
Abstract
Novel rhodesain inhibitors were developed by combining an enantiomerically pure 3-bromoisoxazoline warhead with a 1,4-benzodiazepine scaffold as specific recognition moiety. All compounds were proven to inhibit rhodesain with Ki values in the low-micromolar range. Their activity towards rhodesain was found to be coupled to an in vitro antitrypanosomal activity, with IC50 values ranging from the mid-micromolar to a low-micromolar value for the most active rhodesain inhibitor (R,S,S)-3. All compounds showed a good selectivity against the target enzyme since all of them were proven to be poor inhibitors of human cathepsin L.
Introduction
Human African Trypanosomiasis (HAT), also known as sleeping sickness, is a neglected tropical disease transmitted by bite of a fly (Glossina genus), afflicting millions of people in sub-Saharan Africa, with an estimated number of cases of 30 000/yearCitation1.
HAT is caused by a unicellular protozoan in the class of zooflagellates. The subspecies responsible for the human disease are Trypanosoma brucei gambiese and T. b. rhodesienseCitation2. The first subspecies, widespread in west and central Africa, are able to induce the chronic form of the disease, while T. b. rhodesiense, prevalent in the south-eastern end of Africa causes the rapid onset acute form of HAT.
The first hemolymphatic stage of the disease is characterized by fever, itching, headache, joint pain, while during the late neurological stage, the parasite crosses the blood–brain barrier (BBB) reaching the central nervous system, causing the typical symptoms of the disease such as sensory disturbances and coordination, and finally sleep–wake cycle disorders. If left untreated, sleeping sickness has a fatal outcome.
Because of the lack of an effective vaccine, chemotherapy is at present the unique way to control the disease. There are four currently available drugs to treat HAT: melarsoprol, eflornithine, pentamidine and suramin. Only two which are able to cross the BBB, and therefore are useful in the second stage of the disease, are melarsoprol and eflornithine. However, the arsenical compounds are highly toxic, while eflornithine, is ineffective towards T. b. rhodesiense, is very expensive and requires hospitalization to be administeredCitation3. For these reasons there is an urgent need to develop new drugs endowed with trypanocidal activity and to identify new potential molecular targets.
The cysteine proteases represent very promising targets for the development of new drugs with antiparasitic activity. In this context, rhodesain, a Clan CA papain-family cysteine protease plays an important role in the Trypanosoma life cycle. Rhodesain (TbCatL) is localized in the lysosome and is involved in the degradation of parasite proteins and intracellularly transported host proteinsCitation4. The protease is required to cross the BBB leading to the lethal stage of the sleeping sickness; it is involved in the turnover of variant surface glycoproteins (VSG) of T. brucei coat, and in the degradation of host immunoglobulines, thus evading the host immune response.
At present the main class of rhodesain inhibitorsCitation5 are peptides: vinyl sulfones, peptidyl aldehydes, diazomethyl or halomethyl ketones, peptidyl-α,β-epoxyesters, azadipeptide nitriles and peptides bearing an aziridine-2,3-dicarboxylate warhead. Furthermore, many other chemotypes have been identified as non-peptide rhodesain inhibitors, such as, thiosemicarbazones, isoquinolines, triazine nitriles, Michael acceptorsCitation6 or recently reported non-covalent rhodesain inhibitorsCitation7.
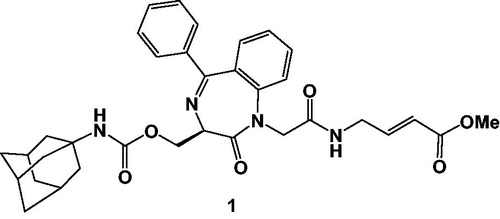
In this research area, our research group has been actively involved in the development of novel non-peptideCitation8 or peptidomimetic cysteine protease inhibitorsCitation9,Citation10, the latter characterized by the presence of a 1,4-benzodiazepine scaffold, introduced into a peptide backbone, in which the condensed aromatic ring could mimic a Phe residue at P2 site, highly preferred by rhodesain, while the hydroxylmethyl group at C3 of the scaffold was used to tie different substituents, able to create additional interactions with the S3 pocket of the target enzyme. In particular, we identified a vinyl ester 1 () which behaves as Michael acceptor, leading to an irreversible alkylation of the target enzyme. Ester 1, which is characterized by the presence of an adamantyl nucleus directly linked to the methyl carbamoyl portion appended to C3 of the BDZ scaffold, showed an impressive second-order rate constant of inhibition value (k2nd = 4 620 000 M−1 min−1), coupled with a good antitrypanosomal activity (IC50 = 4.8 μM)Citation11.
Figure 1. Structure of the Michael acceptor 1.
Starting from this lead compound we now decided to introduce at the C-terminal moiety the 3-bromo isoxazoline nucleus (i.e. compounds (R,R)-2 and (R,S)-2, ), successfully employed by our groupCitation12, to investigate the profile of these inhibitors. Moreover, we have also designed peptidomimetics in which a molecular rigidification has been introduced by incorporating the 3-bromo isoxazoline nucleus into a bicyclic system [i.e. (R,R,R)-3 and (R,S,S)-3), ].
Figure 2. Structure of target compounds.
