
Abstract
N-protected amino acids were reacted with substituted benzothiazoles to give the corresponding N-protected amino acid-benzothiazole conjugates (60–89%). Their structures were confirmed by proton nuclear magnetic resonance (1H NMR), carbon-13 nuclear magnetic resonance (13C NMR), IR and elemental analysis. Their carbonic anhydrase (CA, EC 4.2.1.1) inhibitory activities were determined against two cytosolic human isoforms (hCA I and hCA II), one membrane-associated (hCA IV) and one transmembrane (hCA XII) enzyme by a stopped-flow CO2 hydrase assay method. The new compounds showed rather weak, micromolar inhibitory activity against most of these enzymes.
Introduction
Nitrogen and sulfur-containing heterocycles such as benzothiazoles, benzimidazole and benzoxazoles play an important role for life science and in many industrial fields, such as special and fine chemistryCitation1. Among them, benzothiazoles, in particular, have been studied extensively because of their biological activities as antitumorCitation2, anti-inflammatoryCitation3, antimicrobialCitation4, anticonvulsantCitation5, neuroprotectiveCitation6, kinaseCitation7, microsomal triglyceride transfer proteinCitation8 and carbonic anhydrase inhibitors (CAIs)Citation9. On the other hand, short peptides and their conjugates exhibit a broad spectrum of biological activity. Recently, one of our groups reported biological properties of some peptideCitation10 and their conjugatesCitation11. In the light of our prior studies, the current work expands the studies on synthesis of novel benzothiazole N-protected amino acid conjugates with the aim to combine two important scaffolds in one structure.
CAs (EC 4.2.1.1) are abundant metalloenzymes spread in all life kingdoms with six genetically different families known to date. Up to now, 15 isoforms have been described in human with different cellular localization and organ/tissue distributionCitation12,Citation13. CAs are responsible for the reversible hydration of carbon dioxide to bicarbonate. By catalyzing this simple reaction, CAs participate in many physiological processes such as regulation of respiration and gas exchange, bone resorption, calcification, pH regulation and CO2 homeostasis, electrolyte secretion in a variety of tissues/organs, biosynthetic reactions, tumorigenicityCitation14. The diversity and distribution of different isoforms in the human body, their participation to several metabolic reactions, and as shown in several studies, abnormal levels or activities of this enzyme have been often associated with different human diseases. These factors make many CA isoforms interesting drug targetsCitation14.
It has been almost 70 years since well-known CAIs sulfonamides are in clinical use for the treatment of glaucoma, obesity and epilepsy as diureticsCitation15. Due to the fact that off-target CA inhibition is associated with undesired side effects, such as metallic taste, depression, weight loss, decreased libido, etc.,Citation12 for a long time the main challenge for the CAI’s developers was to synthesize isoform-selective inhibitors. Starting with the “ring and tail approach”Citation16 many efforts has been done not only on improving well-known scaffolds but also for developing a new type of selective inhibitors such as the polyamines,Citation17 phenols,Citation18 dithiocarbamatesCitation19, xanthatesCitation20, coumarinesCitation21 and lately the sulfocoumarinesCitation22,Citation23. Herein, we report the synthesis, characterization and in vitro CA inhibition of a series N-protected amino acid-benzothiazole conjugates, with the hope that they may represent interesting modulators of the activity of these enzymes.
Materials and methods
Chemistry
Anhydrous solvents and all reagents were purchased from Sigma-Aldrich (St. Louis, MO), Acros (abcr GmbH, Karlsruhe, Germany) and Merck (Kenilworth, NJ). All reactions involving air- or moisture-sensitive compounds were performed under an argon atmosphere using dried glassware and syringe techniques to transfer solutions.
All NMR experiments were performed on a Bruker Avance 300 MHz spectrometer equipped with a 5 mm BBO probe head with Z-gradients at a temperature of 298 K. Chemical shifts are reported in parts per million (ppm) and the coupling constants (J) are expressed in Hertz (Hz). Infrared spectra were recorded with attenuated total reflection (ATR) equipment in the range 4000–650 cm−1 on a PerkinElmer spectrum one fourier transform infrared (FTIR) spectrophotometer. All microwave-assisted reactions were carried out in a microwave oven system manufactured by Milestone (Milestone Start S Microwave Labstation for Synthesis, Milestone Inc., Shelton, CT) under an argon atmosphere. The reaction mixtures were transferred into a 10 ml glass pressure microwave tube equipped with a magnetic stir bar. The tube was closed with a silicon septum and the reaction mixture was subjected to microwave irradiation.
