
Abstract
In this study, we investigated the in vitro effect of 16-formyl-17-methoxy dehydroepiandrosterone derivatives on the activity of 5α-reductase type 2 (5α-R2) obtained from human prostate. The activity of different concentrations of these derivatives was determined for the conversion of labelled testosterone to dihydrotestosterone. The results indicated that an aliphatic ester moiety at the C-3 position of these derivatives increases their in vitro potency as inhibitors of 5α-R2 activity compared to finasteride®, which is considered to be a potent inhibitor of 5α-R2. In this case, the augmentation of the lipophilicity of these dehydroepiandrosterone derivatives increased their potency as inhibitors of 5α-R2. However, the presence of cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or cycloheptyl rings as the cycloaliphatic ester moiety at C-3 of the formyl methoxy dehydroepiandrosterone scaffold did not inhibit the activity of this enzyme. This may be due to the presence of steric factors between the enzyme and the spatial structure of these derivatives.
Introduction
The WHO reported a frequency of global prostate and testicular cancer of 250 000 new cases annually. The WHO also indicated that in the Unites States alone, the number of prostate cancer cases per year is 233 159, with a mortality rate of 10.3%. This mortality rate indicates that an improvement in therapies is presently still necessary. These numbers have inspired several research groups to develop new steroidal and non-steroidal molecules with therapeutic potency to relieve this illnessCitation1–5.
Dihydrotestosterone (DHT, ) has been associated with the development of androgen-dependent afflictions such as prostate cancer, benign prostatic hyperplasia, acne and androgen-dependent baldness. Various studies have demonstrated that males with a genetic deficiency of the 5α-reductase type 2 (5α-R2) isozyme have a smaller prostate gland due to a low level of DHTCitation6,Citation7. Lack of DHT induces imperfect development of external genitalia and the prostate, although the epididymis, seminal vesicles and ducts deferens remain normal. However, the external genitalia of these individuals look similar to those of females; furthermore, they do not develop baldness or acneCitation7. Studies also suggest that DHT stimulates the expression of several androgen-response genes in recurrent prostate cancerCitation8. This expression is strongly related to prostate preservation because these genes have the ability to induce apoptosis of the prostate cells and are less involved in cell proliferation throughout prostate developmentCitation9. Furthermore, the increased levels of intracellular DHT cause higher cellular propagation and faulty differentiationCitation10. These facts could be relevant because an overabundance of DHT has been implicated in the pathogenesis of benign prostatic hyperplasiaCitation7–10 (http://www-dep.iarc.fr/WHOdb/WHOdb.htm).
Figure 1. Conversion of testosterone (T) to 5α-dihydrotestosterone (DHT). This reaction is catalysed by the enzyme 5α-reductase, which is present in the prostate glandCitation6.
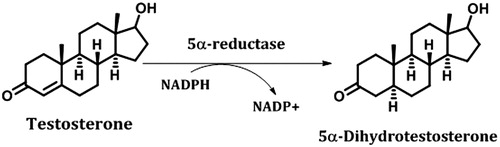
5α-R2 () plays an important role in prostate gland development because it is responsible for intraprostatic DHT levels when the physiological serum testosterone (T) concentration is lowCitation11. The evidences reported in the literature indicate that decreasing of T production during ageing can be a with an increase in prostate tumour growth (benign and malign)Citation10,Citation11. Therefore, 5α-R2 is still an important therapeutic target for the development of better drugs than those currently available, i.e. finasteride® and dutasteride®.
5α-R2 (EC 1.3.99.5) has previously been observed in different speciesCitation6, and the product of the reaction catalysed by this enzyme accumulates in the nuclei of responsive cells and binds to androgen receptors (ARs)Citation12. These AR complexes interact with coregulator proteins (coactivators or corepressors) to adapt transcription of androgen target genes using specific sequences in the DNACitation12.
In this study, we describe the synthesis and biological activity of different steroidal derivatives, which are shown in and . Their biological activities were determined in vitro using human prostate as a source of 5α-R2.
Figure 2. Route of synthesis of dehydroepiandrosterone derivatives.
Reagents and conditions: Vilsmeier–Haack reaction: phosphorus oxychloride (POCl3), dimethylformamide (DMF), chloroform (CHCl3), reflux (). Addition–substitution reaction: sodium methoxyde (NaOMe), methanol (MeOH), nitrogen (N2). Esterification reaction: dicyclohexylcarbodiimide (DCC), dimethylaminopyridine (DMAP).
