIDs. The obtained results clearly revealed that compounds 7a, 11, 15a, 16a, 16b, and 18 showed anti-inflammatory activity similar to or even higher than that of diclofenac sodium. In vitro COX-1/COX-2 inhibition study revealed that among the synthesized compounds, compound 16b demonstrated the highest inhibitory activity with IC50 value of 20.3 μM for COX-1 and 11.6 μM for COX-2. Moreover, these compounds significantly suppressed COX-2 and iNOS protein expression in LPS-stimulated rat PBMCs. On the other hand, compounds 7c, 11, 15a, 16a, 16b, 18, and 19 were found to exhibit analgesic activity nearly equivalent to the reference drug. Collectively, compounds 16b and 18 were proved to display anti-inflammatory activity superior to diclofenac sodium and analgesic activity equivalent to it with a delayed onset of action. Additionally, all of the tested compounds revealed lower ulcerogenic potential and are well tolerated by experimental animals. Consequently, such type of compounds would represent a fruitful matrix that deserves further investigation and derivatization.
Acknowledgements
The authors are deeply grateful to Professor Dr Ahmed Khodair, Prof. of Organic Chemistry, Department of Chemistry, Faculty of Science, Kafr El-Sheikh University, Egypt, for his appreciable help in confirming the chemical structure of compounds 15 utilizing PM6 method implemented in Gaussian 09 program. The authors would like also to thank Sirine Zaatari for technical support.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the contents and writing the paper.
Notes
*A part of this research was presented in abstract form at the 3rd International Conference of Organic Chemistry (ICOC-2014) “Organic Synthesis – Driving Force of Life Development”, 25–26 September 2014, Tbilisi, Georgia. Available at: WWW.CHEMISTRY.GE\CONFERENCES\ICOC-2014.
References
- Nathan C. Points of control in inflammation. Nature 2002;420:846–52
- Vane RJ, Botting RM. Mechanism of action of anti-inflammatory drugs. Scand J Rheumatol Suppl 1996;102:9–21
- Meade EA, Smith WL, DeWitt DL. Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isozymes by aspirin and other non-steroidal anti-inflammatory drugs. J Biol Chem 1993;268:6610–14
- Vane JR, Botting RM. Anti-inflammatory drugs and their mechanism of action. Inflamm Res 1998;47:78–87
- Qiu KM, Yan R, Xing M, et al. Synthesis, biological evaluation and molecular modeling of dihydro-pyrazolyl-thiazolinone derivatives as potential COX-2 inhibitors. Bioorg Med Chem 2012;20:6648–54
- Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Annu Rev Immunol 1996;14:397–440
- Maini RN, Taylor PC. Anti-cytokine therapy for rheumatoid arthritis. Annu Rev Med 2000;51:207–29
- Parente L, Perretti M. Advances in the pathophysiology of constitutive and inducible cyclooxygenases: two enzymes in the spotlight. Biochem Pharmacol 2003;65:153–9
- Allison MC, Howatson AG, Torrance CJ, et al. Gastrointestinal damage associated with the use of nonsteroidal antiinflammatory drugs. N Engl J Med 1992;327:749–54
- Hajjar ER, Cafiero AC, Hanlon JT. Polypharmacy in elderly patients. Am J Geriatr Pharmacother 2007;5:345–51
- Burnier M. The safety of rofecoxib. Expert Opin Drug Saf 2005;4:491–9
- Sharma PK, Kumar S, Kumar P, et al. Synthesis and biological evaluation of some pyrazolylpyrazolines as anti-inflammatory-antimicrobial agents. Eur J Med Chem 2010;45:2650–5
- Jadhav SY, Shirame SP, Kulkarni SD, et al. PEG mediated synthesis and pharmacological evaluation of some fluoro substituted pyrazoline derivatives as antiinflammatory and analgesic agents. Bioorg Med Chem Lett 2013;23:2575–8