Elemental analyses were performed by LECO CHNS-932 elemental analyzer. Melting points were recorded using an Electrothermal-9200 melting point apparatus and are uncorrected.
Synthesis of amino acid-benzothiazole conjugates
Benzyl (2-1H-benzo[d][1,2,3]triazol-1-yl)-2-oxoethyl)carbamate (I), tert-butyl (2-1H-benzo[d][1,2,3]triazol-1-yl)-2-oxoethyl)carbamate (II), (S)-benzyl (1-(1H-benzo[d][1,2,3]triazol-1-yl)-1-oxopropan-2-yl)carbamate (III), (S)-tert-butyl (1-(1H-benzo[d][1,2,3]triazol-1-yl)-1-oxopropan-2-yl)carbamate (IV), (S)-tert-butyl (1-(1H-benzo[d][1,2,3]triazol-1-yl)-1-oxo-3-phenylpropan-2-yl)carbamate (V) were prepared according to the literature procedureCitation11,Citation24.
General method for the synthesis of compounds, 1–3
A mixture of equivalent amounts of the appropriate N-protected aminoacylbenzotriazole and 2-amino-6-(methylsulfonyl)benzo[d]thiazole was subjected to microwave irradiation (100 W, 70 °C) in anhydrous THF (5 ml) for 45 min. After completion of the reaction, all volatiles were removed by rotary evaporator and the obtained crude product was crystallized from ethanol.
Benzyl (2-((6-(methylsulfonyl)benzo[d]thiazol-2-yl)amino)-2-oxoethyl)carbamate, 1
Yield, 0.61 g (light yellow), 67%; mp 234–235 °C; 1H NMR (DMSO-d6, 300 MHz) δ 12.80 (s, 1H, NH), 8.66 (s, 1H, Ar-H), 7.95 (s, 2H, Ar-H), 7.76 (t,1H, NH, J = 6,0 Hz), 7.39–7.31 (m, 5H, Ar-H), 5.08 (s,2H, CH2Ph), 4.05 (d, 2H, CH2NH, J = 6,0 Hz), 3.26 (s,3H, CH3). 13C NMR (DMSO-d6, 300 MHz) δ 170.0 (COCH2NH), 161.8 (SCN), 156.6 (COOCH2Ph), 152.0, 136.9, 135.4, 132.0, 128.3, 128.0, 127.8, 124.8, 122.1, 121.0 (Ar-C), 65.7 (CH2Ph), 44.0 (CH3), 43.6 (CH2NH). υ(C = O)carbamate:1689 cm−1, υ(C = O)amide: 1728 cm−1. Anal. calculated for C18H17N3O5S2: C, 51.54; H, 4.08; N, 10.02; S, 15.29. Found: C, 51.40; H, 4.07; N, 9.98; S, 15.37.
tert-butyl (2-((6-(methylsulfonyl)benzo[d]thiazol-2-yl)amino)-2-oxoethyl)carbamate, 2
Yield, 0.54 g (white), 65%; mp 232–233 °C; 1H NMR (DMSO-d6, 300 MHz) δ 12.73 (s, 1H, NH), 8.46 (s, 1H, Ar-H), 7.86–7.76 (m, 2H, Ar-H), 6.98 (t,1H, NH, J = 5.4 Hz), 3.87 (d, 2H, CH2, J = 5.4 Hz), 3.22 (s,3H, CH3), 1.40 (s, 9H, (CH3)3). 13C NMR (DMSO-d6, 300 MHz) δ 172.4 (COCH2NH), 165.8 (SCN), 155.7 (COOC(CH3)3), 153.1, 133.7, 132.4, 124.2, 121.3, 119.6 (Ar-C), 78.0 (COOC(CH3)3), 44.4 (CH2), 44.2 (CH3), 28.2 ((CH3)3). υ(C = O)carbamate: 1683 cm−1, υ(C = O)amide: 1718 cm−1. Anal. calculated for C15H19N3O5S2: C, 46.74; H, 4.97; N, 10.90; S, 16.64. Found: C, 46.60; H, 4.86; N, 10.82; S, 15.74.