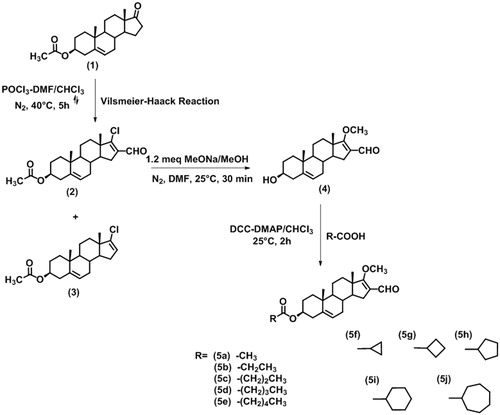
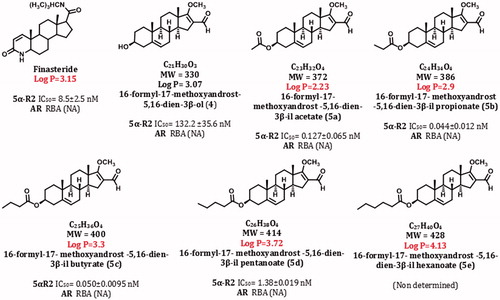
Figure 3. Structures, molecular weight, and biological activities of different dehydroepiandrosterone derivatives.
All steroids exhibited more potency (lower IC50 value) than that of the finasteride to inhibit 5α-R2 enzyme.
IC50 values: concentration of compound required to inhibit 50% of the activity of 5α-reductase type 2 (5α-R2); RBA: relative binding activity to androgen receptor (AR).
Material and methods
Chemical and radioactive materials
Solvents were laboratory grade or better. Melting points were determined using a Fisher–Johns melting point apparatus and are uncorrected. 1H-NMR and 13C-NMR data were collected on Varian Gemini 400 and VRX-300 equipment, respectively. Chemical shifts are given in ppm relative to that of Me4Si (δ = 0) in CDCl3 (the abbreviations of signal patterns are as follows: s, singlet; d, doublet, t, triplet, m, multiplet). Mass spectra (MS) were obtained using a Thermo DFS spectrometer by direct infusion and using FAB ionisation mode.
(1,2,6,7-3H)-Testosterone ([3H] T) with a specific activity of 95 Ci/mmol and Mibolerone (17α-methyl-3H) ([3H] MIB) with a specific activity of 70–87 Ci/mmol were provided by Perkin Elmer Life and Analytical Sciences, respectively (Boston, MA). Non-labelled T, 5α-dihydrotestosterone and MIB were supplied by Steraloids (Wilton, NH). Sigma Chemical Co. (St. Louis, MO) provided NADPH and Lubrol PX. Finasteride was obtained by extraction from Proscar® (Merck, Sharp & Dohme, Kenilworth, NJ). The tablets were crushed and extracted with chloroform, and the solvent was eliminated under vacuum; the crude product was purified by silica gel column chromatography. The melting point of the isolated finasteride (252–254 °C) was identical to that reported in the literature. Bio-gel hydroxyapatite (HAP) was provided by Bio-Rad Laboratories (Hercules, CA).
Synthesis of the steroidal derivatives
General procedure for the preparation of 17-chloro-16-formylandrost-5,16-dien-3β-yl acetateCitation2
17-chloro-16-formylandrost-5,16-dien-3β-yl acetate (2). Yield: 86%; m.p. 135–136 °C, 1H NMR (CDCl3): δ 0.99 (s, H-19, 3H), 1.06 (s, H-18, 3H), 2.03 (s, OCO-CH3, 3H), 4.60 (m, H-3, 1H), 5.39 (d, J = 5.6 Hz, H-6, 1H), 10.09 (s, -CHO, 1H); 13C NMR (CDCl3): δ 14.96 (C-19), 19.20 (CH3- acetoxy), 21.39 (C-18), 73.65 (C-3), 121.82 (C-6), 136.42 (C-16), 139.99 (C-5), 162.24 (C-17-Cl), 170.47 (C=O ester); MS (FAB, m/z): 373 (M + 1, 100%).
Procedure for the preparation of 16-formyl-17-methoxyandrost-5,16-dien-3β-ol (4). To a solution of 17-chloro-16-formylandrost-5,16-dien-3β-yl acetate () (2) (200 mg, 0.53 mmol) in dimethylformamide under nitrogen atmosphere, 0.5 M sodium methoxyde in methanol (1.3 mL, 1.2 meq) was added, and the mixture was stirred for 30 min at room temperature. The reaction mixture was then added into 20 mL of ice, and the solid obtained was filtered by vacuum and washed three times with cold waterCitation13. The yellow crude product was purified by flash column chromatography (FCC, Florisil® 60–100 hexane: AcOEt 65:25)Citation13.
16-formyl-17-methoxyandrost-5,16-dien-3β-ol (4). Yield: 94%; m.p. 167–169 °C, 1H NMR (CDCl3): δ 0.99 (s, H-19, 3H), 1.06 (s, H-18, 3H), 2.03 (s, OCO-CH3, 3H), 4.60 (m, H-3, 1H), 5.39 (d, J = 5.6 Hz, H-6, 1H); 13C NMR (CDCl3): δ 14.96 (C-19), 19.20 (CH3- acetoxy), 21.39 (C-18), 73.65 (C-3), 121.82 (C-6), 136.42 (C-16), 139.99 (C-5), 162.24 (C-17-Cl), 170.47 (C = O ester); and MS (FAB, m/z): 373 (M + 1, 100%).
