
Abstract
Carbonic anhydrases (CAs, EC 4.2.1.1) catalyze the fundamental reaction of CO2 hydration in all living organisms, being actively involved in the regulation of a plethora of patho/physiological conditions. A series of benzothiazole-based sulfonamides were synthesized and tested as possible CA inhibitors. Their inhibitory activity was assessed against the cytosolic human isoforms hCA I and hCA II and the transmembrane hCA IX and hCA XII. Several of the investigated derivatives showed interesting inhibition activity and selectivities for inhibiting hCA IX and hCA XII over the off-target ones hCA I and hCA II. Furthermore, computational procedures were used to investigate the binding mode of this class of compounds, within the active site of hCA IX.
Introduction
The carbonic anhydrases (CAs) are metalloenzymes that catalyze the formation of bicarbonate from carbon dioxide in two stepsCitation1–4. Sixteen isoforms were discovered up to now in humans, and mainly differ in their subcellular localization, catalytic activity and tissue distribution. Five CAs are cytosolic (CA I–III, VII and XIII), two are mitochondrial (CA VA and VB), one is secreted (CA VI) and the others are membrane-bound (CA IV, IX, XII, XIV and XV)Citation5. Many CA subtypes constitute interesting targets for the design of pharmacological agents useful, for example, as antiglaucoma, anticonvulsant, antiurolithic and for the treatment of obesityCitation6. Among the various CA isoforms, CA IX and CA XII were discovered to be overexpressed and associated with many tumors, where they are involved in processes linked with cancer progressionCitation7–9. Hence small molecules targeting CA IX/XII inhibition may be an attractive strategy against cancer. However, due to high homology in CA’s and the ubiquitous nature of CA I and II in humans, selectivity against CA I and II is of utmost importance to prevent off-target related side effects. The classical CA inhibitors (CAIs) are the primary sulfonamides, which coordinate the zinc ion with their terminal deprotonated nitrogen atom and are in clinical use for more than 70 years as diuretics and systemically acting antiglaucoma drugsCitation10. However, the high number of isoforms in humans, as well as their diffuse localization in many tissues/organs usually represented the main hurdle in the design of sulfonamide based CAIs as therapeutic agents. Here, we report a series of novel benzothiazole-based sulfonamides, which showed good CA IX/XII inhibitory activity as well as selectivity for the inhibition of the tumor-associated isoforms over the cytosolic ones CA I and II. The experimental studies were developed together with in silico techniques, aimed to propose a reliable binding disposition for this class of CAIs.
Methods
Chemistry
Melting points (°C) were determined with a MELTEMP II capillary apparatus (LAB Devices, Holliston, MA) without correction. Elemental analyses were performed on a Perkin-Elmer 2400 CHN elemental analyzer and all compounds synthesized were within a 0.4% of theoretical values. IR spectra were recorded, as Nujol mulls, on a Perkin-Elmer Spectrum BX (Waltham, MA). Wave numbers in the IR spectra are given in cm−1. 1H NMR and 13C NMR spectra of the newly synthesized compounds, in DMSO-d6 or CDCl3 solutions, were recorded on a Bruker AC 300 instrument (Bruker, Karlsruhe, Germany) at 298 K. Chemical shifts are reported as δ (ppm) relative to TMS as an internal standard. Coupling constants J are expressed in Hertz (Center of Instrumental Analysis of the University of Thessaloniki). The reactions were monitored by TLC on F254 silica-gel precoated sheets (Merck, Darmstadt, Germany) and each of the purified compounds showed a single spot. Solvents unless otherwise specified were of analytical reagent grade or of the highest quality commercially available. Synthetic starting materials, reagents and solvents were purchased from Aldrich Chemie (Steinheimm, Germany).
General procedure (A) for the synthesis of benzothiazole sulfonamides (2–4, 9)
To a solution of 2-aminobenzothiazole (6 mmol) in anhydrous pyridine (10 ml) an appropriate sulfonyl chloride (6.3 mmol) was added. The reaction mixture was heated under reflux and temperature was maintained at 60° for 1 h or more till the end of the reaction. The reaction mixture poured into the ice and neutralized by 1 N HCl. The precipitate formed was filtered, washed with water and recrystallized from appropriate solvent.