(S)-tert-butyl (1-((6-(methylsulfonyl)benzo[d]thiazol-2-yl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate, 3
Yield, 0.73 g (white), 70%; mp 225–226 °C; 1H NMR (DMSO-d6, 300 MHz) δ 12.90 (s, 1H, NH), 8.66 (s, 1H, Ar-H), 7.95 (s,1H, Ar-H), 7.40–7.17 (m, 6H, Ar-H+ NH), 4.52 (m,1H, COCHCH2), 3.26 (s, 3H, CH3), 3.07-2.87 (m,2H, COCHCH2), 1.32 (s, 9H, (CH3)3). 13C NMR (DMSO-d6, 300 MHz) δ 172.7 (COCHNH), 162.0 (SCN), 155.5 (COOC(CH3)3), 152.1, 137.4, 135.4, 132.0, 129.3, 128.1,126.5, 124.8, 122.1, 120.8 (Ar-C), 78.4 (COOC(CH3)3), 56.2 (COCHCH2), 44.1 (CH3), 36.7 (COCHCH2), 28.1 ((CH3)3). υ(C = O)carbamate: 1664 cm−1, υ(C = O)amide: 1717 cm−1. Anal. calculated for C22H25N3O5S2: C, 55.56; H, 5.30; N, 8.84; S, 13.48. Found: C, 55.44; H, 5.11; N, 8.78; S, 13.14.
General method for the synthesis of compounds, 4–7
A mixture of equivalent amounts of the appropriate N-protected aminoacylbenzotriazole and 4-(6-methylbenzo[d]thiazol-2-yl) aniline was subjected to microwave irradiation (100 W, 70 °C) in anhydrous THF (5 ml) for 45 min. After completion of the reaction, all volatiles were removed by rotary evaporator and the obtained crude product was crystallized from ethanol.
Benzyl (2-((4–(6-methylbenzo[d]thiazol-2-yl)phenyl)amino)-2-oxoethyl)carbamate, 4
Yield, 0.63 g (dark beige), 70%; mp 225–226 °C; 1H NMR (DMSO-d6, 300 MHz) δ 10.4 (s, 1H, NH), 8.04–7.26 (m, 13H, Ar-H + NH), 5.08 (s,2H, CH2Ph), 3.89 (s, 2H, CH2NH), 2.44 (s,3H, CH3). 13C NMR (DMSO-d6, 300 MHz) δ 168.4 (COCH2NH), 165.8 (SCN), 156.6 (COOCH2Ph), 151.8, 141.5, 137.0, 134.4, 128.3, 128.0, 127.8, 127.7, 122.1, 121.7, 119.3 (Ar-C), 65.5 (CH2Ph), 44.2 (CH2NH) 21.0 (CH3). υ(C = O)carbamate:1671 cm−1, υ(C = O)amide:1695 cm−1. Anal. calculated for C24H21N3O3S: C, 66.80; H, 4.91; N, 9.74; S, 7.43. Found: C, 66.58; H, 4.83; N, 9.59; S, 7.35.
tert-butyl (2-((4–(6-methylbenzo[d]thiazol-2-yl)phenyl)amino)-2-oxoethyl)carbamate, 5
Yield, 0.54 g (dark beige), 65%; mp 232–234 °C; 1H NMR (DMSO-d6, 300 MHz) δ 10.3 (s, 1H, NH), 8.04–7.78 (m, 6H, Ar-H), 7.3 (d,2H, Ar-H, J = 8.4 Hz), 7.14 (t, 1H, NH, J = 6.0 Hz), 3.8 (d, 2H, CH2NH, J = 6.0 Hz), 2.25 (s,3H, CH3), 1.37 (s, 9H, C(CH3)3). 13C NMR (DMSO-d6, 300 MHz) δ 168.7 (COCH2NH), 165.8 (SCN), 156.0 (COC(CH3)3), 151.8, 141.6, 135.0,134.4, 128.0, 127.8, 127.6, 122.1, 121.7, 119.2 (Ar-C), 78.1 (C(CH3)3), 43.9 (CH2NH), 28.2 (C(CH3)3), 21.0 (CH3). υ(C = O)carbamate:1679 cm−1, υ(C = O)amide: 1706 cm−1. Anal. calculated for C21H23N3O3S: C, 63.45; H, 5.83; N, 10.57; S, 8.07. Found: C, 63.42; H, 5.82; N, 10.29; S, 7.98.