General procedure for the preparation of esters from carboxylic acid derivatives
To a solution of 16-formyl-17-methoxyandrost-5,16-dien-3β-ol (165 mg, 0.50 mmol) (4), DCC (618 mg, 3.0 mmol) and DMAP (305 mg, 2.50 mmol) in chloroform (15 mL) were added to the corresponding carboxylic acid derivative (3.50 mmol). The resulting solution was stirred at room temperature for 1 h. Ethyl acetate (20 mL) was added, and the precipitated dicyclohexylurea was filtered. Then, 20 mL of chloroform was added to the organic phase, and the solution was washed with 10% aqueous hydrochloric acid (H2O: HCl 7:3, 40 mL × 6), 5% aqueous solution of sodium bicarbonate (twice) and then water (once). The mixture was dried over anhydrous sodium sulphate, and the solvent was removed under vacuum. The crude ester was purified by flash column chromatography (FCC, Florisil® 60–100 hexane: AcOEt 85:15).
16-formyl-17-methoxyandrost-5,16-dien-3β-yl acetate (5a). Yield: 91%; m.p. 150–152 °C, 1H NMR (CDCl3): δ 1.00 (s, H-18, 3H), 1.05 (s, H-19, 3H), 2.03 (s, CH3-acetoxy, 3H), 4.12 (s, 17-OCH3, 3H), 4.60 (m, H-3, 1H), 5.40 (d, J = 4.2 Hz, H-6, 1H), 10.09 (s, -CHO, 1H); 13C NMR (CDCl3): δ 15.54 (C-19), 19.19 (C-18), 20.32 (CH3-acetoxy), 62.30 (CH3O), 73.74 (C-3), 116.12 (C-6), 122.13 (C-16), 139.80 (C-5), 170.50 (C = O ester), 182.21 (C-17), 186.40 (CHO); MS (FAB, m/z): 373 (M + 1, 100%).
16-formyl-17-methoxyandrost-5,16-dien-3β-yl propionate () (5b). Yield: 48%; m.p. 140–141 °C, 1H NMR (CDCl3): δ 0.91 (t, CH3-propiloxy, 3H), 0.99 (s, H-18, 3H), 1.04 (s, H-19, 3H), 2.30 (t, CH2-propyloxy, 2H), 4.11 (s, 17-OCH3, 3H), 4.60 (m, H-3, 1H), 5.39 (d, J = 4 Hz, H-6, 1H), 10.08 (s, -CHO, 1H); 13C NMR (CDCl3): δ 9.30 (CH3-propyloxy), 19.37 (C-19), 20.49 (C-18), 30.33 (CH2-propyloxy), 62.45 (CH3O), 73.68 (C-3), 116.30 (C-6), 122.24 (C-16), 140.04 (C-5), 174.09 (C = O ester), 182.21 (C-17), 186.55 (CHO); MS (FAB, m/z): 373 (M + 1, 100%).
16-formyl-17-methoxyandrosta-5,16-dien-3β-yl butyrate () (5c). Yield: 43% (86.0 mg) white solid; m.p. 166–168 °C, 1H NMR (CDCl3): δ 0.94 (t, CH3-butyroxy, 3H), 0.99 (s, H-18, 3H), 1.04 (s, H-19, 3H), 1.63 (m, 2′-CH2-butyroxy, 2H), 2.24 (t, 1-CH2- butyroxy, 2H), 4.11 (s, 17-OCH3, 3H), 4.60 (m, H-3, 1H), 5.39 (d, J = 4.8 Hz, H-6, 1H), 10.09 (s, -CHO, 1H); 13C NMR (CDCl3): δ 13.77 (CH3- butyroxy), 15.70 (2-CH2- butyroxy), 18.66 (C-19), 19.36 (C-18), 30.33 (1-CH2- butyroxy), 62.44 (CH3O), 73.57 (C-3), 116.30 (C-6), 122.22 (C-16), 140.04 (C-5), 173.22 (C = O ester), 182.07 (C17), 186.48 (CHO); MS (FAB, m/z): 373 (M + 1, 100%).
16-formyl-17-methoxyandrosta-5,16-dien-3β-yl pentanoate (5d). Yield: 34%; m.p. 195–197 °C, 1H NMR (CDCl3): δ 0.92 (t, CH3-pentanoyloxy, 3H), 0.99 (s, H-18, 3H), 1.05 (s, H-19, 3H), 1.39 (m, 3-CH2-pentanoyloxy, 2H), 1.68 (m, 2-CH2-pentanoyloxy, 2H), 2.41 (t, 1-CH2-pentanoyloxy, 2H), 4.12 (s, 17-OCH3, 3H), 3.47 (m, H-3, 1H), 5.39 (d, J = 4.8 Hz, H-6, 1H), 10.09 (s, -CHO, 1H); 13C NMR (CDCl3): δ 15.71 (CH3-pentanoyloxy), 19.32 (C-19), 20.42 (C-18), 25.75 (3-CH2-pentanoyloxy), 28.35 (2-CH2-pentanoyloxy), 34.08 (1-CH2-pentanoyloxy), 60.18 (CH3O), 62.49 (C-3), 116.26 (C-6), 122.18 (C-16), 141.07 (C-5), 156.63 (C = O ester), 182.12 (C17), 186.53 (CHO); MS (FAB, m/z): 373 (M + 1, 100%).