General procedure (B) for the synthesis of benzothiazole sulfonamides (1, 5–8)
To the solution of 2-aminobenzothiazole (0.013 mol) in pyridine (4 ml) and glacial acetic acid (20 ml) appropriate sulfonyl chloride (0.01 mol) was added and heated in water bath for 2 h. After the reaction was completed, reaction mixture was poured to distilled water (20 ml) and precipitate formed filtrated and washed with water.
Synthesis of N-(4-chlorobenzo[d]thiazol 2-yl)-4-chlorobenzosulfonamide (1)
Yield: 53.66%; m.p. 302–304 °C (EtOH). TLC: Rf = 0.62, eluent: CHCl3–EtOH (9.5:0.5). IR (KBr): ν = 1340, 1370 (SO2), 800 (CI). 1H NMR (DMSO-d6, δ, ppm): 7.27–7.33 (t, 3H, H4, H5, H6), 7.51–7.54 (d, 2H, H2′, H6′, J = 9 Hz), 7.94–7.97 (d, 2H, H3′, H5′, J = 9 Hz), 12.67(s, 1H, NH). Anal. Calc. for C13H8Cl2N2O2S2: C: 43.46%; H: 2.24%; N: 7.8%. Found: C: 43.51%; H: 2.40%; N: 8.0%. m/z 358.
Synthesis of N-(5,6-dimethylbenzo[d]thiazol 2-yl)-4-chlorobenzosulfonamide (2)
Yield: 75.25%; m.p. 293–294 °C (dioxan) TLC: Rf = 0.56, eluent: CHCl3–EtOH (9.5:0.5) IR (KBr): ν = 1344 cm−1, 1372 cm−1 νSO2, 800 cm−1 νCI, 3358, 1372 cm−1 νNH (weak). 1H NMR (DMSO-d6, δ, ppm): 2.22 (s, 6H, 5–6 CH3), 7.32–7.27 (t, 2H, H4, H7), 7.53–7.50 (d, 2H, H3′, H5′, J = 9 Hz), 7.96–7.93 (d, 2H, H2′, H6′, J = 9 Hz), 12.66 (s, 1H, NH). Anal. Calc. for C15H13ClN3O2S2: C: 51.06%; H: 3.71; % N: 7.94%. Found: C: 51.11%; H: 3.72%; N: 7.86%. m/z 352.
Synthesis of N-(5,6-dimethylbenzo[d]thiazol 2-yl)-4-nitrobenzosulfonamide (3)
Yield: 30.06%; m.p. 258–259 °C TLC: Rf = 0.43, eluent: CHCl3–EtOH (9.5:0.5). IR (KBr): ν = 1347, 1376 cm−1 (SO2), 1534 (NO2), 3360, 1370 (NH) cm−1. 1H NMR (DMSO-d6, δ, ppm): 2.26 (s, 6H, 5–6CH3), 7.11 (s, 1H, H7), 7.53 (s, 1H, H4), 7.63–7.60 (d, 2H, H2′, H6′, J = 9 Hz), 7.87–7.84 (d, 2H, H3′, H5′, J = 9 Hz). Anal. Calc. for C15H13N4O2S2: C: 49.58%; H: 3.61%; N: 11.56%. Found: C: 49.62%; H: 3.60%; N: 11.43%. m/z 363.
Synthesis of N-(5,6-dimethylbenzo[d]thiazol 2-yl)-benzosulfonamide (4)
Yield: 85.5%; m.p. 288–290 °C (EtOH) TLC: Rf = 0.45, eluent: CHCl3–EtOH (9.5:0.5). IR (KBr): ν = 1305 (SO2), 3360, 1370 (NH) cm−1.1H NMR (DMSO-d6, δ, ppm): 2.26 (s, 6H, 5–6CH3), 7.10 (s, 1H, H4′), 7.62–7.54 (m, 4H, H2′, H3′, H6′, H5′), 7.88–7.85 (d, 2H, H4, H7). Anal. Calc. for C15H14N2O2S2: C: 56.58%; H: 4.43%; N: 8.80%. Found: C: 56.36%; H: 4.39%; N: 8.65%. m/z 318.