(S)-benzyl (1-((4–(6-methylbenzo[d]thiazol-2-yl)phenyl)amino)-1-oxopropan-2-yl)carbamate, 6
Yield, 0.61 g (dark beige), 66%; mp 232–233 °C; 1H NMR (DMSO-d6, 300 MHz) δ 10.4 (s, 1H, NH), 8.05–7.32 (m, 13H, Ar-H + NH), 5.04 (s,2H, CH2Ph), 4.25 (t, 1H, CHCH3, J = 6.9 Hz), 2.45 (s,3H, CCH3), 1.34 (d, 3H, CHCH3, J = 6.9 Hz). 13C NMR (DMSO-d6, 300 MHz) δ) 172.0 (COCHCH3), 165.8 (SCN), 155.8 (COOCH2Ph), 145.8, 141.7, 137.0, 135.0, 134.4, 128.3, 128.0, 127.8, 127.7, 122.1, 121.7, 119.4 (Ar-C), 65.4 (CH2Ph), 50.9 (CHCH3) 21.0 (CCH3), 17.9 (CHCH3). υ(C = O)carbamate:1669 cm−1, υ(C = O)amide: 1691 cm−1. Anal. calculated for C25H23N3O3S: C, 67.40; H, 5.20; N, 9.43; S, 7.20. Found: C, 67.10; H, 5.14; N, 9.47; S, 6.93.
(S)-tert-butyl (1-((4–(6-methylbenzo[d]thiazol-2-yl)phenyl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate, 7
Yield, 0.73 g (dark beige), 63%; mp 219–220 °C; 1H NMR (DMSO-d6, 300 MHz) δ 10.4 (s, 1H, NH), 8.05–7.72 (m, 6H, Ar-H), 7.36–7.18 (m, 7H, Ar-H+ NH), 4.42–4.34 (m,1H, COCHCH2Ph), 3.06–2.84 (m, 2H, COCHCH2Ph), 2.45 (s, 3H, CH3) 1.33 (s, 9H, (CH3)3).13C NMR (DMSO-d6, 300 MHz) δ 171.3 (COCHCH2Ph), 165.8 (SCN), 155.4 (COOC(CH3)3), 151.8, 141.5, 137.8, 135.0, 134.4, 129.2, 128.0,126.3, 122.1, 121.7, 119.5 (Ar-C), 78.1 (COOC(CH3)3), 56.7 (COCHCH2), 37.3 (COCHCH2), 28.1 ((CH3)3) 21.0 (CH3). υ(C = O)carbamate:1672 cm−1, υ(C = O)amide: 1690 cm−1. Anal. calculated for C28H29N3O3S: C, 68.97; H, 5.99; N, 8.62; S, 6.58. Found: C, 68.94; H, 5.97; N, 8.57; S, 6.63.
General method for the synthesis of compounds, 8–12
A mixture of equivalent amounts of the appropriate N-protected aminoacylbenzotriazole and 2-amino-4-methylbenzo[d]thiazole was subjected to microwave irradiation (100 W, 70 °C) in anhydrous THF (5 ml) for 45 min. After completion of the reaction, all volatiles were removed by rotary evaporator and the obtained crude product was crystallized from ethanol.
Benzyl (2-((4-methylbenzo[d]thiazol-2-yl)amino)-2-oxoethyl)carbamate, 8
Yield, 0.77 g (white), 77%; mp 212–213 °C; 1H NMR (DMSO-d6, 300 MHz) δ 12.6 (bs, 1H, NH), 7.79 (d, 1H, Ar-H, J = 7.5 Hz), 7.68 (t, 1H, NH, J = 6.0 Hz), 7.39-7.24 (m, 7H, Ar-H), 5.08 (s,2H, CH2Ph), 4.00 (d, 2H, CH2NH, J = 6.0 Hz), 2.57 (s,3H, CH3). 13C NMR (DMSO-d6, 300 MHz) δ 169.2 (COCH2NH), 160.0 (SCN), 156.6 (COOCH2Ph), 147.6, 136.9, 131.1, 129.8, 128.3, 127.8, 127.7, 126.6, 123.4, 121.0 (Ar-C), 65.6 (CH2Ph), 43.5 (CH2NH), 17.9(CH3). υ(C = O)carbamate: 1681 cm−1, υ(C = O)amide:1710 cm−1. Anal. calculated for 8H17N3O3S: C, 60.83; H, 4.82; N, 11.82; S, 9.02. Found: C, 60.78; H, 4.81; N, 11.68; S, 9.01.
tert-butyl (2-((4-methylbenzo[d]thiazol-2-yl)amino)-2-oxoethyl)carbamate, 9
Yield, 0.66 g (white), 68%; mp 174–175 °C; 1H NMR (DMSO-d6, 300 MHz) δ 10.87 (s, 1H, NH), 7.63 (d, 1H, Ar-H, J = 5.4 Hz), 7.20 (m, 2H, Ar-H), 6.02 (s,1H, CH2NH), 4.30 (s, 2H, CH2NH), 2.60 (s, 3H, CH3), 1.49 (s, 9H, C(CH3)3). 13C NMR (DMSO-d6, 300 MHz) δ 168.9 (COCH2NH), 156.6 (SCN), 156.4 (COC(CH3)3), 147.5, 131.9, 130.7, 126.9, 124.0, 118.8 (Ar-C), 80.7 (C(CH3)3), 44.5 (CH2NH), 28.4 (C(CH3)3), 18.2 (CH3). υ(C = O)carbamate: 1660 cm−1, υ(C = O)amide: 1706 cm−1. Anal. calculated for C15H19N3O3S: C, 56.06; H, 5.96; N, 13.07; S, 9.98. Found: C, 55.91; H, 5.89; N, 13.09; S, 9.67.