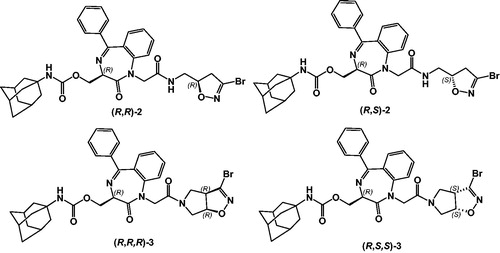
Herein, we report the synthesis and the biological evaluation of compounds (R,R)-2, (R,S)-2, (R,R,R)-3 and (R,S,S)-3 against rhodesain and T. brucei brucei, together with the selectivity towards the target enzyme, verified by testing the new compounds against human cathepsin L, like rhodesain also belonging to papain-family.
Results and discussion
Chemistry
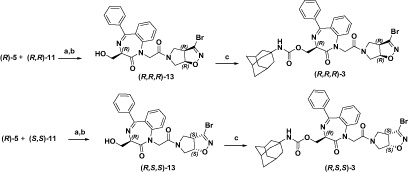
To synthesize compounds (R,R)-2 and (R,S)-2 we first accomplished the synthesis of the key intermediate (R)-5 (Scheme 1) in enantiomerically pure form starting from Garner ester 4, following our previously described procedure (9a).
Scheme 1. Reagents and conditions: (a) LiOH, MeOH, 0 °C, rt, 6 h; (b) i-BuOCOCl, NMM, CH2Cl2, 0 °C, rt, 30 min, then 2-aminobenzophenone, reflux, 20 min, rt, 12 h; (c) HCl/MeOH, reflux, 5 h, then NaHCO3, MeOH, rt, 12 h; (d) TBS-Cl, imidazole, CH2Cl2, 0 °C, rt, 12 h; (e) NaH, BrCH2COOEt, 0 °C, rt, 5 h; (f) LiOH, MeOH, 0 °C, rt, 6 h; (g) lipase-PS, potassium phosphate buffer, pH = 7, acetone, 65′; (h) MsCl, TEA, CH2Cl2, −10 °C, rt, 16 h; (i) NaN3, DMSO, 60 °C, 4 h; (j) Ph3P, THF/H2O, rt, 24 h; (k) DBF, NaHCO3, EtOAc, rt, 48 h; (l) preparative chiral HPLC; and (m) 30% TFA, CH2Cl2, 0 °C, rt, 4 h.
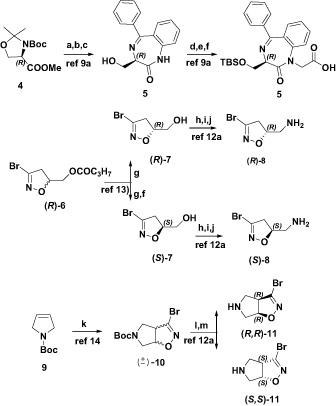
Enantiomerically pure amines (R)-8 and (S)-8 were prepared starting from enantiopure alcohols (R)-7 and (S)-7 (12a) in turn obtained, with a 94% enantiomeric excess, following a previously reported methodologyCitation13.
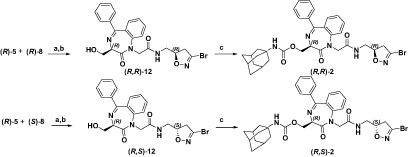
We then coupled the carboxylic acid (R)-5 to the enantiopure amines (R)-8 and (S)-8 (Scheme 2), using carbodiimide EDCI and HOBt as coupling reagent. The coupling products were then desilylated by treatment with TBAF to afford (R,R)-12 and (R,S)-12, which by subsequent reaction with the adamantyl isocyanate gave the desired carbamates (R,R)-2 and (R,S)-2.
Scheme 2. Reagents and conditions: (a) EDCI, HOBt, DIPEA, CH2Cl2, rt, 12 h; (b) TBAF, THF, rt; 6 h; and (c) 1-adamantylNCO, TEA, CH2Cl2, rt, 72 h.
Conversely, for the synthesis of compounds (R,R,R)-3 and (R,S,S)-3, the bicyclic amine (±)-10Citation14 was first prepared via 1,3-dipolar cycloaddition between the dipolarophile 9 and bromonitrileoxide, generated in situ by dehydrohalogenation of the stable precursor dibromoformaldoxime (DBF). Enantiopure amines (R,R)-11 and (S,S)-11 were obtained by resolving the racemic mixture of (±)-10 by chiral HPLC, both with a 99% enantiomeric excess, followed by treatment with 30% TFA in CH2Cl2 (12a).
The condensation of the bicyclic amines (R,R)-11 and (S,S)-11 to the carboxylic acid (R)-5, under the same conditions established for compounds (R,R)-2 and (R,S)-2, allowed us to recover the two diastereoisomers (R,R,R)-3 and (R,S,S)-3 (Scheme 3).
Scheme 3. Reagents and conditions: (a) EDCI, HOBt, DIPEA, CH2Cl2, rt, 12 h; (b) TBAF, THF, rt; 6 h; and (c) 1-adamantylNCO, TEA, CH2Cl2, rt, 72 h.