Synthesis of N-(4-chlorobenzo[d]thiazol 2-yl)-4-methoxybenzosulfonamide (5)
Yield: 57.34%; m.p. 299–301 °C (EtOH) TLC: Rf = 0.44, eluent: CHCl3–EtOH (9.5:0.5). IR (KBr): ν = 1369 (SO2), 1651 (OCH3), 3361, 1370 (NH) cm−1. 1H NMR (DMSO-d6, δ, ppm): 2.23 (s, 3H, 0CH3), 7.33–7.27 (t, 3H, H4, H5, H7), 7.54–7.51 (d, 2H, H2′, H6′, J = 9 Hz), 7.97–7.94 (d, 2H, H3′, H5′, J = 9 Hz), 12.67 (s, 1H, NH). Anal. Calc. for C14H11ClN2O3S2: C: 47.39%; H: 3.12%; N: 7.89%. Found: C: 47.45%; H: 3.05%; N: 8.01%. m/z 354.
Synthesis of N-(6-chlorobenzo[d]thiazol 2-yl)-4-methoxybenzosulfonamide (6)
Yield: 56.99%; m.p. 234–235 °C (EtOH) TLC: Rf = 0.37, eluent: CHCl3–EtOH (9.5:0.5). IR (KBr): ν = 1340(SO2), 1370 (SO2), 1651 (OCH3), 800 (Cl), 3218(NH) cm−1. 1H NMR (DMSO-d6, δ, ppm): 2.22 (s, 3H, OCH3), 7.42–7.46 (dd, 3H, H2′, H6′, H4, JA = 2.1 Hz, JB = 2.4 Hz), 7.71–7.74 (d, 2H, H5, H6, J = 9 Hz), 8.08–8.09 (d, 2H, H3′, H5′, J = 2.1 Hz), 12.35(s, 1H, NH). Anal. Calc. for C14H12ClN2O3S2: C: 47.39%; H: 3.12%; N: 7.89%. Found: C: 47.35%; H: 3.15%; N: 7.94%. M/z 354.
Synthesis of N-(6-chlorobenzo[d]thiazol 2-yl)-4-fluorobenzosulfonamide (7)
Yield: 62.00%; m.p. 235–236 °C (EtOH) TLC: Rf = 0.41, eluent: CHCl3–EtOH (9.5:0.5). IR (KBr): ν = 1340 (SO2), 1370 (SO2), 1000 (F), 800 (Cl) 3219 (NH) cm−1. 1H NMR (DMSO-d6, δ, ppm): 7.45, 7.41 (ddd, 3H, H4, H5, H6, J = 1.2 Hz), 7.70–7.73 (d, 2H, H2′, H6′, J = 9 Hz), 8.08–8.07 (d, 2H, H3′, H5′, J = 1.5 Hz), 12.36 (s, 1H, NH). Anal. Calc. for C13H8ClFN2O2S2: C: 45.55%; H: 2.35%; N: 8.17%. Found: C: 44.45; H: 2.55; N: 8.11%. m/z 342.
Synthesis of N-(6-fluorobenzo[d]thiazol 2-yl)-4-fluorobenzosulfonamide (8)
Yield: 57.77%; m.p. 228–230 (CH2Cl2) °C TLC: Rf = 0.47, eluent: CHCl3–EtOH (9.5:0.5). IR (KBr): ν = 1340(SO2), 1370 (SO2), 1000 (F), 3219 (NH) cm−1. 1H NMR (DMSO-d6, δ, ppm): 7.21–7.28 (m, 2H, H2′, H6′), 7.71–7.86 (m, 5H, H3′, H5′, H4, H5, H6), 12.29 (s, 1H, NH). Anal. Calc. for C13H8F2N2O2S2: C: 47.85%; H: 2.47%; N: 8.58%. Found: C: 47.65%; H: 2.55%; N: 8.61%. m/z 326.