(S)-benzyl (1-((4-methylbenzo[d]thiazol-2-yl)amino)-1-oxopropan-2-yl)carbamate, 10
Yield, 0.76 g (white), 68%; mp 196–197 °C; 1H NMR (DMSO-d6, 300 MHz) δ 12.6 (s, 1H, NH), 7.81-7.20 (m, 9H, Ar-H + NH), 5.05 (s,2H, CH2Ph), 4.39 (quint, 1H, CHCH3, J= 7.2 Hz), 2.58 (s,3H, CCH3), 1.35 (d, 3H, CHCH3, J= 7.2 Hz). 13C NMR (DMSO-d6, 300 MHz) δ 172.7 (COCHCH3), 157.0 (SCN), 156.0 (COOCH2Ph), 147.6, 136.9, 131.1, 129.8, 128.3, 127.8, 126.6, 123.5, 119.1 (Ar-C), 65.5 (CH2Ph), 50.2 (CHCH3) 18.0 (CCH3), 17.5 (CHCH3). υ(C = O)carbamate: 1695 cm−1, υ(C = O)amide: 1717 cm−1. Anal. calculated for C19H19N3O3S: C, 61.77; H, 5.18; N, 11.37; S, 8.68. Found: C, 61.54; H, 5.08; N, 13.36; S, 8.38.
(S)-tert-butyl (1-((4-methylbenzo[d]thiazol-2-yl)amino)-1-oxopropan-2-yl)carbamate, 11
Yield, 0.61 g (white), 60%; mp 209–210 °C; 1H NMR (DMSO-d6, 300 MHz) δ 12.3 (s, 1H, NH), 7.75 (s, 1H, NH), 7.63 (d, 1H, Ar-H, J = 8.4 Hz), 7.30–7.23 (m, 2H, Ar-H), 4.28 (m, 1H, CHCH3), 2.41 (s, 3H, CH3), 1.38 (s, 9H, C(CH3)3) 1.30 (d, 3H, CHCH3, J = 7.2 Hz). 13C NMR (DMSO-d6, 300 MHz) δ 172.9 (COCHNH), 157.0(SCN), 155.2 (COC(CH3)3), 146.5, 132.9, 131.6, 127.4, 121.3, 120.1 (Ar-C), 78.2 (C(CH3)3), 49.8 (COCHCH3), 28.1 (C(CH3)3), 20.9 (CHCH3), 17.4 (CH3). υ(C = O)carbamate:1672 cm−1, υ(C = O)amide: 1706 cm−1. Anal. calculated for C16H21N3O3S: C, 57.29; H, 6.31; N, 12.53; S, 9.56. Found: C, 57.13; H, 6.29; N, 12.41; S, 9.58.
(S)-tert-butyl (1-((4-methylbenzo[d]thiazol-2-yl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate, 12
Yield, 0.88 g (white), 70%; mp 159–160 °C; 1H NMR (DMSO-d6, 300 MHz) δ 12.7 (s, 1H, NH), 7.80–7.18 (m, 9H, Ar-H + NH), 4.51 (m, 1H, COCHCH2), 3.11–2.81 (m, 2H, COCHCH2), 2.58 (s, 3H, CH3) 1.31 (s, 9H, (CH3)3). 13C NMR (DMSO-d6, 300 MHz) δ 172.0 (COCHNH), 156.9 (SCN), 155.4 (COOC(CH3)3), 147.6, 137.6, 131.1, 129.8, 129.3, 128.0, 126.6, 126.4, 123.5, 119.1 (Ar-C), 78.3 (COOC(CH3)3), 56.2 (COCHCH2), 36.9 (COCHCH2), 28.1 ((CH3)3) 17.9 (CH3). υ(C = O)carbamate: 1693 cm−1, υ(C = O)amide: 1700 cm−1. Anal. calculated for C22H25N3O3S: C, 64.21; H, 6.12; N, 10.21; S, 7.79. Found: C, 64.12; H, 6.01; N, 10.22; S, 7.68.