Biological activity
Compounds (R,R)-2, (R,S)-2, (R,R,R)-3 and (R,S,S)-3 () were tested for their inhibitory activity against rhodesain using Cbz-Phe-Arg-AMC as fluorogenic substrate.
Table 1. Activity against rhodesain, hCatL and T. B. Brucei.
First a preliminary screening with an inhibitor concentration of 100 µM was performed and an equivalent volume of DMSO was used as negative control. Compounds capable of inhibiting the enzymatic activity by more than 70% were characterized in detail. Continuous assays were then performed (progress curve method, at seven different concentrations) ranging from those that minimally inhibited to those that fully inhibited the enzyme (), to determine the Ki values () are reported in . All compounds were proven to inhibit rhodesain in the low micromolar range, with the best binding affinity shown by the inhibitor (R,S,S)-3 (Ki = 1.26 µM).
Figure 3. Progress curves of substrate hydrolysis in the presence of the inhibitor (R,S,S)-3. F = fluorescence units. Inhibitor concentrations (from top to bottom): 0, 10, 20, 40, 50, 60, 80 and 100 µM.
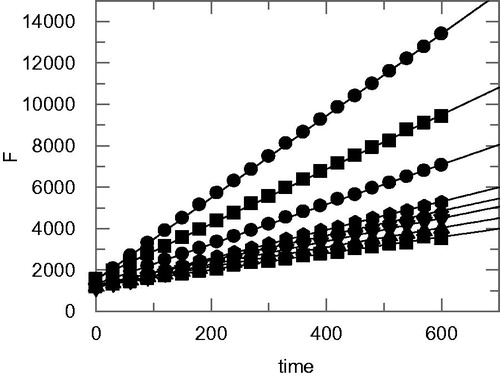
Figure 4. The slopes of the progress curve (b) from were plotted against the inhibitor concentrations and fitted to the 4 parameter IC50 equation. Ki was obtained from the Cheng–Prusoff equation Ki = IC50/(1 + [S]).
![Figure 4. The slopes of the progress curve (b) from Figure 2 were plotted against the inhibitor concentrations and fitted to the 4 parameter IC50 equation. Ki was obtained from the Cheng–Prusoff equation Ki = IC50/(1 + [S]).](/cms/asset/83dd22ab-0462-494c-92c9-a50e17e8f9a8/ienz_a_1108972_f0004_b.jpg)
In case of both couple of diastereoisomers 2 and 3, compounds (S,S) configured on the warhead showed a slightly improved activity towards the target enzyme, e.g. (R,S,S)-3 versus (R,R,R)-3 with Ki values of 1.26 µM and 2.49 µM, respectively.
Selectivity assays were also performed, testing inhibitors against human cathepsin L: none of the synthesized compounds 2–3 passed the initial screening at 100 μM and thus demonstrating a good selectivity against the target enzyme.
The antitrypanosomal activity of the rhodesain inhibitors (R,R)-2, (R,S)-2, (R,R,R)-3 and (R,S,S)-3 was also evaluated on T. brucei brucei TC221 strains, with obtained IC50 values in the range 4.71–13.07 µM. Worthy of note, the most active rhodesain inhibitor (R,S,S)-3 showed the strongest antitrypanosomal activity (IC50 = 4.71 µM) when compared to the other three derivatives, thus giving an evidence of a clear correlation between inhibitory properties and antiparasitic activity.
Given the satisfactory antitrypanosomal properties of (R,S,S)-3 we decided to profile its physicochemical and pharmacokinetic (PK) features as a rough assessment of its drug-like character. For such a purpose, we employed the Qikprop program (Schrödinger, LLC, New York, NY). The results are summarized in . These predictions would indicate that all the predicted parameters fall into the ranges calculated for the 95% on the marketed drugs. Of note, (R,S,S)-3 is predicted to have acceptable gut/blood barrier permeability (see QPPCaco values in ), high potential of passing the blood/brain barrier (see QPlogBB and QPPMDCK values in ) and more than 80% human oral absorption. All these data underscore the potentiality of this compound to be considered as the ideal candidate lead compound for the discovery of potent antitrypanosomal agents endowed with promising PK parameters.
Table 2. Calculated physicochemical and pharmacokinetic properties of compound (R,S,S)-3.
Conclusions
In conclusion, starting from the lead compound 1, we designed two couples of diastereoisomers: a first one (R,R)-2/(R,S)-2 containing a 3-bromo isoxazoline nucleus as novel warhead, and a second one in which the 3-bromo isoxazoline moiety was incorporated into a bicyclic system [i.e. (R,R,R)-3/(R,S,S)-3]. All compounds were proven to inhibit rhodesain with Ki values in the low micromolar range, coupled with a good target selectivity, evaluated by testing compounds against the human cathepsin L.
The most active rhodesain inhibitor (R,S,S)-3 (Ki = 1.26 µM) showed a good match of enzyme inhibitory properties and antitrypanosomal activity, confirmed by an IC50 value of 4.71 µM, comparable to that of the potent Michael acceptor 1 (IC50 = 4.8 µM).