16-formyl-17-methoxyandrosta-5,16-dien-3β-yl hexanoate (5e). Yield: 43%; m.p. 198–199°, 1H NMR (CDCl3): δ 0.89 (t, CH3-hexanoyloxy, 3H), 0.99 (s, H-18, 3H), 1.04 (s, H-19, 3H), 1.30 (m, 3-CH2- hexanoyloxy, 2H), 1.35 (m, 4-CH2- hexanoyloxy, 2H), 1.63 (m, 2-CH2- hexanoyloxy, 2H), 2.26 (t, 1-CH2- hexanoyloxy, 2H), 4.12 (s, 17-OCH3, 3H), 4.60 (m, H-3, 1H), 5.40 (d, J = 4.8 Hz, H-6, 1H), 10.08 (s, -CHO, 1H); 13C NMR (CDCl3): δ 14.07 (CH3- hexanoyloxy), 15.72 (C-19), 19.38 (C-18), 22.46 (4-CH2- hexanoyloxy), 24.88 (2-CH2- hexanoyloxy), 29.84 (3-CH2- hexanoyloxy), 31.44 (1-CH2- hexanoyloxy), 62.48 (CH3O), 73.61 (C-3), 116.30 (C-6), 122.24 (C-16), 140.05 (C-5), 173.47 (C = O ester), 182.31 (C-17), 186.61 (CHO); MS (FAB, m/z): 373 (M + 1, 100%).
General procedure for the preparation of esters from alicyclic carboxylic acids derivatives
The same procedure cited below was used; however, the white crude ester was purified by flash column chromatography (FCC, Florisil® 60–100 hexane: AcOEt 65:35).
16-formyl-17-methoxyandrosta-5,16-dien-3β-yl cyclopropionate () (5f). Yield: 72%; m.p. 130–132 °C, 1H NMR (CDCl3): δ 0.91 (t, CH3-propiloxy, 3H), 0.99 (s, H-18, 3H), 1.04 (s, H-19, 3H), 2.30 (t, CH2-propyloxy, 2H), 4.11 (s, 17-OCH3, 3H), 4.60 (m, H-3, 1H), 5.39 (d, J = 4 Hz, H-6, 1H), 10.08 (s, -CHO, 1H); 13C NMR (CDCl3): δ 8.17 (C-2, C-3), 12.93 (C1), 19.37 (C-19), 20.49 (C-18), 30.33 (CH2-propyloxy), 62.45 (CH3O), 73.68 (C-3), 116.30 (C-6), 122.24 (C-16), 140.04 (C-5), 174.09 (C = O ester), 182.21 (C17), 186.55 (CHO); MS (FAB, m/z): 373 (M + 1, 100%).
16-formyl-17-methoxyandrosta-5,16-dien-3β-yl cyclobutyrate () (5g). Yield: 72%; m.p. 163–165 °C, 1H NMR (CDCl3): 0.98 (s, H-18, 3H), 1.03 (s, H-19, 3H), 4.11 (s, 17-OCH3, 3H), 4.59 (m, H-3, 1H), 5.38 (d, J = 4.9 Hz, H-6, 1H), 10.08 (s, -CHO, 1H); 13C NMR (CDCl3): δ 15.54 (C-19), 18.32 (C-3), 19.21 (C-18), 25.19 (C-2, C-4′), 33.00 (C1), 62.28 (CH3O), 73.36 (C-3), 116.14 (C-6), 122.04 (C-16), 139.91 (C-5), 174.91 (C = O ester), 181.94 (C17), 186.33 (CHO); MS (FAB, m/z): 373 (M + 1, 100%).
16-formyl-17-methoxyandrosta-5,16-dien-3β-yl cyclopentanoate () (5h). Yield: 59%; m.p. 165–168 °C, 1H NMR (CDCl3): 1.00 (s, H-18, 3H), 1.05 (s, H-19, 3H), 4.12 (s, 17-OCH3, 3H), 4.58 (m, H-3, 1H), 5.39 (d, J = 4.9 Hz, H-6, 1H), 10.10 (s, -CHO, 1H); 13C NMR (CDCl3): δ 15.57 (C-19), 19.25 (C-18), 25.82 (C-3, C4′), 30.01 (C-2, C-5′), 44.03 (C1′), 62.32 (CH3O), 73.33 (C-3), 116.14 (C-6), 122.03 (C-16), 139.94 (C-5), 174.24 (C = O ester), 182.03 (C17), 186.40 (CHO); MS (FAB, m/z): 373 (M + 1, 100%).
16-formyl-17-methoxyandrosta-5,16-dien-3β-yl cyclohexanoate () (5i). Yield: 70%; m.p. 175–178 °C, 1H NMR (CDCl3): 1.00 (s, H-18, 3H), 1.05 (s, H-19, 3H), 4.12 (s, 17-OCH3, 3H), 4.60 (m, H-3, 1H), 5.38 (s, H-6, 1H), 10.09 (s, -CHO, 1H); 13C NMR (CDCl3): δ 15.57 (C-19), 19.25 (C-18), 25.45 (C-3, C-5′), 25.62 (C4′), 25.80 (C-2, C-6′), 36.74 (C1), 62.32 (CH3O), 73.17 (C-3), 116.18 (C-6), 122.05 (C-16), 139.95 (C-5), 175.58 (C = O ester), 181.94 (C17), 186.38 (CHO); MS (FAB, m/z): 373 (M + 1, 100%).