(S)-2-(2-ammoniopropanamido)-4-methylbenzo[d]thiazol-3-ium di-2,2,2-trifluoroacetate, 13
(S)-tert-butyl (1-((4-methylbenzo[d]thiazol-2-yl)amino)-1-oxopropan-2-yl)carbamate, 11 (0.3 g; 0.9 mmol) dissolved in CH2Cl2 (3 ml) and treated with TFA (2 ml). The reaction was stirred at room temperature for 30 min. The solvent was then removed under vacuum and the obtained residue was triturated from diethyl ether and petroleum ether mixture (1:1) to obtain compound 13 as dark beige. Yield, 0.38 g, 89%. mp 152–153 °C; 1H NMR (DMSO-d6, 300 MHz) δ 10.3 (bs, 1H, NH), 8.45 (bs, 3H, NH3+), 7.80 (s,1H, Ar-H), 7.67 (d,1H, Ar-H, J = 8.1 Hz), 7.28 (d,1H, Ar-H, J = 7.2 Hz), 4.20 (bs,12H, CHCH3), 2.42 (s,3H, CH3), 1.51 (d, 3H, CHCH3, J = 7.2 Hz). 13C NMR (DMSO-d6, 300 MHz) δ 169.5 (COCHCH3), 158.4 (q, CF3CO, 2J(C-F) = 35.2 Hz) 156.5 (SCN), 146.0, 133.5, 131.5, 127.7, 121.4, 120.3 (Ar-C), 116.0 (q, CF3CO, 1J(C-F) = 293.3 Hz) 48.7 (COCHCH3), 20.9 (COCHCH3), 16.6 (CH3). υ(C = O)TFA:1608 and 1655 cm−1, υ(C = O)amide: 1728 cm−1. Anal. calculated for C15H15F6N3O5S: C, 38.88; H, 3.26; N, 9.07; S, 6.92. Found: C, 38.67; H, 3.24; N, 9.16; S, 6.91.
CA inhibition
An applied photophysics stopped-flow instrument has been used for assaying the CA catalyzed CO2 hydration activity by using the method of KhalifahCitation25. Phenol red (at a concentration of 0.2 mm) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mm Hepes (pH 7.5) as buffer, and 20 mm Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mm for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mm) were prepared in distilled-deionized water and dilutions up to 0.01 nm were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E-I complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM (www.graphpad.com) (GraphPad Software, Inc., La Jolla, CA), and non-linear least-squares methods, values representing the mean of at least three different determinations, as described earlier by usCitation26–28.
Results and discussion
New N-protected amino acid benzothiazole conjugates (1–12) were synthesized by the treatment of appropriate benzothiazole derivatives and N-protected amino acid (Gly, Ala and Phe) derivatives under microwave heating at 70 °C for 45 min with good yields of 60–70%. The compound 13 was synthesized from the reacting of (S)-tert-butyl (1-((4-methylbenzo[d]thiazol-2-yl)amino)-1-oxopropan-2-yl)carbamate, 11 with TFA in CH2Cl2 at room temperature for 30 min with a high yield of 89%. The synthesis of the benzothiazole derivatives 1–13 is summarized in the Scheme 1.
Scheme 1. Synthetic pathways of the new benzothiazole conjugate incorporating N-protected amino acid moieties.
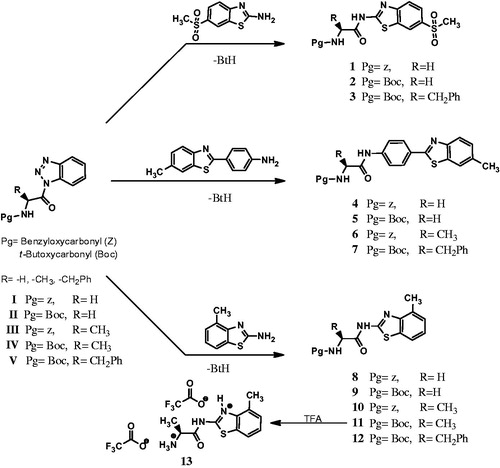
The structures of benzothiazoles (1–13) were elucidated by IR, 1H-NMR, 13C-NMR and microanalytic data. All spectral data were in accordance with the assumed structures. The IR spectra of benzothiazole derivatives, 1–13 have C = Ocarbamate and C = Oamide stretching bands at range of 1660–1693 cm−1(except compound 13) and 1690–1728 cm−1, respectively. The characteristic HNCSN resonance in the 1H NMR spectra and NCS resonance in the Citation13C NMR spectra of benzothiazole derivatives (1–13) were observed around 10.30–12.90 ppm and 156.5–165.8 ppm, respectively.