These data clearly indicate that the 3-bromo isoxazoline moiety, coupled with the 1,4-BDZ scaffold, as recognition moiety, has been successfully employed. The presence of the adamantly nucleus, directly linked to the methyl carbamoyl portion appended to C3 of the BDZ core, is probably fundamental for the physicochemical properties of the inhibitor, making it suitable to cross the parasite membrane. This hypothesis is supported by the interesting PK properties calculated for the most active derivative.
In view of these consideration, compound (R,S,S)-3 could be considered as a lead compound for further development in the design of new inhibitors which can be used as antitrypanosomal agents.
Experimental
Chemistry
General: All reagents and solvents were obtained from commercial suppliers and were used without further purification. Elemental analyses were carried out on a C. Erba Model 1106 (Elemental Analyzer for C, H and N) instrument, and the obtained results are within ±0.4% of the theoretical values. Merck silica gel 60 F254 plates were used for analytical TLC; flash column chromatography was performed on Merck silica gel (200–400 mesh) using a MP-LC BUCHI system. 1H and 13C and NMR spectra were recorded on Bruker Avance 300 MHz NMR spectrometer equipped with a BBI probe and operating at frequencies of 300.13 and 75.47 MHz. We used the residual signal of the deuterated solvent as an internal standard. Splitting patterns are described as singlet (s), doublet (d), doublet of doublet (dd), triplet (t), quartet (q), multiplet (m), or broad singlet (br s). 1H and 13C NMR chemical shifts (δ) are expressed in ppm, and coupling constants (J) are given in Hz. MS analyses were performed on a Varian 320-MS triple-quadrupole mass spectrometer equipped with an electron-spray ionization (ESI) source. Polarimetric analyses were carried out on a Perkin-Elmer Polarimeter 341.
Synthesis of compounds (R,R)-2 and (R,S)-2
N-(((R)-3-bromo-4,5-dihydroisoxazol-5-yl)methyl)-2-((R,Z)-2,3-dihydro-3-(hydroxymethyl)-2-oxo-5-phenylbenzo[e][1,4]diazepin-1-yl)acetamide [(R,R)-12]
Step 1. To a solution of (R)-[3-(tert-butyl-dimethyl-silanyloxymethyl)-2-oxo-5-phenyl-2,3-dihydro-benzo[e][1,4]diazepin-1-yl]-acetic acid (R)-5 (9a) (500 mg, 1.14 mmol) in CH2Cl2 (20 mL), HOBt (185 mg, 1.37 mmol) and EDCI (263 mg, 1.37 mmol) at 0 °C were added. After 5′ the ice bath was removed and ((R)-3-bromo-4,5-dihydroisoxazol-5-yl)methanamine (R)-8 (12a) (203 mg, 1.14 mmol) and DIPEA (177 mg, 234 µL, 1.37 mmol) were added to the resulting mixture and the latter was stirred at room temperature for 12 h. The organic layer was separated, dried (Na2SO4), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography eluting with light petroleum/EtOAc (8/2) to give N-(((R)-3-bromo-4,5-dihydroisoxazol-5-yl)methyl)-2-((R,Z)-2,3-dihydro-3-(tert-butyl-dimethyl-silanyloxymethyl)-2-oxo-5-phenylbenzo[e][1,4]diazepin-1-yl)acetamide (636 mg, 93%).
Step 2. To a solution of N-(((R)-3-bromo-4,5-dihydroisoxazol-5-yl)methyl)-2-((R,Z)-2,3-dihydro-3-(tert-butyl-dimethyl-silanyloxymethyl)-2-oxo-5-phenylbenzo[e][1,4]diazepin-1-yl)acetamide (636 mg, 1.06 mmol) in dry THF (20 mL) TBAF (1.6 mL of 1 M solution in THF, 1.60 mmol) was added dropwise. The mixture was stirred at room temperature until disappearance of the starting material (TLC monitoring) and then it was diluted with EtOAc (40 mL) and washed with water (2 × 100 mL). The organic layer was separated, dried (Na2SO4), filtered, concentrated under reduced pressure and purified by flash column chromatography eluting with EtOAc to give the deprotected title compound (R,R)-12 (504 mg, 98%) Rf (EtOAc/light petroleum, 8:2) = 0.18. 1H NMR (300 MHz, CDCl3) δ: 2.93 (bs, 1H), 3.03 (dd, J = 7.3, 17.2 Hz, 1H), 3.33 (dd J = 10.2, 17.2 Hz, 1H), 3.40–3.56 (m, 2H), 3.88 (t, J = 7.4 Hz, 1H), 4.07 (d, J = 16.0 Hz, 1H), 4.19–4.29 (m, 1H), 4.36–4.45 (m, 1H), 4.73 (d, J = 16.0 Hz, 1H), 4.76–4.84 (m, 1H), 6.64 (bs, 1H), 7.18–7.68 (m, 9H).