16-formyl-17-methoxyandrosta-5,16-dien-3β-yl cycloheptanoate () (5j). Yield: 68%; m.p. 190–193 °C, 1H NMR (CDCl3): 1.00 (s, H-18, 3H), 1.05 (s, H-19, 3H), 4.12 (s, 17-OCH3, 3H), 4.57 (m, H-3, 1H), 5.39 (s, H-6, 1H), 10.10 (s, -CHO, 1H); 13C NMR (CDCl3): δ 15.54 (C-19), 19.22 (C-18), 26.36 (C-3, C-6′), 30.18 (C-4, C-5′), 30.67 (C-2, C-7′), 45.16 (C1), 62.28 (CH3O), 73.16 (C-3), 116.14 (C-6), 122.01 (C-16), 139.93 (C-5), 176.48 (C = O ester), 181.96 (C17), 186.34 (CHO); MS (FAB, m/z): 373 (M + 1, 100%).
Human and animal tissues and procedures
Four hours after a 58-year-old man died from myocardial infarction, his normal prostate was extirpated in the Pathology Department of the General Hospital in Mexico City. The Ethical Committee of the General Hospital in Mexico City approved this protocol.
The tissue was rinsed and immediately chilled in ice-cold 150 mM NaCl and stored at −20 °C. The frozen human prostate was thawed on ice, rinsed, and minced in buffer A (20 mM sodium phosphate, pH 6.5, containing 0.32 M sucrose, 0.1 mM DTT (Sigma-Aldrich, Mexico City, Mexico) using an IKA® A11 basic tissue mill (IKA Laboratory Equipment, Mexico City, Mexico). Unless otherwise specified, the following procedures were carried out at 4 °C.
5α-R2 enzyme isolated from human prostate
Human prostate was used in this experiment because this tissue is an abundant source of 5α-R2 for the study of the effect(s) of 5a-j, which were designed for the inhibition of the activity of this enzyme in humans.
Human prostate tissue was homogenised in two volumes of buffer A using an Ultra-Turrax IKA tissue homogeniser, T18 basic (Wilmington, NC). The homogenates were centrifuged (1500 × g; 60 min)Citation10 in a SW 60 Ti rotor (Beckman Instruments, Palo Alto, CA). The pellets were resuspended in buffer A and stored at −70 °C. This suspension had a final concentration of 5 mg protein/mL, as determined by the Bradford methodCitation14, and was used as the source of 5α-R2 isozyme.
Cytosol of rat prostate as source of AR
Prostates were extirpated from 50 adult rats (8 months old; 500 g). In this study, we used rats because their prostate gland is large; there is no difference in the binding activity of the AR present in the cytosol of rats and humansCitation15.
The rat prostates were blotted, weighed, and soaked in ice-cold TEMD (40 mM Tris-HCl, 3 mM EDTA, 20 mM sodium molybdate, 0.5 mM DTT, 10% glycerol; pH 8) prior to use. Unless otherwise specified, all procedures were carried out at 0 °C on a bed of chopped ice. Tissues were homogenised in one volume of TEMD plus protease inhibitors (2 mM phenylmethylsulfonyl fluoride, 10 μg antipain/mL, 5 mM leupeptin)Citation16 using a tissue homogeniser immersed in a bed of chopped ice. The homogenates were centrifuged (140 000 × g; 60 min)Citation5 in a SW 60 Ti rotor. The final protein concentration of the supernatant containing the cytosolic fraction, as determined by the Bradford method, was 6 mg protein/200 μL. This supernatant was stored at −70 °C until its use.
Biological activity of steroidal derivatives
Experiments in vitro
The activity of the 5α-R2 was determined by following the conversion of T to DHT, as previously describedCitation1,Citation5. The reagent mixture (final volume, 1 mL) contained 1 mM DTT in 40 mM sodium phosphate buffer (pH 6.5), 2 nM [1,2,6,7-3H] T, and 2 mM NADPH. The reaction, which was carried out in duplicate, was started when this mixture was added to the enzymatic fraction (50 μg/80 μL from the membrane fraction of human prostate). After incubation (37.5 °C; 60 min), the reaction was stopped by adding dichloromethane (1 mL) and mixing; this was considered to be the end point. As a control, this procedure was carried out without the addition of the enzyme fraction.