As it is seen in none of the N-protected amino acid-benzothiazole conjugates 1–13 showed affinity toward to membrane-associated isoform hCA IV and compounds 12 and 13 are inactive on four hCAs tested in this study. In addition to that compounds 1, 3, 7–11 are only inhibiting the most abundant cytosolic isoform hCA I with inhibition constants ranging between 45.4 and 88.8 μm (they are thus quite weak inhibitors). Compound 4 showed an interesting profile by having an affinity toward another cytosolic isoform hCA II with a KI of 10.2 μm. Compounds 5 and 6 had an affinity toward cytosolic isoforms, hCA I, of 85.9, 71.1 μm – and hCA II, of 6.6 and 5.0 μm, respectively. Compound 2 is the only compound with inhibitory effects on the transmembrane isoform hCA XII with a KI 8.4 μm.
Table 1. hCA I, II, IV and XII inhibition data with N-protected amino acid-benzothiazole conjugates 1–13, by a stopped-flow CO2 hydrase assayCitation25.
The presence of methylsulfonyl group on the benzothiazole part of the molecules for compounds 1–3 (45.4–67.5 μm) led to an increasing inhibitory power when compared with the compounds 8,9,12 (86.8 > 100 μm) which did not incorporate that group. This effect could be the result of oxygen atoms in sulfonyl group which are providing additional stability to the molecule by creating extra interactions with amino acid residues in the active site of the enzyme. It should be also noted that when compounds 2 and 3 were compared, the presence of rather bulky benzyl groups on the sixth position of the molecule resulted in losing selectivity toward hCA XII with 8.4 μm (2) to > 100 μm (3). A similar behavior was observed for compounds 5 and 7, with an additional benzyl group leading to the loss of selectivity, this time toward hCA II with 6.6 μm (5) to > 100 μm (7).
Conclusion
In this study, 13 new N-protected amino acid-benzothiazole conjugates were tested against two cytosolic (hCA I and hCA II), one membrane-associated (hCA IV) and one transmembrane (hCA XII) CA isoforms. hCA I considered as a potential drug target treatment of pathogenesis of proliferative diabetic retinopathy and diabetic macular edema, as it mediates hemorrhagic retinal and cerebral vascular permeability through activation of prekallikrein and generation of the highly active serine protease factor XIIa 8Citation12,Citation29. Considering these phenomena even these new N-protected amino acid-benzothiazole conjugates which do not possess very strong affinity to hCA I but show selectivity toward this isoform, might be considered as interesting starting molecules for developing hCA I selective scaffolds with biomedical applicationsCitation30–35.
Declaration of interest
We thank Inönü University, Turkey (BAPB) and University Degli Studi di Firenze, Italy, for financial support.
References
- Pareek PK, Mithlesh S, Kriplani P, et al. Rapid synthesis and biological activities of some new benzothiazol-2-ylhexahydro-S-triazine derivatives. Phosphorus Sulfur Silicon Relat Elem 2010;185:1338–45
- Wells G, Bradshaw TD, Diana P, et al. Antitumour benzothiazoles. Part 10: the synthesis and antitumour activity of benzothiazole substituted quinol derivatives. Bioorg Med Chem Lett 2000;10:513–15
- Gupta A, Rawat S. Synthesis and anti-inflammatory study of novel fluorobenzothiazole derivatives. J Chem Pharm Res 2010;2:244–58
- Küçükbay H, Çetinkaya E, Durmaz R. Synthesis and antimicrobial activity of substituted benzimidazole, benzothiazole and imidazole derivatives. Arzneimittelforschung 1995;45:1331–4
- Rana A, Siddiqui N, Khan SA, et al. N-{[(6-substituted-1,3-benzothiazole-2-yl)amino]carbonothioyl}-2/4-substituted benzamides: synthesis and pharmacological evaluation. Eur J Med Chem 2008;43:1114
- Anzini M, Chelini A, Mancini A, et al. Synthesis and biological evaluation of amidine, guanidine, and thiourea derivatives of 2-amino(6-trifluoromethoxy)benzothiazole as neuroprotective agents potentially useful in brain diseases. J Med Chem 2010;53:734–44
- Yin Y, Lin L, Ruiz C, et al. Benzothiazoles as Rho-associated kinase (ROCK-II) inhibitors. Bioorg Med Chem Lett 2009;19:6686–90