- Youssef AM, White MS, Villanueva EB, et al. Synthesis and biological evaluation of novel pyrazolyl-2,4-thiazolidinediones as anti-inflammatory and neuroprotective agents. Bioorg Med Chem 2010;18:2019–28
- Vijesh AM, Isloor AM, Shetty P, et al. New pyrazole derivatives containing 1,2,4-triazoles and benzoxazoles as potent antimicrobial and analgesic agents. Eur J Med Chem 2013;62:410–15
- Ragab FA, Abdel Gawad NM, Georgey HH, et al. Synthesis of novel 1,3,4-trisubstituted pyrazoles as anti-inflammatory and analgesic agents. Eur J Med Chem 2013;63:645–54
- Fioravanti R, Bolasco A, Manna F, et al. Synthesis and biological evaluation of N-substituted-3,5-diphenyl-2-pyrazoline derivatives as cyclooxygenase (COX-2) inhibitors. Eur J Med Chem 2010;45:6135–8
- El-Sayed MA, Abdel-Aziz N, Abdel-Aziz I. AA, et al. Design, synthesis, and biological evaluation of substituted hydrazone and pyrazole derivatives as selective COX-2 inhibitors: molecular docking study. Bioorg Med Chem 2011;19:3416–24
- Singh P, Mittal A. Current status of COX-2 inhibitors. Mini Rev Med Chem 2008;8:73–90
- Ranatunge RR, Garvey DS, Janero DR, et al. Synthesis and selective cyclooxygenase-2 (COX-2) inhibitory activity of a series of novel bicyclic pyrazoles. Bioorg Med Chem 2004;12:1357–66
- Bekhit AA, Hymete A, El-Din ABA, et al. Pyrazoles as promising scaffold for the synthesis of anti-inflammatory and/or antimicrobial agent: a review. Mini Rev Med Chem 2010;10:1014–33
- Bandgar BP, Gawande SS, Bodade RG, et al. Synthesis and biological evaluation of a novel series of pyrazole chalcones as anti-inflammatory, antioxidant and antimicrobial agents. Bioorg Med Chem 2009;17:8168–73
- Bekhit AA, Fahmy HT, Rostom SA, et al. Synthesis and biological evaluation of some thiazolylpyrazole derivatives as dual anti-inflammatory antimicrobial agents. Eur J Med Chem 2010;45:6027–38
- Bekhit AA, Abdel-Aziem T. Design, synthesis and biological evaluation of some pyrazole derivatives as anti-inflammatory-antimicrobial agents. Bioorg Med Chem 2004;12:1935–45
- Khalil NA, Ahmed EM, El-Nassan HB, et al. Synthesis and biological evaluation of novel pyrazoline derivatives as anti-inflammatory and antioxidant agents. Arch Pharm Res 2012;35:995–1002
- Bandgar BP, Adsul LK, Chavan HV, et al. Synthesis, biological evaluation, and docking studies of 3-(substituted)-aryl-5-(9-methyl-3-carbazole)-1H-2-pyrazolines as potent anti-inflammatory and antioxidant agents. Bioorg Med Chem Lett 2012;22:5839–44
- Bekhit AA, Ashour HMA, Guemei A. Novel pyrazole derivatives as potential promising anti-inflammatory antimicrobial agents. Arch Pharm (Weinheim) 2005;338:167–74
- Bekhit AA, Ashour HM, Bekhit A-D, et al. Synthesis of some pyrazolyl benzenesulfonamide derivatives as dual anti-inflammatory antimicrobial agents. J Enzyme Inhib Med Chem 2009;24:296–309
- Bekhit AA, Ashour HM, Abdel Ghany YS, et al. Synthesis and biological evaluation of some thiazolyl and thiadiazolyl derivatives of 1H-pyrazole as anti-inflammatory antimicrobial agents. Eur J Med Chem 2008;43:456–63
- Bekhit AA, Ashour HM, Bekhit A-D, et al. Synthesis and biological evaluation of novel pyrazole derivatives as anti-inflammatory antimicrobial agents. Med Chem 2009;5:103–17
- Rostom SA, el-Ashmawy IM, Abd el Razik HA, et al. Design and synthesis of some thiazolyl and thiadiazolyl derivatives of antipyrine as potential non-acidic anti-inflammatory, analgesic and antimicrobial agents. Bioorg Med Chem 2009;17:882–95
- Tominaga Y, Honkawa Y, Hara M, et al. Synthesis of pyrazolo[3,4-d]pyrimidine derivatives using ketene dithioacetals. J Heterocycl Chem 1990;27:775–83