To extract and purify the DHT formed by the activity of 5α-R2, after the individual reaction mixtures had been agitated (1 min) using a Type 16700 mixer (Barnstead Thermoline, Proveedor Científico, México, D.F., Mexico), the dichloromethane phase of each was placed into individual tubes. This extraction was repeated four additional times. Each pooled dichloromethane extract was evaporated to dryness under a nitrogen stream and then suspended in methanol (50 μL); each preparation was spotted onto HPTLC Keiselgel 60 F254 plates. T, DHT, and a mixture of both standards were applied to the plates in distinct lanes on either side of the spotted preparation samples. The plates were developed in chloroform-acetone (9:1) and were air-dried; the chromatography was repeated two additional times. DHT was detected using phosphomolybdic acid reagent (Sigma-Aldrich), and T was detected by fluorescence (UV lamp; 254 nm). The radioactivity on the plate was scanned using a Bioscanner AR2000 (Bioscan, Washington, DC). The zones that showed chromatographic behaviour identical to that of the standards (retention factor, Rf) were quantified as T or DHT. The DHT transformation yields were calculated from the lanes, taking into account the entire radioactivity in the TLC, and were plotted using SigmaPlot 12 software (Systat Software Inc., San Jose, CA). For the control incubations, the chromatographic separations and identifications were carried out in the same manner.
The 5α-R2 activity was calculated, taking into consideration the recovery, blank values, and the specific activity of [3H] T.
The Rf of the T standard was 0.56. The radioactive zone that exhibited chromatographic behaviour identical to that of the standard T corresponded to 70% of the radioactivity in the plate. The radioactive compound contained in the zone of the experimental chromatogram, which had an Rf identical to that of the DHT standard (Rf: 0.67), was identified as the transformed DHT; it corresponded to 20–27% of the total radioactivity accounted for in the TLC. This result was considered to be 100% of the activity of 5α-R2 for the development of inhibition plots. Unmodified [3H]T, identified (Rf: 0.56) from control incubations (i.e. those that had not contained tissue), had identical chromatographic behaviour to the non-labelled standard T.
The radioactivity contained in the zone corresponding to the DHT standard (Rf: 0.67) of the control chromatogram was 1% of the total radioactivity accounted for in the plate and was considered to be an error; it was subtracted from the experimental chromatograms.
Effect of steroids derivatives on activity of 5α-R2
The effect of steroids 5a–j on the activity of 5α-R2 was determined under the same conditions in vitro as described in section 2.3.3.1 but in the presence of six different concentrations (1 × 10−11, 1 × 10−10 M, … 1 × 10−3 M) of each of the 5a–j derivatives. Extraction and purification of the DHT formed from these incubations were carried out as described in “Experiments in vitro” section.
Control tubes (without inhibitors) were prepared with the same incubation medium and under the same conditions (“Experiments in vitro” section) and were labelled T plus 5α-R2. DHT transformations in the presence of 5a–j were calculated from the lanes, taking into account the entire radioactivity in the plate, and were plotted using SigmaPlot 12 software for inhibition curves.
Determination of the 50% inhibitory concentration of steroids 5a-j on the activity of 5α-R2
To determine the 50% inhibitory concentration (IC50) of steroids 5a-j on the activity of 5α-R2, the inhibition plots obtained in “Experiments in vitro” section were analysed with SigmaPlot 12 software.
AR competitive binding assay
For the AR competitive binding studies, a series of tubes containing 1 nM [3H] MIB plus a range of increasing concentrations (1 × 10−10, 1 × 10−9 M, … 1 × 10−7 M) of cold MIB or of one of the steroids 5a–j () in ethanol, acetone or in the absence of competitor were preparedCitation5.
To prevent the interaction of MIB with either glucocorticoid or PR receptors that may be present in the cytosol from rat prostates, 200 nM triamcinolone (Sigma-Aldrich) in ethanol was added to the incubation mixturesCitation5. For each preparation, the solvent was removed under vacuum, and the cytosol containing AR (5 mg protein/200 μL), together with 300 μL of TEM (pH 8) containing protease inhibitors, was placed into individual tubes. The tubes were incubated (18 h; 4 °C), and the bound MIB-AR was separated from free radiolabeled MIB using HAP (Bio-Rad, Mexico, D.F., Mexico), as previously describedCitation17. The ethanolic fraction (0.8 mL) obtained by this method was added to Ultima Gold scintillation liquid (10 mL) (Packard Instruments, Mexico, D.F., Mexico) and counted in a Tri Carb 2100 TR scintillation counter (Packard Instruments, Meriden, CT).
The IC50 value of each compound was calculated according to the plots of concentration versus percentage of binding using SigmaPlot 12. The relative binding affinity (RBA) values of 5a-j for AR were calculated using the following equation:
IC50: concentration of compound required to inhibit 50% the activity of 5α-R2; RBA: relative binding activity to AR.
Calculation of log p values
The calculation of the log P parameter was carried out using the programme ACD/LABS version 9. This value is calculated on the basis of the molecular structure of the tested steroid and describes the relationship between the lipophilicity of the drug, represented by log P, and the minimum concentration of the drug (log1/c) necessary to produce biological activity. A higher value of log P indicates a greater solubility of the compound in the lipoprotein cell membrane with a corresponding increase in the pharmacological activity. This relationship is reliable up to a log P of 5.