Synthesis of N-(5,6-dimethylbenzo[d]thiazol 2-yl)-4-fluorobenzosulfonamide (9)
Yield: 71.70%; m.p. 268–269 °C (EtOH). TLC: Rf = 0.41, eluent: CHCl3–EtOH (9.5:0.5). IR (KBr): ν = 1305 (SO2), 1000 (F), 3360, 1370 (NH) cm−1. 1H NMR (DMSO-d6, δ, ppm): 2.26 (s, 6H, 5, 6CH3), 7.11 (s, 1H, H4), 7.36–7.41 (dd, 2H, H2′, H6′, JB = 2.1 Hz), 7.54 (s, 1H, H7), 7.89–7.94 (dd, 2H, H3′, H5′ JB = 2.1 Hz), 12.97 (s, 1H, NH). Anal. Calc. for C15H13FN2O2S2: C: 53.56%; H: 3.90%; N: 8.33%. Found: C: 53.65%; H: 3.95%; N: 8.51%. m/z 336.
Synthesis of N-(6-fluorobenzo[d]thiazol 2-yl)-4-methoxybenzosulfonamide (10)
Yield: 53.70%; m.p. 219–221 °C, TLC: Rf = 0.31, eluent: CHCl3–EtOH (9.5:0.5). IR (KBr): ν = 1339(SO2) (SO2), 1650 (OCH3), 1049 (F), 3203(NH) cm−1. 1H NMR (DMSO-d6, δ, ppm): 2 2.20 (s, 3H, 4–0CH3), 7.31–7.24 (dt, 3H, H2′, H6′, H4, JA = 2.4 Hz), 7.76–7.71 (dd, 2H, H5, H6), 7.86–7.89 (dd, 2H, H3′, H5′, J = 2.4 Hz), 12.4(s, 1H, NH). Anal. Calc. for C14H11FN2O3S2: C: 49.69%; H: 3.28%; N: 8.28%. Found: C: 49.65%; H: 3.35%; N: 8.33%. m/z 338.
Synthesis of N-(4-methylbenzo[d]thiazol 2-yl)-4-fluorobenzosulfonamide (11)
Yield: 67.70%; m.p. 270–271 °C (EtOH) TLC: Rf = 0.68, eluent: CHCl3–EtOH (9.5:0.5). IR (KBr): ν = 1305 (SO2), 1038 (F), 3168 (NH) cm−1. 1H NMR (DMSO-d6, δ, ppm): 2.20 (s, 3H, 4CH3), 7.19–7.24 (m, 5H, H2′, H6′, H5, H6, H7), 7.75–7.78 (d, 2H, H3′, H5′, J = 9 Hz), 12.35 (s, 1H, NH). Anal. Calc. for C14H11FN2O2S2: C: 52.16%; H: 3.44%; N: 8.69%. Found: C: 52.20%; H: 3.35%; N: 8.76%. m/z 322.
Synthesis of N-(4-methylbenzo[d]thiazol 2-yl)-4-methylbenzosulfonamide (12)
Yield: 60.90%; m.p. 267–269 °C (EtOH) TLC: Rf = 0.60, eluent: CHCl3:EtOH (9.5:0.5). 1H NMR (DMSO-d6, δ, ppm): 2.19 (d, 6H, 4′CH3, 4CH3), 7.18–7.22 (m, 5H, H3′, H5′, H5, H6, H7), 7.74–7.76 (d, 2H, H2′, H5′, J = 6 Hz), 12.35 (s, 1H, NH). Anal. Calc. for C15H14N2O2S2: C: 56.58%; H: 4.43%; N: 8.8%. Found: C: 56.40%; H: 4.35%; N: 8.63%. m/z 318.