N-(((S)-3-bromo-4,5-dihydroisoxazol-5-yl)methyl)-2-((R,Z)-2,3-dihydro-3-(hydroxymethyl)-2-oxo-5-phenylbenzo[e][1,4]diazepin-1-yl)acetamide [(R,S)-12]
Compound (R,S)-12 was prepared as described for its diastereoisomer (R,R)-12, starting from (R)-5 (9a) (500 mg, 1.14 mmol) and amine (S)-8 (12a) (203 mg, 1.14 mmol), followed by deprotection with TBAF (1.6 mL of 1 M solution in THF, 1.6 mmol). (Overall yield 90%, 497 mg); Rf (EtOAc/light petroleum, 8:2) = 0.18; 1H NMR (300 MHz, CDCl3) δ: 3.01 (bs, 1H), 3.08 (dd, J = 7.2, 17.2 Hz, 1H), 3.35 (dd J = 10.3, 17.2 Hz, 1H), 3.42–3.51 (m, 1H), 3.60–3.78 (m, 1H), 3.87 (t, J = 7.4 Hz, 1H), 4.09 (d, J = 16.0 Hz, 1H), 4.18–4.26 (m, 1H), 4.34–4.43 (m, 1H), 4.72 (d, J = 16.0 Hz, 1H), 4.74–4.82 (m, 1H), 6.60 (bs, 1H), 7.20–7.67 (m, 9H).
((R-1-(2-(((R)-3-bromo-4,5-dihydroisoxazol-5-yl)methylamino)-2-oxoethyl)-2-oxo-5-phenyl-2,3-dihydro-1H-benzo[e][1,4]diazepin-3-yl)methyl adamantan-1-ylcarbamate [(R,R)-2]
To a solution of compound (R,R)-12 (504 mg, 1.04 mmol), in dry CH2Cl2 (20 mL), under nitrogen atmosphere, adamantyl isocyanate (369 mg, 2.08 mmol) and TEA (210 mg, 289 µL, 2.08 mmol) were added and the resulting mixture was stirred for 72 h at room temperature. After this time the mixture was washed with water (2 × 100 mL), dried over Na2SO4 and the solvent was removed under reduced pressure. The crude material was purified by flash column chromatography eluting with EtOAc/light petroleum 8:2 to give the title compound (R,R)-2. (262 mg, 38% yield). Rf (EtOAc/light petroleum, 8:2) = 0.61; Anal. Calcd. for C33H36BrN5O5: C 59.26, H 5.28, N 10.80; found: C 59.01, H 5.39, N 10.44; = −35.0 (c = 0.25, CHCl3); MS [M + 1]+: 662.1; 1H NMR (300 MHz, CDCl3) δ: 1.56–1.74 (m, 8H), 1.86–1.96 (m, 4H), 2.02–2.08 (m, 3H), 2.97 (dd, J = 8.0 and 17.3 Hz, 1H), 3.20 (dd, J = 10.5 and 17.3 Hz, 1H), 3.47–3.55 (m, 2H), 3.9 (t, J = 6.3 Hz, 1H), 4.22 (d, J = 16.0 Hz, 1H), 4.70 (d, J = 16.0 Hz, 1H), 4.71–4.80 (m, 1H), 4.81–4.88 (m, 2H), 4.95 (bs,1H), 6.66 (bs, 1H), 7.26–7.64 (m, 9H); 13C NMR (75 MHz, CDCl3): δ = 29.93, 36.57, 41.96, 42.01, 44.14, 52.68, 62.18, 65.53, 80.35, 121.53, 124.89, 128.60, 129.33, 129.46, 130.01, 130.86, 132.47, 134.48, 138.01, 138.41, 153.47, 166.78, 169.04, 170.16.
((R-1-(2-(((S)-3-bromo-4,5-dihydroisoxazol-5-yl)methylamino)-2-oxoethyl)-2-oxo-5-phenyl-2,3-dihydro-1H-benzo[e][1,4]diazepin-3-yl)methyl adamantan-1-ylcarbamate [(R,S)-2]
Compound (R,S)-2 was synthesized as described for its diastereoisomer (R,R)-2 starting from compound (R,S)-12 (497 mg, 1.02 mmol), adamantyl isocyanate (361 mg, 2.04 mmol) and TEA (206 mg, 284 µL, 2.04 mmol). (243 mg, 36%); Rf (EtOAc/light petroleum, 8:2) = 0.61; Anal. Calcd. for C33H36BrN5O5: C 59.26, H 5.28, N 10.80; found: C 59.08, H 5.51, N 10.51; = -15.0 (c = 0.25, CHCl3); MS [M + 1]+: 662.2. 1H NMR (300 MHz, CDCl3) δ: 1.55–1.79 (m, 9H), 1.85–1.98 (m, 4H), 2.01–2.09 (m, 2H), 3.02 (dd, J = 7.4 and 17.6 Hz, 1H), 3.26 (dd, J = 11.0 and 17.6 Hz, 1H), 3.43–3.54 (m, 1H), 3.62–3.80 (m, 1H), 3.88–3.96 (m, 1H), 4.10 (d, J = 15.4 Hz, 1H), 4.72–4.91 (m, 4H), 5.16 (bs, 1H), 6.89 (bs, 1H), 7.32–7.64 (m, 9H); 13C NMR (75 MHz, CDCl3): δ = 29.90, 36.55, 41.98, 42.03, 44.10, 52.63, 62.20, 65.50, 80.32, 121.50, 124.90, 128.62, 129.30, 129.49, 130.05, 130.83, 132.48, 134.50, 138.03, 138.40, 153.50, 166.80, 169.05, 170.12.