- Claudino RF, Kassuya CA, Ferreira J, et al. Pharmacological and molecular characterization of the mechanisms involved in prostaglandin E2-induced mouse paw edema. J Pharmacol Exp Ther 2006;318:611–18
- Abbas SE, Awadallah FM, Ibrahin NA, et al. New quinazolinone-pyrimidine hybrids: synthesis, anti-inflammatory, and ulcerogenicity studies. Eur J Med Chem 2012;53:141–9
- Güvenç A, Küpeli Akkol E, Süntar I, et al. Biological activities of Pseudevernia furfuracea (L.) Zopf extracts and isolation of the active compounds. J Ethnopharmacol 2012;144:726–34
- Lee SH, Son MJ, Ju HK, et al. Dual inhibition of cyclooxygenases-2 and 5-lipoxygenase by deoxypodophyllotoxin in mouse bone marrow-derived mast cells. Biol Pharm Bull 2004;27:786–8
- Rodríguez RE, Rodríguez FD, Sacristán MP, et al. Antinociceptive activity of glycosidic enkephalin analogues. Psychopharmacology (Berl) 1990;101:222–5
- Salaheldin AM. Cyanoacetylation of 5-Aminopyrazole: synthesis of 2-(1-Aryl-4-substituted pyrazolo[3,4-d]pyrimidin-6-yl)acetonitrile Derivatives. Z Naturforsch 2009;64b:840–6
- Shams HZ, Mohareb RM, Helal MH, et al. Novel synthesis and antitumor evaluation of polyfunctionally substituted heterocyclic compounds derived from 2-cyano-N-(3-cyano-4,5,6,7-tetrahydrobenzo[b]thiophen-2-yl)-acetamide. Molecules 2010;16:52–73
- Fadda AA, Elattar KM. Synthesis of novel azo disperse dyes derived from 4-aminoantipyrine and their applications to polyester fabrics. Am J Org Chem 2012;2:52–7
- Bernard M, Hulley E, Molenda H, et al. [Azoles. 17. Beta-(4-pyrazol)acrylic and propionic acids and their anti-inflammatory activity]. Pharmazie 1986;41:560–2
- Ibrahim HM, Makhseed S, Abdel-Motaleb RM, et al. Utility of cyanoacetamides as precursors to pyrazolo[3,4-d]pyrimidine-4-ones, 2-aryl-6-substituted 1,2,3-triazolo[4,5-d]pyrimidines and pyrazolo[1,5-a]pyrimidine-3-carboxamides. Heterocycles 2007;71:1951–66
- Behbehani H, Ibrahim HM, Makhseed S, et al. 2-Aminothiophenes as building blocks in heterocyclic synthesis: synthesis and antimicrobial evaluation of a new class of pyrido[1,2-a]thieno[3,2-e]pyrimidine, quinoline and pyridin-2-one derivatives. Eur J Med Chem 2012;52:51–65
- Gouda MA, Berghot MA, Shoeib AI, et al. Synthesis and antimicrobial of new anthraquinone derivatives incorporating pyrazole moiety. Eur J Med Chem 2010;45:1843–8
- Elgemeie GH, Widyan K, Kurz T. Novel synthesis of fluorinated cyanoketene N,S acetals and their conversions to fluorinated pyrazole derivatives. Phosphorus Sulfur Silicon 2006;181:299–304
- Frisch MJ, Trucks W, Schlegel GHB, et al. Gaussian 09, Revision B.01. Wallingford, CT: Gaussian, Inc.; 2010
- Ammar YA, El-Sharief AMS, Al-Sehemi AG, et al. Cyanoacetanilides intermediates in heterocyclic synthesis. Part 2: preparation of some hitherto unknown ketene dithioacetal, benzoazole and pyridone derivatives. J Chinese Chem Soc 2005;52:553–8
- Goodwin DC, Landino LM, Marnett LJ. Effects of nitric oxide and nitric oxide-derived species on prostaglandin endoperoxide synthase and prostaglandin biosynthesis. FASEB J 1999;13:1121–36
- Kim SF, Huri DA, Snyder SH. Inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science 2005;310:1966–70
- Mollace V, Muscoli C, Masini E, et al. Modulation of prostaglandin biosynthesis by nitric oxide and nitric oxide donors. Pharmacol Rev 2005;57:217–52
- Vassalle C, Domenici C, Lubrano V, et al. Interaction between nitric oxide and cyclooxygenase pathways in endothelial cells. J Vasc Res 2003;40:491–9
- Sibilia V, Pagani F, Rindi G, et al. Central ghrelin gastroprotection involves nitric oxide/prostaglandin cross-talk. Br J Pharmacol 2008;154:688–97