Synthesis of N-(4-chlorobenzo[d]thiazol 2-yl)-4-methylbenzosulfonamide (13)
Yield: 55.10%; m.p. 300–302, 267–269 °C (EtOH) TLC: Rf = 0.62, eluent: CHCl3–EtOH (9.5:0.5). IR (KBr): ν = 1305 (SO2), 817 (Cl), 3161, 3126 (NH) cm−1. 1H NMR (DMSO-d6, δ, ppm): 2.21 (s, 3H, CH3), 7.26–7.32 (t, 3H, H5, H6, H7), 7.51–7.54 (d, 2H, H3′, H5′, J = 9 Hz), 7.94–7.97 (d, 2H, H2′, H6′, J = 9 Hz), 12.74 (S, 1H, NH). Anal. Calc. for C14H11ClN2O2S2: C: 49.69%; H: 3.27%; N: 8.27%. Found: C: 49.75%; H: 3.32%; N: 8.31%. m/z 338.
Synthesis of N-(5,6-dimethylbenzo[d]thiazol 2-yl)-4-fluorobenzosulfonamide (14)
Yield: 54.73%; m.p. 290–291 °C (EtOH) TLC: eluent. IR (KBr): ν = 1306, 1374 cm−1 (SO2), 3352 (NH) cm−1. 1H NMR (DMSO-d6, δ, ppm): 2.17 (s, 3H, 4′CH3), 2.31 (s, 6H, 5–6CH3), 7.69–7.53 (d, 6H, H4, H7, H2′, H3′, H5′, H6′), 12.23 (s, 1H, NH). Anal. Calc. for C16H16N2O2S2: C: 57.81%; H: 4.85%; N: 8.43%. Found: C: 57.60%; H: 4.95%; N: 8.53%. m/z 332.
CA inhibition
An Applied Photophysics stopped-flow instrument has been used for assaying the CA catalyzed CO2 hydration activity. Phenol red (at a concentration of 0.2 mM) has been used as an indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5) as a buffer, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The unanalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled–deionized water and dilutions up to 0.01 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 15 min to 72 h at room temperature (15 min) or 4 °C (all other incubation times) prior to assay, in order to allow for the formation of the E–I complex. Data from were obtained after 15 min incubation as there were no differences of inhibitory power when the enzyme and inhibitors were kept for longer periods in incubationCitation11. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3 and the Cheng–Prusoff equation, as reported earlierCitation12, and represent the mean from at least three different determinations. All CA isofoms were recombinant ones obtained in-house as reported earlierCitation9,13–15.
Table 1. Inhibition data of human CA isoforms hCA I, II, IX and XII with compounds 1–14 and the standard sulfonamide inhibitor AAZ.
Molecular modeling
Docking of compound 7
The crystal structure of CA IX (PDB code 3IAI) was taken from the Protein Data BankCitation16. The ligand was built using Maestro, version 9.0 (Schrödinger Inc., Portland, OR, 2009) and was subjected to a conformational search (CS) of 1000 steps, using a water environment model (generalized-Born/surface-area model) by means of Macromodel, version 9.7 (Schrödinger Inc.). The algorithm used was based on the Monte Carlo method with the MMFFs force field and a distance-dependent dielectric constant of 1.0. The ligand was then energy minimized using the conjugated gradient method until a convergence value of 0.05 kcal/Å mol was reached, using the same force field and parameters used for the CS. Automated docking was carried out by means of the AUTODOCK 4.0 programCitation17. AUTODOCK Tools utilitiesCitation18 were used in order to identify the torsion angles in the ligands, to add the solvent model and assign the Gasteiger atomic charges to protein and ligands. The regions of interest used by AUTODOCK were defined by considering the reference ligand as the central group of a grid box of 10 Å in the x, y and z directions. A grid spacing of 0.375 Å and a distance dependent function of the dielectric constant were used for the energetic map calculations. By using the Lamarckian genetic algorithm, the docked compounds were subjected to 200 runs of the AUTODOCK search using 2 500 000 steps of energy evaluation and the default values of the other parameters. The resulting docking poses were clusterized using a threshold of 2.0 Å and for each cluster one representative docking pose was subjected to MD simulation.