Results
Chemistry
The synthetic strategy for compounds 5a–j is summarised in . The dehydroepiandrosterone derivatives 5a–5j were synthesised using a previously published procedure with slight modificationsCitation13. The new compounds 5a–j were obtained with moderate yields, and their structures and purities were confirmed by spectroscopic and spectrometric methods and melting point (see experimental section).
The yields of the products after the various steps in the syntheses were as follows: 86% for derivative 2 resulting from the Vilsmeier–Haack reaction, 94% for the free alcohol resulting from 2 being treated with sodium methoxyde-methanol in dimethylformamide, and 34%–91% for 5a–5j when the 16-formyl-17-methoxy-3β-ol was reacted with phosphorus oxychloride and dimethylformamide and chloroform.
All compounds were isolated in their crystalline form, and the melting points showed a maximum difference of two degrees.
Activity of compounds 5a–5j as inhibitors of 5α-R2
shows the IC50 values of 4 and 5a–d required to inhibit 50% of the activity of 5α-R2. The steroids 4 and 5a–d showed higher potency than that of the finasteride (the compound used as a referenceCitation18) to inhibit 5α-R2 enzyme. Compared with 4, 5a, 5c and 5d, 5b steroid (IC50 = 0.044 ± 0.012 nM) it was the most potent of them and exhibited an effectiveness 200 times greater than finasteride (IC50 = 8.5 ± 2.5 nM) to inhibit 5α-R2 activity. Nevertheless, derivatives 5f–j did not inhibit the activity of this enzyme ().
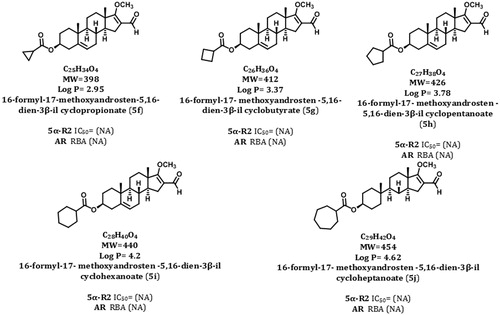
Figure 4. Structures, molecular weights, and biological activities of different dehydroepiandrosterone derivatives.
None of these compounds was capable to inhibit the activity of 5α-R2 enzyme.
IC50 values: concentration of compound required to inhibit 50% of the activity of 5α-reductase type 2 (5α-R2); RBA: relative binding activity to androgen receptor (AR).
Competitive binding of compounds 5a–5j to the AR
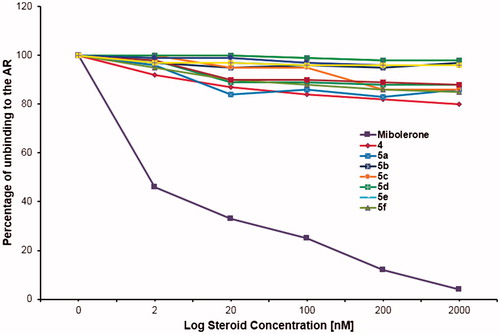
The results of the competitive binding assays () indicated that these dehydroepiandrosterone derivatives (5a–5j) did not replace labelled MIB of the AR.
Figure 5. Competitive binding of compounds 5a–5j to the androgen receptors (AR) Unlabelled mibolerone (MIB) competed with the labelled MIB by the binding to the AR; however, the novel steroids did not remove the labelled MIB from the AR. These results support the idea that these steroids did not bind to the AR.
Discussion
5a–5j steroids were designed to inhibit the activity of 5α-R2 and this effect it can be demonstrated throughout this study. Furthermore it was tried, in order to increase its therapeutic value as anti-prostate cancer drug, adding a formyl group at the C-16 position of these derivatives. This formyl could convert 5a–5j in a potential substrate of transformylase enzymes. These enzymes are involved in the purine synthesis, which are essential for the cancer cells divisionCitation19. Therefore, we hypothesise these steroidal derivatives could be inhibitors of the activity of these transformylasesCitation20 and thus could stop the prostate cancer cell growthCitation21. Nevertheless, the formyl group present at the C-16 5a–5j is toxic to the body so that an ester moiety was added in C-3 with the purpose to mask this formyl group. The pharmacological results obtained in this experiment demonstrated that the compounds 5a–5j were not toxic when applied to the hamsters for 6 consecutive days. Future investigation using these derivatives will be needed to determine their effect on the nucleotide synthesis pathway.
The lipophilicity that we added to steroids 5a–5d could be also advantageous for improving the ability of these molecules to cross through cell membranes. However, it is well known the fact the presence of non-specific esterases in the bloodstreamCitation22, thus these esters derivatives could be hydrolysed by the esterases present in the blood, and the resultant hydrolysed molecules may contain an alcohol moiety at C-3, similar to that present in steroid 4. This possibility was considered in this study, and we demonstrated that steroid 4 is also an inhibitor of 5α-R2.
In conclusion, since these esters can be regarded as prodrugs, could be used to mask side effects and toxicity caused by the formyl groupCitation23. Therefore, when these ester groups are hydrolysed by plasma esterases, the active drug is released causing the desired response.