Synthesis of compounds (R,R,R)-3 and (R,S,S)-3
(R,4Z)-1-(2-((3aR,6aR)-3-bromo-3a,4,6,6a-tetrahydropyrrolo[3,4-d]isoxazol-5-yl)-2-oxoethyl)-3-(hydroxymethyl)-5-phenyl-1H-benzo[e][1,4]diazepin-2(3H)-one [(R,R,R)-13]
Compound (R,R,R)-13 was prepared as described for compound (R,R)-12, starting from acid (R)-5 (9a) (500 mg, 1.14 mmol) and amine (R,R)-11(12a) (218 mg, 1.14 mmol); followed by deprotection with TBAF (1.6 mL of 1M solution in THF, 1.6 mmol). (Overall yield 91%, 516 mg); Rf (EtOAc/light petroleum, 8:2) = 0.11. 1H NMR (300 MHz, CDCl3) δ: 3.55–3.63 (m, 1H), 3.82–4.05 (m, 5H), 4.12–4.19 (d, J = 16.1 Hz, 1H), 4.26–4.35 (m, 2H), 4.70–4.77 (d, J = 16.1, 1H), 5.34–5.41 (m, 1H), 7.17–7.65 (m, 9H).
(R,4Z)-1-(2-((3aS,6aS)-3-bromo-3a,4,6,6a-tetrahydropyrrolo[3,4-d]isoxazol-5-yl)-2-oxoethyl)-3-(hydroxymethyl)-5-phenyl-1H-benzo[e][1,4]diazepin-2(3H)-one [(R,S,S)-13]
Compound (R,S,S)-13 was synthesized as described for compound (R,R)-12, starting from acid (R)-5 (9a) (500 mg, 1.14 mmol) and amine (S,S)-11 (12a) (218 mg, 1.14 mmol), followed by subsequent deprotection with TBAF (1.6 mL of 1M solution in THF, 1.6 mmol). (Overall yield 89%, 505 mg); Rf (EtOAc/light petroleum, 8:2) = 0.11. 1H NMR (300 MHz, CDCl3) δ: 3.38–3.48 (m, 1H), 3.69–3.83 (m, 2H), 3.84–3.98 (m, 3H), 4.11–4.18 (d, J = 15.2 Hz, 1H), 4.22–4.34 (m, 2H), 4.74–4.81 (d, J = 15.2 Hz, 1H), 5.28–5.36 (m, 1H), 7.20–7.66 (m, 9H).
((R,4Z)-1-(2-((3aR,6aR)-3-bromo-3a,4,6,6a-tetrahydropyrrolo[3,4-d]isoxazol-5-yl)-2-oxoethyl)-2,3-dihydro-2-oxo-5-phenyl-1H-benzo[e][1,4]diazepin-3-yl)methyl adamantylcarbamate [(R,R,R)-3]
The title compound has been synthesized following the procedure reported for compound (R,R)-2, starting from (R,R,R)-13 (516 mg, 1.04 mmol), adamantly isocyanate (369 mg; 2.08 mmol) and TEA (210 mg, 289 µL, 2.08 mmol). (224 mg, 32%); Rf (EtOAc/light petroleum, 8:2) = 0.46; Anal. calcd for C34H36BrN5O5: C 60.54, H 5.38, N 10.38; found C 60.71, H 5.67, N 10.22; = −58.8 (c = 0.36, CHCl3); MS [M + 1]+: 674.1; 1H NMR (300 MHz, CDCl3) δ: 1.56–1.67 (m, 8H), 1.88–1.93 (m, 4H), 2.00–2.07 (m, 3H), 3.59–3.66 (m, 1H), 3.80–4.03 (m, 5H), 4.13 (d, J = 16.1 Hz, 1H), 4.28 (d, J = 16.1 Hz, 1H), 4.72–4.83 (m, 2H), 5.12 (bs, 1H), 5.31–5.40 (m, 1H), 7.12–7.66 (m, 9H); 13C NMR (75 MHz, CDCl3): δ = 29.55, 36.44, 42.54, 48.61, 50.81, 51.18, 53.97, 62.38, 63.29, 84.83, 122.20, 124.60, 128.31, 128.91, 129.52, 130.22, 130.67, 132.12, 138.34, 138.83, 139.66, 156.25, 166.77, 169.15, 169.42.
((R,4Z)-1-(2-((3aS,6aS)-3-bromo-3a,4,6,6a-tetrahydropyrrolo[3,4-d]isoxazol-5-yl)-2-oxoethyl)-2,3-dihydro-2-oxo-5-phenyl-1H-benzo[e][1,4]diazepin-3-yl)methyl adamantylcarbamate [(R,S,S)-3]
The title compound has been synthesized following the procedure reported for compound (R,R)-2, starting from (R,S,S)-13 (505 mg, 1.01 mmol), adamantly isocyanate (358 mg; 2.02 mmol) and TEA (204 mg, 281 µL, 2.02 mmol). (221 mg, 31%); Rf (EtOAc/light petroleum, 8:2) = 0.46; Anal. calcd for C34H36BrN5O5: C 60.54, H 5.38, N 10.38; found C 60.83, H 5.54, N 10.13; = −15.5 (c = 0.18, CHCl3);MS [M + 1]+: 674.2; 1H NMR (300 MHz, CDCl3) δ: 1.59–1.68 (m, 8H), 1.93–1.97 (m, 4H), 2.04–2.10 (m, 3H), 3.61–3.69 (m, 1H), 3.85–4.08 (m, 5H), 4.15 (d, J = 15.8 Hz, 1H), 4.31 (d, J = 15.8 Hz, 1H), 4.75–4.88 (m, 2H), 5.18 (bs, 1H), 5.32–5.46 (m, 1H), 7.16–7.67 (m, 9H); 13C NMR (75 MHz, CDCl3): δ = 29.53, 36.42, 42.55, 48.60, 50.79, 51.20, 53.96, 62.35, 63.28, 84.80, 122.18, 124.58, 128.29, 128.92, 129.50, 130.20, 130.69, 132.10, 138.37, 138.80, 139.64, 156.27, 166.78, 169.16, 169.40.