MD simulations
All simulations were performed using AMBER 14 (AMBER 2015, University of California, San Francisco, CA). MD simulations were carried out using the parm10 force field at 300 K. The complexes were placed in a rectangular parallelepiped water box. An explicit solvent model for water, TIP3P, was used, and the complexes were solvated with a 15 Å water cap. Sodium ions were added as counter ions to neutralize the system. Prior to MD simulations, two steps of energy minimization were carried out. In the first stage, we kept the protein and ligand fixed with a position restraint of 100 kcal/(mol Å2) and we just energy minimized the positions of the water molecules. In the second stage, we applied a restraint of 30 kcal/(mol Å2) only on the α carbons of the receptor. The two energy minimization stages consisted of 10 000 steps. The first 1000 steps were steepest descent, and the last 9000 were conjugate gradient. Molecular dynamics trajectories were run using the energy minimized structure as the input, and particle mesh Ewald electrostaticsCitation19 and periodic boundary conditions were used in the simulation. The time step of the simulations was 2.0 fs with a cutoff of 12 Å for the non-bonded interaction. SHAKE was employed to keep all bonds involving hydrogen atoms rigid. Constant-volume periodic boundary MD was carried out for 500 ps, during which the temperature was raised from 0 to 300 K. Then, 9.5 ns of constant pressure periodic boundary MD was carried out at 300 K using the Langevin thermostat to maintain constant the temperature of our system. General Amber force field parameters were assigned to the ligand, while partial charges were calculated using the AM1-BCC method as implemented in the Antechamber suite of AMBER 14.
Energy evaluation
We extracted from the last 8 ns of MD of the ligand–receptor complexes, 80 snapshots (at time intervals of 100 ps) for each species (complex, receptor and ligand). Electrostatic, van der Waals and internal energies were obtained using the SANDER module in AMBER 14. Polar energies were obtained from the PBSA module of the AMBER 14 program (using the Poisson–Boltzman method) applying dielectric constants of 1 and 80 to represent the gas and water phases, respectively. Nonpolar energies were determined using the MOLSURF program (San Francisco, CA).
Results and discussion
Chemistry
Fourteen compounds were designed based on 5-benzothiazole matrix and 5-arylsoulfonyl chlorides. Compounds were synthesized starting from 2-aminobenzothiazole-6-substituted derivatives, via a condensation with arylsulfonyl chlorides, in the presence of a catalytic amount of 4-dimethylamino-pyridine and trimethylamine (Scheme 1)Citation20. The reactions proceeded smoothly and in good yields for the majority of the compounds, in range 31–86%. Compounds were purified by recrystallization. The chemical structures of the synthesized compounds were satisfactory confirmed by their spectral data (IR, 1H NMR), as well as by elemental analysis. In IR spectra were observed absorptions at 1340–1370 cm−1 (C–SO2) due to participation in coordination with the aromatic ring, 1460 cm−1 (C=C arom.) as well as at 3360–3370 cm−1 (SO2–NH–). In the nuclear magnetic resonance spectra (1H NMR; d ppm), the signals of protons were confirmed on the bases of their chemical shifts and coupling constants.
Scheme 1. General synthetic scheme of compounds 1 –14.
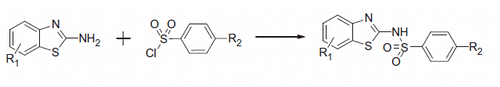
CA inhibition
All the compounds herein reported were tested by a stopped flow carbon dioxide hydration assay for their in vitro inhibitory activities against four physiological relevant hCA isoforms such as the cytosolic I, II, and the membrane bound IX and XII.