Supplementary material available online
Supplementary Figures S1-12
Supplementary_Figures_S1_S12.pdf
Download PDF (1.5 MB)Declaration of interest
The authors declare that there are no real or perceived conflicts of interest arising from intellectual, personal, or financial circumstances of this research. Additionally, all authors are aware and approve of the contents and order of authorship of the manuscript. This study was supported by grants from Consejo Nacional de Ciencia y Tecnología (CONACYT project number 165049) and from Dirección General de Asuntos del Personal Académico (DGAPA project number IN 211312).
Related Research Data
References
- Arellano Y, Bratoeff E, Garrido M, et al. New ester derivatives of dehydroepiandrosterone as 5α-reductase inhibitors. Steroids 2011;76:1241–6
- Bratoeff E, Sánchez A, Arellano Y, et al. In vivo and in vitro effect of androstene derivatives as 5α-reductase type 1 enzyme inhibitors. J Enzyme Inhib Med Chem 2013;28:1247–54
- Garrido M, Cabeza M, Cortés F, et al. Cytotoxic effect of novel dehydroepiandrosterone derivatives on different cancer cell lines. Eur J Med Chem 2013;68:301–11
- Cabeza M, Trejo KV, González C, et al. Steroidal 5α-reductase inhibitors using 4-androstenedione as substrate. J Enzyme Inhib Med Chem 2011;26:712–19
- Bratoeff E, García P, Heuze Y, et al. Molecular interactions of progesterone derivatives with 5α-reductase types 1 and 2 and androgen receptors. Steroids 2010;75:499–505
- Russell DW, Wilson JD. Steroid 5 alpha-reductase: two genes/two enzymes. Annu Rev Biochem 1994;63:25–61
- Imperato-McGinley J, Zhu YS. Androgens and male physiology the syndrome of 5alpha-reductase-2 deficiency. Mol Cell Endocrinol 2002;198:51–9
- Dadras SS, Cai X, Abasolo I, Wang Z. Inhibition of 5alpha-reductase in rat prostate reveals differential regulation of androgen-response gene expression by testosterone and dihydrotestosterone. Gene Expr 2001;9:183–94
- Wright AS, Douglas RC, Thomas LN, et al. Androgen-induced regrowth in the castrated rat ventral prostate: role of 5alpha-reductase. Endocrinology 1999;140:4509–15
- Bosland MC. Chapter 2: the role of steroid hormones in prostate carcinogenesis. JNCI Monographs 2000;2000:39–66
- Raynaud JP. Prostate cancer risk in testosterone-treated men. Proceedings of the 17th International Symposium of the Journal of Steroid Biochemistry and Molecular Biology Recent Advances In Steroid Biochemistry And Molecular Biology (Seefeld, Tyrol); 2006 May 31–June 3;102:261–6; Austria
- Lee HJ, Chang C. Recent advances in androgen receptor action. Cell Mol Life Sci 2003;60:1613–22
- Njar VCO, Klus GT, Brodie AMH. Nucleophilic vinylic “addition-elimination” substitution reaction of 3-acetoxy-17-chloro-16-formylandrosta-5,16-diene: a novel and general route to 17-substituted steroids. Part 1. Synthesis of novel 17-azolyl-Δ16-steroids; inhibitors of 17α-hydroxylase/17,20-lyase (17α -lyase). Bioorg Med Chem Lett 1996;6:2777
- Bradford MM. A rapid and sensitive method for the quantisation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–54
- Altschul SF, Wootton JC, Gertz EM, et al. Protein database searches using compositionally adjusted substitution matrices. FEBS J 2005;272:5101–9
- Hendry IWJ, Danzo BJ. Structural conversion of cytosolic steroid receptors by an age-dependent epididymal protease. J Steroid Biochem 1985;23:883–93
- Mukherjee A, Kirkovsky LI, Kimura Y, et al. Affinity labelling of the androgen receptor with nonsteroidal chemoaffinity ligands. Biochem Pharmacol 1999;58:1259–67
- Bull HG, Garcia-Calvo M, Andersson S, et al. Mechanism-based inhibition of human steroid 5α-reductase by finasteride: enzyme-catalyzed formation of NADP-dihydrofinasteride, a potent bisubstrate analog inhibitor. J Am Chem Soc 1996;118:2359–65
- Warren MS, Marolewski AE, Benkovic SJ. A rapid screen of active site mutants in glycinamide ribonucleotide transformylase. Biochemistry 1996;35:8855–62
- Desharnais J, Hwang I, Zhang Y, et al. Design, synthesis and biological evaluation of 10-CF3CO-DDACTHF analogues and derivatives as inhibitors of GAR Tfase and the de novo purine biosynthetic pathway. Bioorg Med Chem 2003;11:4511–21
- Christopherson RI, Lyons SD, Wilson PK. Inhibitors of de novo nucleotide biosynthesis as drugs. Acc Chem Res 2002;35:961–71
- Graham LP. Medicinal chemistry. Oxford: Oxford University Press; 2001, 226–53
- Heck HD, Casanova M, Starr TB. Formaldehyde toxicity-new understanding. Crit Rev Toxicol 1990;20:397–426