Biology
Enzyme assays
The preliminary screening was performed with 100 µM inhibitor concentration using an equivalent amount of DMSO as negative control. The enzyme was recombinantly expressed as previously describedCitation4. Product release from substrate hydrolysis (Cbz-Phe-Arg-AMC, 10 µM) was determined continuously over a period of 10 min. Compounds showing at least 70% inhibition at 100 µM were subjected to detailed assays. These were performed in a 50 mM sodium acetate buffer, pH 5.5 containing 10 mM DTT with Cbz-Phe-Arg-AMC (10 µM) as substrateCitation15. The Km value used to calculate Ki values from IC50 values was determined as 0.9 µM (rhodesain)Citation16. Inhibitor solutions were prepared from stocks in DMSO. Each assay was performed twice in 96-well-plates in a total volume of 200 µL. Fluorescence of the product AMC of the substrate hydrolyses was measured using an Infinite 200 PRO microplate reader (Tecan, Männedorf, Switzerland) at room temperature with a 380 nm excitation filter and a 460 nm emission filter. The dissociation constants Ki were obtained from progress curves (10 min) at various concentrations of inhibitor by fitting the progress curves to the 4-parameter IC50 equation:
with y [dF/min] as the substrate hydrolysis rate, ymax as the maximum value of the dose–response curve that is observed at very low inhibitor concentrations, ymin as the minimum value that is obtained at high inhibitor concentrations, and s denotes the Hill coefficientCitation17, and correction to zero substrate concentration from Ki = IC50/(1 + [S]
).
Assays with cathepsin L was performed as described previouslyCitation18, Cbz-Phe-Arg-AMC was used as substrate (5 µM).
Drug screening on T. b. brucei cultures
Trypomastigote forms of T. b. brucei laboratory strain TC 221 were cultured in Baltz medium according to standard conditionsCitation19. Trypanocidal activity was determined using the Alamar Blue assayCitation20,Citation21 in a 96-well plate format and a MR 700 Microplate ELISA Reader. Trypanosomes were added to culture medium containing various concentrations of test compound and 1% solvent to give a cell concentration of 104 cells mL−1 in a final volume of 200 µL. Positive and negative controls comprised wells containing medium, 1% solvent and trypanosomes, and wells with test compounds but without trypanosomes, respectively. After 24 h, 20 µL of Alamar Blue were added to each well and the plates were incubated again for a further 24 h. Absorbance were then measured at 550 nm with a reference wavelength of 630 nm. IC50 values were calculated by linear interpolation as describedCitation22. Experiments were repeated twice.
Computational chemistry
A molecular model of (R,S,S)-3 was constructed and energy minimized using the Schrodinger Maestro 12 program running on a Dell Precision with Fedora Core. Minimizations were performed using the OPLS 2005 force field, an enhanced version of Jorgensen’s OPLS force field, 4 with 5000 steps of steepest descent followed by conjugate-gradient energy calculations with a convergence of 0.0005 kJÅ−1 mol−1. Subsequently, the constructed (R,S,S)-3 was subjected to QikProp software calculations (QikProp, version 3.4 (2011); Schrödinger, LLC, New York, NY). In addition to predicting molecular properties, QikProp provides ranges for comparing each compound’s property with those of 95% of known drugs. This software was also used because it allows for flagging reactive functional groups that may cause false positives in biological assays.
Declaration of interest
The authors report no declarations of interest.