The inhibition data of benzothiazole-based sulfonamides 1–14 and the sulfonamide acetazolamide (AAZ) (as a standard inhibitor) against hCA I, II, IX and XII are presented in . Isoform hCA I was moderately or poorly inhibited by these derivatives. Six compounds (7–10, 13, 14) showed KIs in the range of 3.6–40.2 μM, whereas the remaining ones did not inhibit significantly the enzyme up until 50 μM (). Isoform II was poorly inhibited by compounds 2, 3, 5–8, 10, 13 in the micromolar range, whereas the other derivatives did not inhibit significantly the enzyme. The transmembrane isoform hCA IX was effectively inhibited in the low micromolar range by almost all derivatives reported here, with KIs in the range of 0.078–0.95 μM. And finally, a similar situation was also observed for the inhibition of hCA XII, with almost all analyzed compounds that showed effective inhibitory action, with KIs in the range of 0.064–0.85 μM ().
Molecular modeling studies
In order to identify the possible binding mode of these analogs, the interaction of compound 7 into CA IX was evaluated by means of docking and molecular dynamic (MD) simulations. As shown in the docking results suggested six possible binding modes; the binding mode D suggested a coordination of one of the two sulfonamide oxygens of 7 with the catalytic zinc ion, whereas for all the other predicted binding modes there was no direct interactions between the ligand and the zinc ion. However, in all cases the compound showed H-bonds and lipophilic interactions.
Figure 1. Possible binding dispositions of 7 into CA IX obtained through docking calculations.
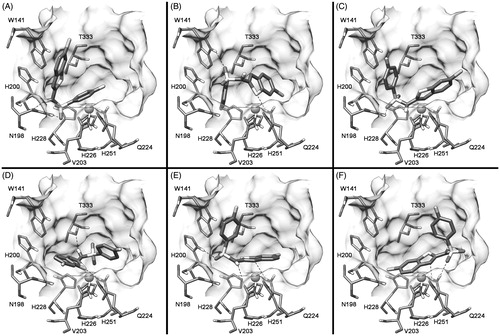
In order to further analyze the stability of these possible binding modes, the six complexes were then subjected to 10 ns of MD simulation with explicit water molecules, as described in the “Methods” section. Analyzing the root-mean-square deviation (RMSD) of the heavy atoms of the ligand during the different MD simulations, it was observed that some binding modes (A, C and F) were unstable, as for these MD trajectories the compound after about 2 ns moved away from the binding site cavity. On the contrary, as shown in , the binding modes B, D and E were stable and where thus further taken into account.
Figure 2. Analysis of the CA IX-7 MD simulations. The plots show the RMSD of the ligand heavy atoms from the starting model structures during the simulations.
The three stable CA IX-7 complexes (binding modes B, D and E, respectively) were further analyzed through the MM-PBSA methodCitation21, which proved to accurately estimate the ligand–receptor energy interactionCitation22,Citation23. This approach averages the contributions of gas phase energies and solvation free energies calculated for snapshots of the complex molecule as well as the unbound components extracted from MD trajectories, according to the procedure fully described in the “Methods” section. The MM-PBSA results () suggested that the binding mode E was the most reliable, as it was more stable of about 7 kcal mol−1 with respect to the other two binding modes.
Table 2. MM-PBSA results for the CA IX-7 complexes. Data are expressed as kcal mol−1.
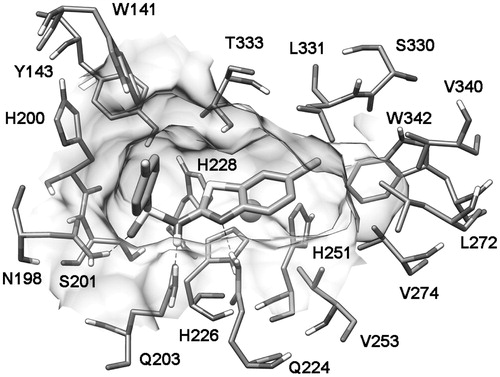
shows the average minimized structure of the CA IX-7 complex obtained from the binding mode E MD simulation. The N-benzothiazolylsulfonamide part of the molecule showed three H-bonds with the side chains of N198, Q203 and Q224; the p-fluoro phenyl group was directed toward the entrance of the binding site cavity, whereas the 6-chloro benzothiazolyl scaffold was inserted inside the binding site cavity and showed lipophilic interactions with V253, V274, L331 and W342.