- Vu CB, Milne JC, Carney DP, et al. Discovery of benzothiazole derivatives as efficacious and enterocyte-specific MTP inhibitors. Bioorg Med Chem Lett 2009;19:1416–20
- Korman J. Carbonic anhydrase inhibitors. I. Benzothiazole derivatives. J Org Chem 1958;23:1768–71
- Ibrahim MA, Panda SS, Oliferenko AA, et al. Macrocyclic peptidomimetics with antimicrobial activity: synthesis, bioassay, and molecular modeling studies. Org Biomol Chem 2015;13:9492–503
- Panda SS, Ibrahim MA, Küçükbay H, et al. Synthesis and antimalarial bioassay of quinine – peptide conjugates. Chem Biol Drug Des 2013;82:361–6
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81
- Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77
- Supuran CT. Carbonic anhydrases: from biomedical applications of the inhibitors and activators to biotechnological use for CO(2) capture. J Enzyme Inhib Med Chem 2013;28:229–30
- Scozzafava A, Menabuoni L, Mincione F, et al. Carbonic anhydrase inhibitors. Synthesis of water-soluble, topically effective, intraocular pressure-lowering aromatic/heterocyclic sulfonamides containing cationic or anionic moieties: is the tail more important than the ring? J Med Chem 1999;42:2641–50
- Carta F, Temperini C, Innocenti A, et al. Polyamines inhibit carbonic anhydrases by anchoring to the zinc-coordinated water molecule. J Med Chem 2010;53:5511–22
- Innocenti A, Vullo D, Scozzafava A, et al. Carbonic anhydrase inhibitors: interactions of phenols with the 12 catalytically active mammalian isoforms (CA I-XIV). Bioorg Med Chem Lett 2008;18:1583–7
- Carta F, Aggarwal M, Maresca A, et al. Dithiocarbamates: a new class of carbonic anhydrase inhibitors. Crystallographic and kinetic investigations. Chem Commun (Camb) 2012;48:1868–70
- Carta F, Akdemir A, Scozzafava A, et al. Xanthates and trithiocarbonates strongly inhibit carbonic anhydrases and show antiglaucoma effects in vivo. J Med Chem 2013;56:4691–700
- Maresca A, Temperini C, Vu H, et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–62
- Tars K, Vullo D, Kazaks A, et al. Sulfocoumarins (1,2-benzoxathiine-2,2-dioxides): a class of potent and isoform-selective inhibitors of tumor-associated carbonic anhydrases. J Med Chem 2013;56:293–300
- Tanc M, Carta F, Bozdag M, et al. 7-Substituted-sulfocoumarins are isoform-selective, potent carbonic anhydrase II inhibitors. Bioorg Med Chem 2013;15:4502–10
- Khatib ME, Jauregui L, Tala SR, et al. Solution-phase synthesis of chiral O-acyl isodipeptides. Med Chem Comm 2011;2:1087–92
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73
- Casini A, Scozzafava A, Mincione F, et al. Carbonic anhydrase inhibitors: water-soluble 4-sulfamoylphenylthioureas as topical intraocular pressure-lowering agents with long-lasting effects. J Med Chem 2000;43:4884–92
- Fabrizi F, Mincione F, Somma T, et al. A new approach to antiglaucoma drugs: carbonic anhydrase inhibitors with or without NO donating moieties. Mechanism of action and preliminary pharmacology. J Enzyme Inhib Med Chem 2012;27:138–47
- Supuran CT, Ilies MA, Scozzafava A. Carbonic anhydrase inhibitors. Part 29. Interaction of isozymes I, II and IV with benzolamide-like derivatives. Eur J Med Chem 1998;33:739–52
- Gao BB, Clermont A, Rook S, et al. Extracellular carbonic anhydrase mediates hemorrhagic retinal and cerebral vascular permeability through prekallikrein activation. Nature Med 2007;13:181–8
- Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72
- Capasso C, Supuran CT. Anti-infective carbonic anhydrase inhibitors: a patent and literature review. Expert Opin Ther Pat 2013;23:693–704
- Capasso C, Supuran CT. An overview of the alpha-, beta- and gamma-carbonic anhydrases from bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem 2015;30:325–32
- Supuran CT, Capasso C. The η-class carbonic anhydrases as drug targets for antimalarial agents. Expert Opin Ther Targets 2015;19:551–63
- Capasso C, Supuran CT. Sulfa and trimethoprim-like drugs – antimetabolites acting as carbonic anhydrase, dihydropteroate synthase and dihydrofolate reductase inhibitors. J Enzyme Inhib Med Chem 2014;29:379–87
- Supuran CT. Carbonic anhydrase inhibitors: an editorial. Expert Opin Ther Pat 2013;23:677–9