Related Research Data
References
- http://www.who.int/neglected_diseases/diseases/en/
- Barrett MP, Burchmore RJS, Stich A, et al. The trypanosomiases. Lancet 2003;362:1469–80
- http://www.who.int/mediacentre/factsheets/fs259/en/
- Caffrey CR, Hansell E, Lucas KD, et al. Active site mapping, biochemical properties and subcellular localization of rhodesain, the major cysteine protease of Trypanosoma brucei rhodesiense. Mol Biochem Parasitol 2001;118:61–73
- Ettari R, Tamborini L, Angelo IC, et al. Inhibition of rhodesain as a novel therapeutic modality for human African trypanosomiasis. J Med Chem 2013;56:5637–58
- McShan D, Kathman S, Lowe B, et al. Identification of non-peptidic cysteine reactive fragments as inhibitors of cysteine protease rhodesain. Bioorg Med Chem Lett 2015;25:4509-12
- Jefferson T, McShan D, Warfield J, Ogungbe IV. Screening and identification of inhibitors of Trypanosoma brucei cathepsin L with antitrypanosomal activity. Chem Biol Drug Des 2015. [Epub ahead of print]. doi:10.1111/cbdd.12628
- Micale N, Ettari R, Schirmeister T, et al. Novel 2H-isoquinolin-3-ones as antiplasmodial falcipain-2 inhibitors. Bioorg Med Chem 2009;17:6505–11
- (a) Micale N, Kozikowski AP, Ettari R, et al. Novel peptidomimetic cysteine protease inhibitors as potential antimalarial agents. J Med Chem 2006;49:3064–7. (b) Ettari R, Nizi E, Di Francesco ME, et al. Development of peptidomimetics with a vinyl sulfone warhead as irreversible falcipain-2 inhibitors. J Med Chem 2008;51:988–96. (c) Ettari R, Nizi E, Di Francesco ME, et al. Nonpeptidic vinyl and allyl phosphonates as falcipain-2 inhibitors. ChemMedChem 2008;3:1030–3. (d) Ettari R, Micale N, Schirmeister T, et al. Novel peptidomimetics containing a vinyl ester moiety as highly potent and selective falcipain-2 inhibitors. J Med Chem 2009;52: 2157–60
- (a) Ettari R, Zappalà M, Micale N, et al. Synthesis of novel peptidomimetics as inhibitors of protozoan cysteine proteases falcipain-2 and rhodesain. Eur J Med Chem 2010;45:3228–33. (b) Ettari R, Zappalà M, Micale N, et al. Peptidomimetics containing a vinyl ketone warhead as falcipain-2 inhibitors. Eur J Med Chem 2011;46:2058–65. (c) Ettari R, Micale N, Grazioso G, et al. Synthesis and molecular modeling studies on derivatives of a highly potent peptidomimetic vinyl ester as falcipain-2 inhibitor. ChemMedChem 2012;7:1594–600. (d) Grazioso G, Legnani L, Toma L, et al. Mechanism of falcipain-2 inhibition by a,b-unsaturated benzo[1,4]diazepin-2-one methyl ester. JCAMD 2012;26:1035–43
- Bova F, Ettari R, Micale N, et al. Constrained peptidomimetics as antiplasmodial falcipain-2 inhibitors. Bioorg Med Chem 2010;18:4928–38
- (a) Ettari R, Tamborini L, Angelo IC, et al. Development of novel inhibitors of rhodesain with a 3-bromo-isoxazoline warhead. ChemMedChem 2013;8:2070–6. (b) Ettari R, Pinto A, Tamborini L, et al. Synthesis and biological evaluation of papain-family cathepsin L-like cysteine protease inhibitors containing a 1,4-benzodiazepine scaffold, as antiprotozoan agents. ChemMedChem 2014;9:1817–25. (c) Ettari R, Pinto A, Previti S, et al. Development of novel dipeptide-like rhodesain inhibitors containing the 3-bromoisoxazoline warhead in a constrained conformation. Bioorg Med Chem 2015. [Epub ahead of print]. doi:10.1016/j.bmc.2015.09.029
- De Amici M, Magri P, De Micheli C, et al. Nitrile oxides in medicinal chemistry. Chemoenzymic synthesis of chiral heterocyclic derivatives.. J Org Chem 1992;57:2825–9
- Conti P, De Amici M, Pinto A, et al. Synthesis of 3-hydroxy- and 3-carboxy-?2-isoxazoline amino acids and evaluation of their interaction with GABA receptors and transporters. Eur J Org Chem 2006;24:5533–42
- Breuning A, Degel B, Schulz F, et al. Michael acceptor based antiplasmodial and antitrypanosomal cysteine protease inhibitors with unusual amino acids. J Med Chem 2010;53:1951–63
- Vicik R, Hoerr V, Glaser M, et al. Aziridine-2,3-dicarboxylate inhibitors targeting the major cysteine protease of Trypanosoma brucei as lead trypanocidal agents. Bioorg Med Chem Lett 2006;16:2753–7
- Ludewig S, Kossner M, Schiller M, et al. Enzyme kinetics and hit validation in fluorimetric protease assays. Curr Top Med Chem 2010;10:368–82
- Vicik R, Busemann M, Gelhaus C, et al. Aziridide-based inhibitors of cathepsin L: synthesis, inhibition activity, and docking studies. ChemMedChem 2006;1:1126–41
- Baltz T, Baltz D, Giroud C, Crocket J. Cultivation in semi-defined medium of animal infective forms of Trypanosoma brucei, T. equiperdum, T. evansi, T. rhodesiense and T. gambiense. EMBO J 1985;4:1273–7
- Räz B, Iten M, Grether-Bühler Y, et al. The Alamar Blue assay to determine drug sensitivity of African trypanosomes (T.b. rhodesiense and T.b. gambiense) in vitro. Acta Trop 1997;68:139–47
- Merschjohann K, Sporer F, Steverding D, Wink M. In vitro effect of alkaloids on bloodstream forms of Trypanosoma brucei and T. congolense. Planta Med 2001;67:623–7
- Huber W, Koella JC. A comparison of three methods of estimating EC50 in studies of drug resistance of malaria parasites. Acta Trop 1993;55:257–61