Figure 3. Minimized average structures of compound 7 docked into CA IX.
Conclusions
A series of benzothiazole-based sulfonamides were prepared and the obtained 14 new compounds were assayed for the inhibition of the cytosolic, widespread human hCA I and II isoforms, and the trans-membrane, tumor-associated ones hCA IX and XII. Most of the benzothiazole-based sulfonamides did not inhibit significantly hCA I and II, but showed nanomolar inhibitory action against the transmembrane isoforms. Since many hypoxic tumors over-express hCA IX/XII, the present findings constitute an interesting extension to the knowledge of selective inhibitors of CA isoforms involved in serious pathologies.
Declaration of interest
The authors report no declarations of interest. Emine Terzi is a recipient of a TUBITAK 2228-A MSc grant.
Related Research Data
References
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77.
- Supuran CT, Scozzafava A. Carbonic anhydrases as targets for medicinal chemistry. Bioorg Med Chem 2007;15:4336–50.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81.
- Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2010;20:3467–74.
- Supuran CT. Carbonic anhydrase inhibitors and activators for novel therapeutic applications. Future Med Chem 2011;3:1165–80.
- Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68.
- Thiry A, Dogne JM, Masereel B, Supuran CT. Targeting tumor-associated carbonic anhydrase IX in cancer therapy. Trends Pharmacol Sci 2006;27:566–73.
- Tuccinardi T, Ortore G, Rossello A, et al. Homology modeling and receptor-based 3D-QSAR study of carbonic anhydrase IX. J Chem Inf Model 2007;47:2253–62.
- Vullo D, Innocenti A, Nishimori I, et al. Carbonic anhydrase inhibitors. Inhibition of the transmembrane isozyme XII with sulfonamides – a new target for the design of antitumor and antiglaucoma drugs? Bioorg Med Chem Lett 2005;15:963–9.
- Masini E, Carta F, Scozzafava A, Supuran CT. Antiglaucoma carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Pat 2013;23:705–16.
- Maresca A, Temperini C, Vu H, et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–62.
- Temperini C, Innocenti A, Scozzafava A, et al. The coumarin-binding site in carbonic anhydrase accommodates structurally diverse inhibitors: the antiepileptic lacosamide as an example and lead molecule for novel classes of carbonic anhydrase inhibitors. J Med Chem 2010;53:850–4.
- Alterio V, Hilvo M, Di Fiore A, et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc Natl Acad Sci USA 2009;106:16233–8.
- Casey JR, Morgan PE, Vullo D, et al. Carbonic anhydrase inhibitors. Design of selective, membrane-impermeant inhibitors targeting the human tumor-associated isozyme IX. J Med Chem 2004;47:2337–47.
- Casini A, Scozzafava A, Mincione F, et al. Carbonic anhydrase inhibitors: water-soluble 4-sulfamoylphenylthioureas as topical intraocular pressure-lowering agents with long-lasting effects. J Med Chem 2000;43:4884–92.
- Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res 2000;28:235–42.
- Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 2009;30:2785–91.
- Sanner MF. Python: a programming language for software integration and development. J Mol Graph Model 1999;17:57–61.
- Essmann U, Perera L, Berkowitz ML, et al. A smooth particle mesh Ewald method. J Chem Phys 1995;103:8577–93.
- Moreno-Diaz H, Villalobos-Molina R, Ortiz-Andrade R, et al. Antidiabetic activity of N-(6-substituted-1,3-benzothiazol-2-yl)benzenesulfonamides. Bioorg Med Chem Lett 2008;18:2871–7.
- Kollman PA, Massova I, Reyes C, et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res 2000;33:889–97.
- Tuccinardi T, Manetti F, Schenone S, et al. Construction and validation of a RET TK catalytic domain by homology modeling. J Chem Inf Model 2007;47:644–55.
- Tuccinardi T, Granchi C, Iegre J, et al. Oxime-based inhibitors of glucose transporter 1 displaying antiproliferative effects in cancer cells. Bioorg Med Chem Lett 2013;23:6923–7.